

Abstract
Many higher-chlorinated biphenyls, persistent and predominant in foods, are active as promoters in hepatocarcinogenesis. Lower-chlorinated biphenyls, predominating in indoor and outdoor air, are more readily metabolized and therefore shorter lived, ‘episodic’ contaminants. Thus inhalation of such lower chlorinated biphenyls may expose humans to reactive, possibly genotoxic/carcinogenic intermediates. Lower chlorinated biphenyls may be metabolized via arene-oxides to mono- and dihydroxylated intermediates and further to (semi)quinones, highly reactive intermediates. Covalently bound lower chlorinated biphenyls have been detected in rodent tissues in vivo. We recently showed using the modified Solt-Farber foci assay that several mono- to tetrachlorinated biphenyls have initiating activity in the livers of rats. In a follow-up study of PCB3 (4-chlorobiphenyl) metabolites only one monohydroxy- and one quinoid- metabolite showed initiating activity, indicating that the metabolic activation of PCB3 proceeds via hydroxylation and oxidation to the 3,4-quinone, the ultimate carcinogen. Since cancer initiation is based on genotoxic event(s), we hypothesized that PCB3 and/or its metabolite(s) are mutagenic in rat liver in vivo. To investigate this, BigBlue® rats, transgenic for the lacI reporter gene, were exposed to PCB3 and 4-hydroxy-PCB3 (4-HO-PCB3). In male rats the mutant frequency (MF) of lac I in the liver was significantly elevated and the mutation spectrum differed significantly from the control. 4-HO-PCB3 caused a non-significant (p = 0.115) doubling of the MF compared to the control. These studies prove that lower halogenated biphenyls may be metabolically activated in vivo to genotoxic and initiating intermediates.
Keywords: PCB, carcinogenesis, in vivo, mutation, transgenic rat, 4-monochlorobiphenyl, metabolic activation
Background and Routes of Exposure
PCBs were commercially manufactured for about 50 years and used as dielectrics in transformers and capacitors, as cooling fluids in hydraulic systems, in the formulation of lubricating and cutting oils, in pesticides and flame retardants, and as plasticizers in paints, copying paper, adhesives, sealants and plastics (WHO, 1976). Although the U.S. sales and distribution of PCBs ended in the late 1970's, a significant portion of PCBs purchased by industry are still in use, mostly within capacitors and transformers. The stability of these compounds, once the major reason for their widespread commercial use, has led to their worldwide distribution in the environment, as first reported by Jensen in 1966 (Jensen, 1966). Although the production of PCBs peaked in the 1970s and has steadily declined thereafter as most countries throughout the world have banned their production, these compounds remain in our environment and are routinely found in human serum, breast milk and adipose samples.
Regular monitoring of environmental PCBs in water, fish, and sediment of the Great Lakes and other regions in the USA started in the 1980s (Schneider et al., 2001). PCBs have also been found in at least 500 of the 1,598 National Priorities List sites identified by the Environmental Protection Agency (EPA) (http://www.atsdr.cdc.gov/tfacts17.html). High PCB levels in fish have resulted in fish advisories for rivers and lakes in nearly all states of the USA. The PCBs in these fish samples are mostly higher chlorinated congeners. It is therefore assumed that diet, especially fish, is the major source of the higher chlorinated biphenyls that are found in human tissue samples.
Non-atmospheric sources of PCBs are carefully monitored and regulated. Air as a source of lower chlorinated biphenyls, however, was nearly completely ignored until a decade ago. Systematic measurements of atmospheric PCBs started only in the 1990s. The first urban monitoring site was installed in Chicago in 1995. The level of PCB contamination in the air is strongly influenced by temperature. In Chicago air concentrations between 100 - 300 pg/m3 in winter and up to 5,000 -16,000 pg/m3 on hot summer days were reported (1996-2002 IADN Data; http://epa.gov/glnpo/monitoring/air/iadn/reports/IADN_1996.pdf).
