

Abstract
Using comparative genomic hybridisation, we have analysed genetic imbalance in a series of 86 ependymomas from children and adults. Tumours were derived from intracranial and spinal sites, and classified histologically as classic, anaplastic or myxopapillary. Ependymomas showing a balanced profile were significantly (P<0.0005) more frequent in children than adults. Profiles suggesting intermediate ploidy were common (44% of all tumours), and found more often (P<0.0005) in tumours from adults and the spinal region. Loss of 22q was the most common specific abnormality, occurring in 50% of spinal (medullary) ependymomas and 26% of tumours overall. Genetic profiles combining loss of 22q with other specific abnormalities – gain of 1q, loss of 6q, loss of 10q/10, loss of 13, loss of 14q/14 – varied according to site and histology. In particular, we showed that classic ependymomas from within the cranium and spine have distinct genetic profiles. Classic and anaplastic ependymomas with gain of 1q tended to occur in the posterior fossa of children and to behave aggressively. Our extensive data on ependymomas demonstrate significant associations between genetic aberrations and clinicopathological variables, and represent a starting point for further biological and clinical studies.
British Journal of Cancer (2002) 86, 929–939. DOI: 10.1038/sj/bjc/6600180 www.bjcancer.com
© 2002 Cancer Research UK
Keywords: ependymoma, CGH, gain 1q, loss 22, intermediate ploidy
Ependymomas are gliomas that exhibit degrees of ependymal differentiation (Burger and Scheithauer, 1995; Ellison, 1998). Typically, they develop in relation to the ventricular system and the cauda equina. They account for only 4–8% of gliomas, but are the third most common central nervous system (CNS) tumour of childhood, after astrocytomas and medulloblastomas. About 90% of paediatric ependymomas are intracranial, but in adults most are intraspinal (Gilles et al, 1995; Hamilton and Pollack, 1997; Bouffet et al, 1998; Gjerris et al, 1998; Wiestler et al, 2000). The World Health Organisation (WHO) pathological classification (Wiestler et al, 2000) recognises classic (WHO grade 2), anaplastic (grade 3) and myxopapillary (grade 1) variants, plus the subependymoma (grade 1). Classic and anaplastic variants mainly occur as posterior fossa tumours in children and young adults, and myxopapillary tumours nearly always present in the cauda equina of adults (Hamilton and Pollack, 1997; Ellison, 1998; Packer, 2000).
In children with intracranial ependymoma, event-free survival after 5 years is less than 50% (Bouffet et al, 1998; Robertson et al, 1998; Horn et al, 1999; Packer, 2000; Grill et al, 2001). Various factors have been reported to influence prognosis, though clinical research in this area has produced many conflicting results. However, gross surgical resection and the use of radiotherapy have been consistently associated with enhanced event-free and overall survival (Nazar et al, 1990; Sutton et al, 1990; Healey et al, 1991; Vanuytsel et al, 1992; Ferrante et al, 1994; Pollack et al, 1995; Bouffet et al, 1998; Robertson et al, 1998; Horn et al, 1999; Grill et al, 2001). Histological features of anaplasia, such as mitoses, microvascular proliferation and necrosis, serve as indicators of biological behaviour in other gliomas, including diffuse astrocytic tumours and oligodendrogliomas (Cohadon et al, 1985; Burger and Green, 1987; Ellison, 1998). However, the biological significance of these morphological features in ependymoma remains unclear; clinicopathological studies have provided conflicting evidence on the prognostic value of dividing ependymomas into classic (WHO grade 2) and anaplastic (grade 3) variants (Ross and Rubinstein, 1989; Sutton et al, 1990; Schiffer et al, 1991; Gerszten et al, 1996; Bouffet et al, 1998; Figarella-Branger et al, 2000).
With very few definite clinical or pathological markers of biological behaviour, the identification of genetic abnormalities responsible for the generation and maintenance of the malignant phenotype in ependymomas will be crucial, if there are improvements to be made in the management of patients with this disease. Not only may such defects serve as markers of aggressive disease, allowing more efficient use of existing therapies, but they may also suggest biological targets for novel therapeutic approaches. Cytogenetic studies have shown that chromosomal abnormalities are relatively common in ependymomas (Stratton et al, 1989; Neumann et al, 1993; Hamilton and Pollack, 1997; Mazewski et al, 1999; Vagner-Capodano et al, 1999). However, there are few large studies of genetic abnormalities in this disease, and their role in tumour behaviour therefore remains unclear.
We have employed comparative genomic hybridisation (CGH) to study tumours from a large population of patients with ependymoma. Specifically, our aims were to identify associations between patterns of genetic imbalance and the principal histopathological variants of ependymomas (classic/anaplastic and myxopapillary), which occur at distinct sites in adults and children. Such findings may enable future research to be targeted at genetic markers with potential prognostic significance.
MATERIALS AND METHODS
Tumour samples
Tumours (n=86) came from patients (n=77) treated in three regional UK neurosurgical units (Newcastle, Nottingham, Southampton, UK) between 1985 and 1999. Eight patients contributed more than one sample (seven patients – two samples, one patient – three samples) from consecutive resections of their tumour. In 16 other cases, the single sample analysed was not from the first tumour resection. In all three centres, a portion of each tumour was snap frozen and stored in liquid nitrogen, while the remainder was fixed in buffered formalin for histological examination. Frozen tumour samples were transported on dry ice to a single institution (Wessex Regional Genetics Laboratory) for CGH analysis.
Surgical resection had been the primary therapy in all cases, although most patients had received adjuvant chemotherapy and/or radiotherapy at some stage (Table 1). The male : female ratio was 1 : 1. Across the entire group, age at presentation ranged from 8 months to 69 years, with a median of 29 years (Table 1). Adults (median age: 39 years) contributed 58 samples, and children (<16 years; median age: 6 years) contributed 28 samples. The primary site for tumours was: supratentorial – 10 (13%), posterior fossa – 31 (40%), spinal cord – 14 (18%), cauda equina – 22 (29%). All tumours from children were intracranial, except one myxopapillary example, which presented in the filum terminale of a boy aged 15 years. No patients with spinal tumours had metastatic disease at presentation, but three intracranial tumours were found, during staging investigations, to be associated with metastases in the CSF pathways.
Table 1. CGH data for adult intracranial ependymomas.
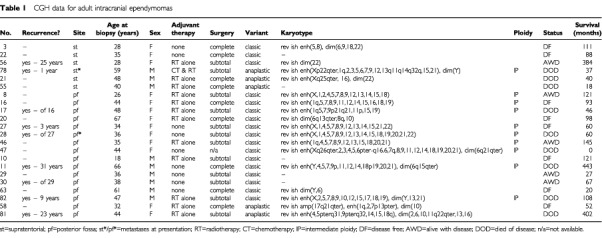
Histological assessment of each tumour was undertaken by two neuropathologists (R Allibone and D Ellison), using criteria from the WHO classification of central nervous system tumours (Wiestler et al, 2000); of a total of 86 tumours, 44 (51%) were designated classic ependymomas, 24 (28%) anaplastic and 18 (21%) myxopapillary. All frozen tumour samples for CGH analysis were examined histologically to confirm the neoplastic nature of at least 80% of the tissue.
Comparative genomic hybridisation
CGH was performed on frozen tumour tissue using an indirect technique modified from those reported by Kallioniemi et al (1994) and our own group (Nicholson et al, 1999). Genomic DNA was labelled by nick translation with biotin, and sex-matched control DNA with digoxigenin (both from Roche Diagnostics Ltd). Probe mixtures containing 2 μg of labelled tumour and sex matched control DNA were hybridised with 50 μg Cot 1 DNA (Gibco–BRL) and 20 μg herring sperm carrier (Sigma) at 37°C for 3 days to sex-matched normal target lymphocyte metaphase spreads prepared in the Wessex Regional Genetics Laboratory. Biotin-labelled probes were detected with two rounds of avidin-FITC interspersed with one of biotinylated anti-avidin (Vector Laboratories) and digoxigenin-labelled probes were detected with mouse anti-digoxigenin, rabbit anti-mouse, and finally goat anti-rabbit TRITC (Sigma). Labelled detected metaphases were then mounted in antifade solution containing 1.5 μg ml−1 4,6′-diamino-2-phenylindole (DAPI) counterstain (Vector laboratories). Each CGH experiment included a control hybridisation using normal DNA.
Image capture and CGH analysis
Hybridisations were viewed under a fluorescence microscope and three colour images were captured by a cooled charged couple device camera (Photometrics), in conjunction with Macprobe 4.1 software (Perceptive Scientific International Ltd, Chester, UK). Metaphases were karyotyped and green : red fluorescence intensity ratios calculated along the length of each chromosome. Mean ratio profiles, together with profiles corresponding to ±1 standard deviation (s.d.), were constructed from 5–10 metaphases per tumour. Gains or losses of material in the tumour were inferred by deviation of the mean ratios beyond thresholds set at 1.15 and 0.85 respectively, providing the s.d. profile deviated to the same side of the midline. Selected cases with apparent imbalance on chromosomes 19 and/or 22 were chosen for CGH experiments with reverse labelling, because of concerns about artefacts in these regions. All abnormal results were confirmed by these experiments, using tumour DNA labelled with digoxigenin and controls with biotin. Gains associated with ratios greater than 1.5 were interpreted as amplifications.