The sources of these atmospheric PCBs are almost completely undefined. Occupational exposure to atmospheric PCBs may be significantly higher. Indeed, inhalation exposure is considered to be a major route of occupational exposure to PCBs, and it was estimated that in capacitor workers, for example, a maximum of 80% of adipose PCBs may have been absorbed by inhalation exposure (Wolff, 1985). Recently even higher levels of PCBs were measured in indoor air in buildings, especially schools, constructed in the 1970s where joint sealants containing 4-9% PCBs were used. Concentrations up to 13,000 ng/m3 were measured in some classrooms of a contaminated school (Kohler et al., 2002), which is more than an order of magnitude above the NIOSH guidelines of 1 μg/m3 for occupational settings. Other possible sources for indoor PCBs are believed to be data screen terminals (Digernes and Astrup, 1982), ceiling tiles (Anonymus, 1988) and fluorescent lights (Harris, 1985). It was reported that the concentration of PCBs in indoor air can be at least an order of magnitude higher than outdoor air (Balfanz et al., 1993; Vorhees et al., 1997; Wallace et al., 1996), although the sources are not completely clear. A recent publication from Great Britain found a daily average total PCB intake of 0.49 μg/person/day for adults of which 30.6% (4.2 - 63%) was derived from inhalation exposure (Harrad et al., 2006). The average intake of toddlers was 0.222 μg/toddler/day, with ∼12.6% from inhalation. It is very well possible that under certain circumstances the intake from inhalation exposure may currently be the major source of PCB intake for humans.
Major PCB congeners in Chicago air are PCB 5/8 and 15/17 (co-elute), 18, 28, 31, 33, 44, 52, and 95, to name a few (see Figure 1). Hexa- or higher chlorinated PCB congeners are only a minor fraction in air samples. PCB28 and PCB52 were the prevailing congeners in indoor air of contaminated schools (Kohler et al., 2002; Schwenk et al., 2002). Elevated levels of PCB28 and PCB52 were measured in the blood of teachers from contaminated schools compared to non-contaminated schools (Kohler et al., 2002; Schwenk et al., 2002), whereas the mean blood levels of higher chlorinated biphenyls, i.e. PCB138, 153 and 180 were almost identical between groups. Inhalation exposure may also be the reason for higher levels of PCB28 and PCB52 in mothers' milk from women who smoked and/or were living in industrialized areas compared to non-smokers and women living in non-industrialized areas (Angulo et al., 1999). Environmental levels of monochlorinated PCB congeners are usually not determined because of technical reasons, but a recent report identified PCB1 and PCB3 as the major congeners in indoor air in a child care facility (Davis et al., 2002). PCB3 was by far the dominant congener emitted in the emission gases of cement plants (Ishikawa et al., 2007). Unlike higher chlorinated biphenyls, lower, airborne PCBs do not bioaccumulate to the same extent since they are more readily metabolized and excreted. The true level of human exposure and contamination with lower chlorinated biphenyls may therefore be grossly underestimated.
Figure 1.

Mean Chicago ΣPCB signal (1996-2002 IADN Data, IIT. Figure created by Dr. Keri Hornbuckle, University of Iowa Dept. of Environmental Engineering). Congeners with a fraction > 0.03 of the ΣPCB are considered to be major components (dotted line).
Implications in Carcinogenesis
Studies indicate that PCB mixtures with a high chlorine content are more potent in inducing nodular hyperplasia and hepatocarcinomas than mixtures with less chlorination (Silberhorn et al., 1990), especially in male rodents. In a comprehensive chronic toxicity and carcinogenicity study, the effects of four Aroclor products (1016, 1242, 1254, and 1260) were investigated at multiple dietary concentrations, ranging from 25 to 200 ppm, for 24 months in male and female Sprague-Dawley rats. Statistically significant increases in hepatocellular carcinomas was noted in male rats only for the higher chlorinated mixture Aroclor 1260, while all four commercial products produced an elevated incidence of hepatocellular carcinomas in female rats. It should be noted that Aroclor 1016 averages only 3 chlorines per biphenyl. These data indicate that commercial mixtures of chlorinated biphenyls are complete carcinogens, especially in the female rat (Mayes et al., 1998).
The process of chemical-induced carcinogenesis is believed to occur over several stages, such as initiation, promotion, and progression (Figure 2). Initiation is believed to be caused by a genotoxic event, whereas promotion is characterized by an increase in the number of initiated cells through long-term epigenetic mechanisms. Progression is that final stage of carcinogenesis associated with chromosomal/karotypic changes.
Figure 2.

Pathway from normal cell to malignant cancer.
Although their potency varies, the various halogenated biphenyls have been uniformly reported to have promoting activity in various liver models (reviewed by Glauert et al., 2001; Silberhorn et al., 1990). Generally those compounds which are inducers of cytochrome P-450 (the higher halogenated biphenyls) were more potent as promoters in rat two-stage hepatogenesis (Deml et al., 1985). It has been widely assumed that PCBs and related compounds do not have initiating activity in hepatocarcinogenesis, but very little work has been done.
Metabolic Activation of Halogenated Biphenyls
Although more highly halogenated biphenyls are resistant to metabolic conversion, and tend to persist in plants, animals and in the environment in general, they do nonetheless have slow and measurable rates of metabolism. In general, the fewer halogens, the more rapid the metabolic transformation. In other words, PCBs are not only efficacious inducers of xenobiotic metabolism (Parkinson et al., 1980), but may also become substrates for the induced enzymes. Associated with their metabolism may be the metabolic activation to electrophiles and the ensuing reaction with cellular substituents, such as proteins, lipids, and DNA.