Fluorescence in situ hybridisation (FISH)
In a number of tumours the pattern of loss and gain was suggestive of intermediate ploidy (Rosenberg et al, 1997). This term is used where an increased copy number applies to some, but not all, chromosomes, and overall ploidy lies between one level and the next (usually diploidy and triploidy). Fluorescence in situ hybridisation (FISH) was performed on extracted nuclei from selected cases to confirm this. Loss/gain thresholds were then skewed to normalise for individual diploid chromosomes and the results interpreted with respect to the diploid state.
The FISH methodology allows an accurate and reproducible analysis of formalin fixed, paraffin wax embedded tumour material, and has been described in detail elsewhere (Nicholson et al, 2000). Briefly, cytospin preparations of nuclei were produced from sections (2–3×15 μm) of tumour. Digoxigenin-labelled or biotin-labelled plasmid probes to the centromeric regions of chromosomes 4, 6, 7, 8, 10, and 17 were applied singly or in pairs to preparations of nuclei. Probe hybridisation was conducted overnight at 37°C in a humidified chamber followed by stringency washes at 43°C using 0.1–0.5×SSC plus 30% formamide to remove non-specifically bound probe. Probes were visualised using a mixture of Texas red-labelled avidin and FITC-conjugated anti-digoxigenin antibody.
Data analysis
Relationships among variables were assessed using standard statistical techniques: 2×2 contingency tables (Fisher's exact test), Log Rank analysis to produce Kaplan–Meier survival curves, and multivariate Cox analysis. Survival analysis of patients with spinal tumours was impossible because three of only five patients in the uncensored category had died in the immediate post-operative period.
RESULTS
The results of CGH analysis are detailed by age/site in Tables 1, 2, 3, 4, alongside clinical parameters, and by histology in Figure 1. Balanced karyotypes were particularly prevalent among childhood tumours (P<0.0005), and were not found in myxopapillary variants. Ependymomas showing imbalance had a mean of six abnormalities. The progressive acquisition of genetic abnormalities was demonstrated by only one of eight ependymomas for which successive biopsies were available. This tumour acquired gain of 1q and loss of 16q, having shown a balanced profile at first surgery. This change accompanied progression of the tumour's histological features from classic to anaplastic.
Table 2. CGH data for childhood intracranial ependymomas.
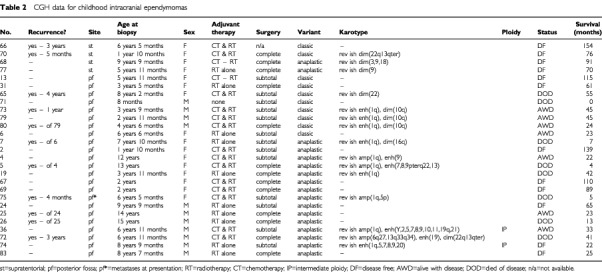
Table 3. CGH data for spinal classic and anaplastic ependymomas.
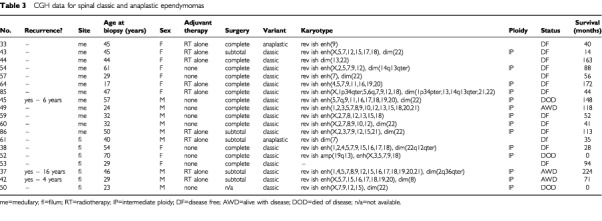
Table 4. CGH data for spinal myxopapillary ependymomas.
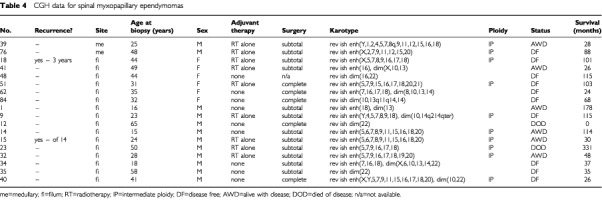
Figure 1.

CGH ideograms divided according to histological variant/site: (A) Intracranial classic tumours; (B) Spinal classic tumours; (C) Anaplastic tumours; (D) Myxopapillary tumours. Loss and gain bars are on the left and right sides of each chromosome respectively.
A high proportion of tumours (44%) demonstrated particular patterns of gain, and occasionally loss, across multiple chromosomes that were highly suggestive of intermediate ploidy. This interpretation was confirmed by FISH in a number of representative cases (Figure 2). Certain chromosomes, 2/5/7/9/12/15/18/X, were commonly involved in these patterns. Such aberrations were more frequent in myxopapillary and classic tumours than in anaplastic variants (P=0.0199), in spinal relative to intracranial ependymomas (P<0.0005), and in tumours from adults vs children (P<0.0005).
Figure 2.

CGH profiles and FISH from case 38. Standard thresholds (A) imply a severely hypodiploid karyotype, but none of the profiles follows the mid-line. This is characteristic of intermediate ploidy cases, and FISH with centromere probes (B) confirms that there were two copies of chromosome 8 (green) in all cells and three copies of chromosome 17 (red) in many. Skewing the midline to the left to give a chromosome 8 profile midway between the gain and loss thresholds (i.e. normal) produces a profile (C) that implies gain of a large number of chromosomes, although distal 22q is still clearly lost. By this interpretation chromosomes 4 and 16 could have two extra copies compared to a single extra copy of the other gained chromosomes, or could be gained in all cells while a smaller proportion of cells have the other gains.
The acquisition of certain genetic abnormalities: gain 1q, loss 6q, loss 10q/10, loss 13, loss14q/14 and loss 22 appeared distinct from the gains and losses of whole chromosomes that reflect intermediate ploidy, and the characteristics of tumours with these abnormalities were analysed. Loss of 22 was the commonest abnormality, detected in 20 tumours (26%). This change was found in all histological variants, though relatively less in the anaplastic tumours. However, loss of 22 was significantly (P=0.0154) associated with a spinal rather than an intracranial location. Gain of 1q and loss of 6q were particular features of classic/anaplastic ependymomas of the posterior fossa. Nearly all (11 of 13) tumours showing gain of 1q had come from the posterior fossa, and eight of the 13 were designated as anaplastic. Of all the posterior fossa anaplastic ependymomas with gain of 1q (n=8), all but one was from a child. All tumours with loss of 6q were from the posterior fossa. In contrast, seven of nine ependymomas showing loss of 13 and all showing loss of 14q/14 were spinal. Myxopapillary ependymomas were associated with both loss of 13 and loss of 14q/14. Loss of 10q/10 was present across histological variants, but significantly (P=0.0192) more often in myxopapillary tumours. All of the spinal tumours with losses on chromosome 10 were myxopapillary (P=0.006).
From data on the selected genetic abnormalities above, we tested the hypothesis that non-myxopapillary spinal ependymomas have different genetic profiles from those of myxopapillary and intracranial ependymomas. We compared the profiles of classic ependymomas from intracranial and spinal sites (Figure 1A,B), finding significant differences (P<0.0001). Both adults and children contributed intracranial tumours to this cohort (to ensure adequate numbers for the analysis), but all spinal tumours in this analysis were from adults (only one spinal tumour in the series was from a child). We also compared the profiles of classic plus anaplastic ependymomas from intracranial and spinal sites, again finding distinct profiles (P<0.0001). All tumours in this cohort were from adults. Finally, our analysis of spinal ependymomas from adult patients revealed significantly different profiles for classic and myxopapillary ependymomas (P<0.0001).
High level gain (CGH ratio >1.5 : 1) was infrequent, and mostly implied gain of several copies of the whole, or most of, a chromosome arm. It occurred in only six tumours, five of which were anaplastic (Table 1). Half of these tumours, showing gain of 1q, were from children. Single, focal high level gains were located at 6q27 and 13q33q34 in one child's tumour, and at 17q21qter and 19q13 in the remaining two tumours.
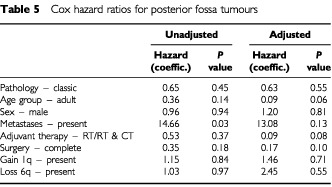
Because of the association between gain of 1q and anaplastic ependymomas, we examined the effect of histological diagnosis and gain of 1q on the survival of patients with intracranial tumours. Survival curves comparing intracranial tumours split into classic and anaplastic groups and those comparing intracranial tumours with and without gain of 1q showed clear differences (Figures 3 and 4), and the difference (Figure 5) between patients with anaplastic ependymomas showing gain of 1q and others with intracranial tumours was even more significant (P=0.0032). However, when the effect of multiple variables on the survival of patients with posterior fossa tumours was analysed (Cox proportional hazard ratios), trends towards a poor outcome were seen for children rather than adults, presence of metastases, no adjuvant therapy, and subtotal surgical resection in multivariate analyses (Table 5), but none of these effects reached statistical significance.
Figure 3.
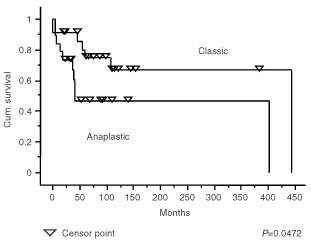
Kaplan–Meier plots showing a significant difference (P=0.0472) in the survival of patients with intracranial classic ependymomas and intracranial anaplastic ependymomas.
Figure 4.
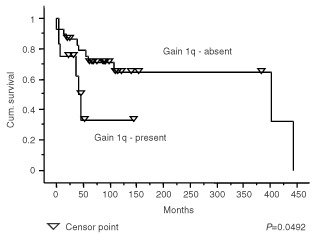
Kaplan–Meier plots showing a significant difference (P=0.0492) in the survival of patients with intracranial ependymomas with and without gain of 1q.