Influence of CYPs on Metabolic Activation of Lower Chlorinated Biphenyls
It has long been recognized that biphenyl and halogenated biphenyls, particularly the lower halogenated congeners, are hydroxylated in vivo and in vitro (see review by: Safe, 1989) and hydroxylated PCBs have been found in human blood and environmental tissue and even soil samples (Bergman et al., 1994; Ueno et al., 2007). These hydroxylation reactions are primarily catalyzed by isozymes of cytochrome P-450. Our experiments with PCB3 (4-chlorobiphenyl) and rat liver microsomes showed that five mono- and three di-hydroxy metabolites were formed (McLean et al., 1996a). Using rat liver microsomes from animals that had been treated with different CYP inducers we observed that CYP 1A (methylcholanthrene-treated animals) was more efficient in PCB3 metabolism and produced two catechols (2′,3′- and 3′,4′-) and the hydroquinone (2′,5′-) in the non-chlorinated ring in a ratio of about 2:2:1, respectively. CYP 2B (Phenobarbital-treated rats) produced a very different pattern of dihydroxy metabolites, with 3′,4′- and 2′,5′- metabolites in a ratio of 2:1 and no detectable 2′,3′-metabolites (McLean et al., 1996a). This shows that the metabolic environment, i.e. which CYPs are expressed and at which level, may be of major importance with respect to the potential formation of initiating PCB metabolites.
The metabolism of PCB3 by cytochrome P-450 seems to involve arene oxide intermediates (McLean et al., 1996b). These are converted to mono-hydroxy metabolites and in further steps, probably through a secondary arene oxide intermediates, from the mono- to the di-hydroxy forms (Figure 3). Arene oxides are strong electrophiles which may react with critical cellular targets. The dihydroxybiphenyls can be further oxidized by various enzymes like peroxidases, prostaglandin synthase and cytochromes P-450, to the corresponding quinone with the formation of a semiquinone intermediate. Both, quinones and semiquinones, are also reactive metabolites.
Figure 3.
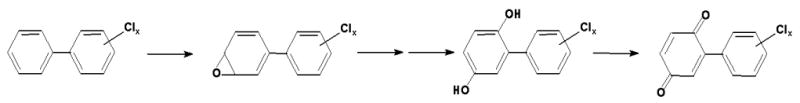
Metabolic activation of PCBs to arene oxides, hydroxylated and quinonoid metabolites
In Vivo Distribution and Binding
One important question concerns the distribution of lower chlorinated biphenyls in the tissues of exposed animals and whether these compounds are activated to covalently bound metabolites. We performed an experiment in which we injected radiolabeled PCB3 and PCB77 into phenobarbital- and ß-naphthoflavone-treated mice. Large levels of these PCBs were located in the blood, especially the plasma, with PCB77 ≫ PCB3 (Tampal et al., 2003). However, a much larger portion of PCB3 than PCB77 was covalently bound to hemoglobin. Significant amounts of both PCBs were discovered in various tissues, with liver > kidneys > lung (Pereg et al., 2001). With respect to subcellular localization of the PCBs, PCB77 in lungs and PCB3 in all tissues was found in cytosol > organelles > microsomes. This was different for PCB77 in liver and kidneys, where the order was organelles > cytosol > microsomes. Further analysis showed that significant amounts of PCB3 and PCB77 were covalently bound to nuclear proteins in the liver of these animals, with PCB3 ≫ PCB77. Increased covalent binding of these PCBs to liver DNA compared to control was observed as well, but the analysis of variance did not reveal any significant differences between groups (p = 0.241). We concluded from these studies that both congeners are metabolically activated in vivo to species that bind covalently to proteins and probably DNA in the nuclei of liver cells which could indicate that they may cause heritable changes and thereby initiation of hepatocarcinogenesis.
Other Forms of Reactivity of PCB Metabolites
Direct DNA adduction was not the only mechanism of genotoxicity. These mono- or dichlorinated diOH-PCBs or corresponding PCB quinones are able to redox cycle (McLean et al., 2000), to form 8-oxodG (Oakley et al., 1996), to produce DNA strand breaks (Srinivasan et al., 2001), and to bind to sulfhydryl groups (Amaro et al., 1996), including those in glutathione and topoisomerase (Bender et al., 2007; Srinivasan et al., 2002). Binding to –SH causes GSH depletion, oxidative stress and toxicity in cells (Srinivasan et al., 2001). Most of these studies were done with PCB3, some also with congeners with 2 or 3 chlorines in the same ring (PCB12, PCB38). All of the congeners or their synthesized metabolites were active in these assays. These congeners were chosen as model compounds. The new emphasis on air pollution made us aware that most airborne PCB congeners have chemical structures that could very well give them a similar metabolic activation pathway and reactivity as our test compounds, but few studies were found that addressed the reactivity and toxicity of these airborne congeners especially with respect to their possible carcinogenicity.