Figure 5.
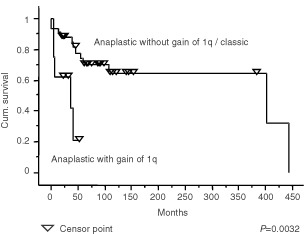
Kaplan–Meier plots showing a significant difference (P=0.0032) in the survival of patients with intracranial anaplastic ependymomas with gain of 1q and patients with intracranial classic ependymomas or anaplastic ependymomas without gain of 1q.
Table 5. Cox hazard ratios for posterior fossa tumours.
DISCUSSION
This is the largest CGH study of ependymomas to be reported so far, and demonstrates that particular genetic profiles in these tumours reflect the principal division in their biology; specifically, that classic and anaplastic ependymomas occur mainly in the posterior fossa of children and young adults, and the myxopapillary variant occurs mainly in the region of the cauda equina in adults (Hamilton and Pollack, 1997; Ellison, 1998). In addition, we provide evidence to support the proposal that classic (WHO grade 2) ependymomas from the region of the spinal cord are genetically distinct from intracranial classic (grade 2) and anaplastic (grade 3) tumours (Ebert et al, 1999; Hirose et al, 2001).
Many previous cytogenetic or CGH studies of ependymomas have targeted tumours in children, and this may account for the paucity of genetic data on myxopapillary ependymomas; only one child from our series, a boy aged 15 years, presented with a myxopapillary spinal tumour. The consensus from previous studies is that about 40% of childhood ependymomas show no chromosomal imbalances (Reardon et al, 1999; Ward et al, 2001). This mirrors our data, which show a balanced chromosomal profile in 41% of childhood ependymomas, in contrast to only 9% of adult tumours. In this respect, a distinction is also seen between intracranial and spinal tumours; balanced profiles are evident in 32% of intracranial ependymomas, but in only one spinal tumour (3%). Another recent CGH study demonstrated balanced profiles in 21% of intracranial ependymomas, but none of the spinal tumours (Hirose et al, 2001). A common pattern of abnormalities across spinal (64%) and adult (56%) tumours in our study is gain across multiple whole chromosomes. This pattern suggests intermediate ploidy, and we confirmed this in a number of tumours using FISH. Such widespread imbalance was shown by the only myxopapillary ependymoma in one previous CGH study (Reardon et al, 1999), and by many spinal tumours in another (Hirose et al, 2001). Overall, the data indicate that spinal ependymomas, which present almost entirely in adult patients, and intracranial childhood tumours differ significantly in their genetic profiles. The former is characterised by widespread copy number aberrations, generally without evidence of rearrangement, and the latter by restricted specific gains or losses. The corollary of this is that low-grade tumours, which in practice are easier to treat, have more widespread abnormalities than the high-grade tumours, a situation that also pertains to neuroblastomas (Plantaz et al, 1997).
Loss of 22q has been the commonest abnormality in several genetic studies of ependymoma (Ransom et al, 1992; Mazewski et al, 1999; Vagner-Capodano et al, 1999), though gain of 1q or loss of 6q assumes predominance in others (Reardon et al, 1999; Ward et al, 2001). If the changes suggestive of intermediate ploidy are discounted, loss of 22q is the most frequent genetic abnormality in our series of ependymomas, occurring in 26% of ependymomas, which is in line with data from other CGH studies (Reardon et al, 1999; Hirose et al, 2001; Ward et al, 2001). However, the frequency of loss of 22q in ependymomas varies greatly (up to 71%) in previous studies that have used a variety of methods (Ransom et al, 1992; Neumann et al, 1993; Kramer et al, 1998; Vagner-Capodano et al, 1999; Zheng et al, 2000). This variability is likely to reflect ascertainment bias, because the frequency of loss of 22 in ependymomas varies according to histological variant, anatomic site, and age of the patient. The discrepancy between its occurrence in adults and children has been recorded in a large cytogenetic series (Mazewski et al, 1999), and several lines of evidence link loss of 22 to spinal ependymomas. There is an increasing susceptibility to gliomas in patients with neurofibromatosis type 2 (NF2), the gene for which, NF2, is at 22q12 (Wiestler et al, 2000). Most gliomas in NF2 (80%) are ependymomas, and three quarters of NF2 gliomas are spinal (Rodriguez and Berthrong, 1966). Mutations in the NF2 gene are uncommon in sporadic ependymomas (von Haken et al, 1996), and appear to be restricted to spinal tumours. In a study of 62 ependymomas (Ebert et al, 1999), twelve showed allelic loss on 22q and six of them also had NF2 mutations. All six ependymomas with mutations were classic variants from around the spinal cord.
Gain of 1q is a relatively frequent abnormality in ependymomas, and has been emphasised in CGH studies of paediatric tumours, where it has been reported in up to 22% of cases (Reardon et al, 1999). In our series, ependymomas with gain of 1q represent 17% of the total, but are significantly associated with childhood, posterior fossa location, and anaplastic histological features. Evidence for several extra copies of 1q is found in three of the tumours in our series, all anaplastic ependymomas from children. A similar phenomenon was reported in the study by Ward et al (2001), who also described three tumours with a combination of gain of 1q and loss of 16q as the only abnormalities. We found this combination once, in the anaplastic recurrence of a classic ependymoma that originally showed a balanced CGH profile. This combination strongly suggests a der(16)t(1;16), which is a well recognised secondary chromosomal change in a variety of tumours. This is the only instance of a change in genetic profile between two successive biopsies. Gain of 1q and loss of 10q as sole abnormalities occur twice in our cohort of intracranial tumours, both times in anaplastic ependymomas from children, and in one anaplastic tumour with other genetic abnormalities, which included amplification of 17q21qter. Further investigation is required to determine the significance of the association between gain of 1q and loss of 10q or loss of 16q in ependymomas. However, this phenomenon appears to occur in anaplastic childhood tumours with an aggressive behaviour. The association between gain of 1q and anaplastic ependymomas prompted us to look at the survival of patients with posterior fossa tumours divided according to histological variant and the presence of 1q. While numbers of tumours in the analysis are small, the designation of a tumour as anaplastic in the presence of gain of 1q was associated with a significantly worse survival curve. However, it is important to note that these variables were not significant prognostic indicators in a multivariate analysis, and this may reflect the need for a greater number of patients in such analyses.
Loss of 6q has been found in ependymomas with variable frequency (Reardon et al, 1999; Hirose et al, 2001; Ward et al, 2001). We demonstrate an overall frequency of 6%, matching the results of Ward et al (2001), but this is lower than results of up to 22% in some CGH/cytogenetic studies (Reardon et al, 1999). Again, the analysis of small numbers of patients, or of patients with tumours from a certain site, may account for this. For example, loss of 6q in our study attained its greatest frequency (26%) in adults with posterior fossa ependymomas, which was also a feature of another CGH study (Zheng et al, 2000).
Abnormalities of chromosomes 10 have featured prominently in the study of genetic abnormalities in gliomas. Loss of part or most of chromosome 10 is common in glioblastomas, and many of them also harbour mutations of the PTEN tumour suppressor gene at 10q23 (Bostrom et al, 1998; Duerr et al, 1998). Abnormalities of chromosome 10 appear less commonly in ependymomas, but among spinal tumours in our series clearly differentiate classic tumours from myxopapillary tumours. Microsatellite analysis has shown loss of 10q to be uncommon in ependymomas, and no mutation of the PTEN gene has been reported (Duerr et al, 1998; Ebert et al, 1999; Tong et al, 1999).
In summary, our study provides genetic data on a large range of ependymomas. We show that distinct genetic profiles characterise intracranial and spinal tumours, and tumours categorised by histological variant. In particular, we demonstrate that classic ependymomas from intracranial and spinal sites should be distinguished on the basis of genetic information. While our data suggest that gain of 1q is a possible marker for aggressive biological behaviour, further research is indicated to define clinically useful ways to incorporate histological and genetic assessments in the classification of ependymomas.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Miss Sarah Beal of the Wessex Regional Genetics Laboratory, Salisbury, UK, and Mr Phil Steart and Mrs Jean Buontempo of the Department of Cellular Pathology, Southampton General Hospital, UK. We have also benefited from the support of neurosurgeons at all three centres, particularly Mr Jonathan Punt, Consultant Neurosurgeon, Nottingham. This work has been supported in part by grants from the Wessex Cancer Trust, the Wessex Medical Trust, and the Wolfson Foundation.
References
- BostromJCobbersJMWolterMTabatabaiGWeberRGLichterPCollinsVPReifenbergerG1998Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q Cancer Res 582933 [PubMed] [Google Scholar]
- BouffetEPerilongoGCaneteAMassiminoM1998Intracranial ependymomas in children: a critical review of prognostic factors and a plea for cooperation Med Pediatr Oncol 30319–329; discussion329331 [DOI] [PubMed] [Google Scholar]
- BurgerPCGreenSB1987Patient age, histologic features, and length of survival in patients with glioblastoma multiforme Cancer 5916171625 [DOI] [PubMed] [Google Scholar]
- BurgerPCScheithauerBW1995Tumours of the Central Nervous System Vol 10Atlas of Tumour PathologyArmed Forces Institute of Pathology [Google Scholar]
- CohadonFAouadNRougierAVitalCRivelJDartiguesJF1985Histologic and non-histologic factors correlated with survival time in supratentorial astrocytic tumors J Neuro-oncol 3105111 [DOI] [PubMed] [Google Scholar]
- DuerrEMRollbrockerBHayashiYPetersNMeyer-PuttlitzBLouisDNSchrammJWiestlerODParsonsREngCvon DeimlingA1998PTEN mutations in gliomas and glioneuronal tumors Oncogene 1622592264 [DOI] [PubMed] [Google Scholar]
- EbertCvon HakenMMeyer-PuttlitzBWiestlerODReifenbergerGPietschTvon DeimlingA1999Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas Am J Pathol 155627632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EllisonDW1998Non-astrocytic gliomasInNeuropathologyEllison DW, Love S (eds)Chapter 36London: Mosby [Google Scholar]
- FerranteLMastronardiLSchettiniGLunardiPFortunaA1994Fourth ventricle ependymomas. A study of 20 cases with survival analysis Acta Neurochirurgica 1316774 [DOI] [PubMed] [Google Scholar]
- Figarella-BrangerDCivatteMBouvier-LabitCGouvernetJGambarelliDGentetJCLenaGChouxMPellissierJF2000Prognostic factors in intracranial ependymomas in children J Neurosurg 93605613 [DOI] [PubMed] [Google Scholar]
- GersztenPCPollackIFMartinezAJLoKHJanoskyJAlbrightAL1996Intracranial ependymomas of childhood. Lack of correlation of histopathology and clinical outcome Pathol Res Pract 192515522 [DOI] [PubMed] [Google Scholar]
- GillesFHSobelELTavareCJLevitonAHedley-WhiteET1995Age-related changes in diagnoses, histological features, and survival in children with brain tumors: 1930–1979. The Childhood Brain Tumor Consortium Neurosurgery 3710561068 [DOI] [PubMed] [Google Scholar]
- GjerrisFAgerlinNBorgesenSEBuhlLHaaseJKlinkenLMortensenACOlsenJHOvesenNReske NielsenESchmidtK1998Epidemiology and prognosis in children treated for intracranial tumours in Denmark 1960–1984 Childs Nerv Syst 14302311 [DOI] [PubMed] [Google Scholar]
- GrillJLe DeleyMCGambarelliDRaquinMACouanetDPierre-KahnAHabrandJLDozFFrappazDGentetJCEdanCChastagnerPKalifaC2001Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology J Clin Oncol 1912881296 [DOI] [PubMed] [Google Scholar]
- HamiltonRLPollackIF1997The molecular biology of ependymomas Brain Path 7807822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HealeyEABarnesPDKupskyWJScottRMSallanSEBlackPMTarbellNJ1991The prognostic significance of postoperative residual tumor in ependymoma Neurosurgery 28666672 [DOI] [PubMed] [Google Scholar]
- HiroseYAldapeKBollenAJamesCDBratDLambornKBergerMFeuersteinBG2001Chromosomal abnormalities subdivide ependymal tumors into clinically relevant groups Am J Pathol 15811371143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HornBHeidemanRGeyerRPollackIPackerRGoldweinJTomitaTSchombergPAterJLuchtman-JonesLRivlinKLambornKPradosMBollenABergerMDahlGMcNeilEPattersonKShawDKubalikMRussoC1999A multi-institutional retrospective study of intracranial ependymoma in children: identification of risk factors J Pediatr Hematol Oncol 21203211 [DOI] [PubMed] [Google Scholar]
- KallioniemiOPKallioniemiAPiperJIsolaJWaldmanFMGrayJWPinkelD1994Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors Genes Chrom Cancer 10231243 [DOI] [PubMed] [Google Scholar]
- KramerDLParmiterAHRorkeLBSuttonLNBiegelJA1998Molecular cytogenetic studies of pediatric ependymomas J Neurooncol 372533 [DOI] [PubMed] [Google Scholar]
- MazewskiCSoukupSBallardEGotwalsBLampkinB1999Karyotype studies in 18 ependymomas with literature review of 107 cases Cancer Genet Cytogenet 11318 [DOI] [PubMed] [Google Scholar]
- NazarGBHoffmanHJBeckerLEJenkinDHumphreysRPHendrickEB1990Infratentorial ependymomas in childhood: prognostic factors and treatment J Neurosurg 72408417 [DOI] [PubMed] [Google Scholar]
- NeumannEKalousekDKNormanMGSteinbokPCochraneDDGoddardK1993Cytogenetic analysis of 109 pediatric central nervous system tumors Cancer Genet Cytogenet 714049 [DOI] [PubMed] [Google Scholar]
- NicholsonJWickramasingheCRossFCrollaJEllisonD2000Imbalances of chromosome 17 in medulloblastomas determined by comparative genomic hybridisation and fluorescence in situ hybridisation Mol Pathol 53313319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NicholsonJCRossFMKohlerJAEllisonDW1999Comparative genomic hybridization and histological variation in primitive neuroectodermal tumours Br J Cancer 8013221331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PackerRJ2000Ependymomas in children J Neurosurg 93721723 [DOI] [PubMed] [Google Scholar]
- PlantazDMohapatraGMatthayKKPellarinMSeegerRCFeuersteinBG1997Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization Am J Pathol 508189 [PMC free article] [PubMed] [Google Scholar]
- PollackIFGersztenPCMartinezAJLoKHShultzBAlbrightALJanoskyJDeutschM1995Intracranial ependymomas of childhood: long-term outcome and prognostic factors Neurosurgery 37655667 [DOI] [PubMed] [Google Scholar]
- RansomDTRitlandSRKimmelDWMoertelCADahlRJScheithauerBWKellyPJJenkinsRB1992Cytogenetic and loss of heterozygosity studies in ependymomas, pilocytic astrocytomas, and oligodendrogliomas Genes Chrom Cancer 5348356 [DOI] [PubMed] [Google Scholar]
- ReardonDAEntrekinRESublettJRagsdaleSLiHBoyettJKepnerJLLookAT1999Chromosome arm 6q loss is the most common recurrent autosomal alteration detected in primary pediatric ependymoma Genes Chrom Cancer 24230237 [DOI] [PubMed] [Google Scholar]
- RobertsonPLZeltzerPMBoyettJMRorkeLBAllenJCGeyerJRStanleyPLiHAlbrightALMcGuire-CullenPFinlayJLStevensJrKRMilsteinJMPackerRJWisoffJ1998Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children's Cancer Group J Neurosurg 88695703 [DOI] [PubMed] [Google Scholar]
- RodriguezHABerthrongM1966Multiple primary intracranial tumors in von Recklinghausen's neurofibromatosis Arch Neurol 14467475 [DOI] [PubMed] [Google Scholar]
- RosenbergCSchutTBMostertMCTankeHJRaapAKOosterhuisJWLooijengaLH1997Comparative genomic hybridization in hypotriploid/hyperdiploid tumors Cytometry 29113121 [DOI] [PubMed] [Google Scholar]
- RossGWRubinsteinLJ1989Lack of histopathological correlation of malignant ependymomas with postoperative survival J Neurosurg 703136 [DOI] [PubMed] [Google Scholar]
- SchifferDChioAGiordanaMTMigheliAPalmaLPolloBSoffiettiRTriboloA1991Histologic prognostic factors in ependymoma Child's Nerv Syst 7177182 [DOI] [PubMed] [Google Scholar]
- StrattonMRDarlingJLantosPLCooperCSReevesBR1989Cytogenetic abnormalities in human ependymomas Int J Cancer 44579581 [DOI] [PubMed] [Google Scholar]
- SuttonLNGoldweinJPerilongoGLangBSchutLRorkeLPackerR1990Prognostic factors in childhood ependymomas Pediatr Neurosurg 165765 [DOI] [PubMed] [Google Scholar]
- TongCYNgHKPangJCHuiABKoHCLeeJC1999Molecular genetic analysis of non-astrocytic gliomas[see comments]Histopathology 34331341 [DOI] [PubMed] [Google Scholar]
- Vagner-CapodanoAMZattara-CannoniHGambarelliDFigarella-BrangerDLenaGDufourHGrisoliFChouxM1999Cytogenetic study of 33 ependymomas Cancer Genet Cytogenet 1159699 [DOI] [PubMed] [Google Scholar]
- VanuytselLJBessellEMAshleySEBloomHJBradaM1992Intracranial ependymoma: long-term results of a policy of surgery and radiotherapy Int J Rad Oncol Biol Phys 23313319 [DOI] [PubMed] [Google Scholar]
- von HakenMSWhiteECDaneshvar-ShyestherLSihSChoiEKalraRCogenPH1996Molecular genetic analysis of chromosome arm 17p and chromosome arm 22q DNA sequences in sporadic pediatric ependymomas Genes Chrom Cancer 173744 [DOI] [PubMed] [Google Scholar]
- WardSHardingBWilkinsPHarknessWHaywardRDarlingJLThomasDGWarrT2001Gain of 1q and loss of 22 are the most common changes detected by comparative genomic hybridisation in paediatric ependymoma Genes Chrom Cancer 325966 [DOI] [PubMed] [Google Scholar]
- WiestlerODSchifferDCoonsSWPraysonRARosenblumMK2000EpendymomaInTumours of the Nervous SystemKleihues P, Cavenee WK (eds)World Health Organization Classification of Tumourspp7281Lyon: IARC Press [Google Scholar]
- ZhengPPPangJCHuiABNgHK2000Comparative genomic hybridization detects losses of chromosomes 22 and 16 as the most common recurrent genetic alterations in primary ependymomas Cancer Genet Cytogenet 1221825 [DOI] [PubMed] [Google Scholar]