Solt-Farber Studies with PCB Congeners
The Solt-Farber protocol may be used to test whether PCBs or their congeners can initiate carcinogenesis in the livers of exposed rats (Farber et al., 1977). Using this protocol, we first tested 3 mono- to tri-chlorinated congeners, i.e. PCB 3, PCB12 (3,4-dichlorobiphenyl) and PCB38 (3,4,5-trichlorobiphenyl) (Espandiari et al., 2003). In this study the initiating activity of PCBs was investigated in animals receiving partial hepatectomy, followed by a single dose of 100 mg/kg of the initiator diethylnitrosamine (DEN, positive control), the test agent (PCB), or vehicle (corn oil). Two weeks post-dosing the initiated cells were growth selected by treating the animals with daily doses of 2-acetylaminofluorene (2-AAF, for 3 days), followed by a single dose of CCl4 and then 3 additional daily treatments with 2-AAF. Negative controls without selection-treatment with 2-AAF and CCl4 were conducted in parallel. All animals in the positive control group receiving DEN showed visible nodules, while no nodules were apparent in animals in the negative control groups (vehicle and groups not receiving selection agents). Furthermore, no nodules were apparent in the groups receiving the dichloro- or trichloro-biphenyls as initiators. However, the monochloro-congener (PCB3) induced grossly visible nodules in 50% of the rats treated. Histological staining of liver sections further revealed the presence of foci and nodules in H+E-stained sections as well as γ-glutamyl transpeptidase (GGT)-positive foci and nodules in all DEN-treated animals (5500 foci/liver) and in 80% of PCB3-treated animals (1500 foci/liver). The number of foci and focal volume in DEN treated animals was 425 foci/cm3 and 34%, respectively, while PCB3 treated animals had 200 foci/cm3 occupying 3% of the total liver. No GGT-positive foci were detected in vehicle or non selected groups. In another group of experiments PCB15 (4,4′-dichlorobiphenyl), PCB77, PCB52 and a combination of PCB77 & PCB52 were tested (Espandiari et al., 2003). All 4 treatment groups increased the number of foci per cm3 in the liver, with DEN > PCB15 ≫ PCB52 > PCB77 > PCB52/77. Only PCB15 and DEN also significantly increased the focal volume (% of liver). We conclude from these experiments that lower chlorinated biphenyls with 1 to 4 chlorines are able to initiate hepatocarcinogenesis in the rat, but from these few congeners tested we are not able to assign a structure-activity relationship. Also, further testing is needed to explain why the combination of two active congeners (PCB52/77) resulted in a reduction of the number of foci induced and/or whether enzyme induction plays a role in this effect.
Solt-Farber Studies with PCB3 Metabolites
As a next step, we performed a series of experiments with synthetic PCB3 metabolites in an effort to determine the metabolic activation pathway and the ultimate initiating agent. For these experiments the fasting/refeeding protocol and 20 mg/kg DEN as positive control was used. Test compounds included the 2-OH-, 3-OH-, 4-OH-, 2,3-diOH-, 3,4-diOH-, 2,5-diOH-, 2,3-quinone, 3,4-quinone, and 2,5-quinone metabolites of PCB3. To summarize the results: the 4-OH- and 3,4-quinone metabolites of PCB3 significantly increased the number of γ-glutamyl transpeptidase (GGT)-positive foci/cm3, the number of foci per liver and the focal volume (% of liver). In fact, 100 μmol/kg 3,4-quinone of PCB3 was more active than 20 mg/kg DEN with respect to foci number. None of the other PCB3 metabolites had a significant effect on either foci number or foci volume (Espandiari et al., 2004). We conclude that the 3,4-ortho-quinone of PCB3 is an ultimate initiating metabolite. It is noteworthy that formation of 3,4-quinone-derived protein adducts in the liver and brain of rats treated with PCB52 were reported (Lin et al., 2000). All together this implies that PCB congeners like PCBs 5, 8, 17, 18, 31, 33, 44, 52, 84, 92, 95, 101, and 110, all major congeners found in Chicago air and with a free 3,4-position, may be activated through the same pathway. Clearly more experiments are needed to understand the metabolic activation pathway, the structure-activity requirements, and the interactions between different congeners in a mixture during PCB-initiated hepatocarcinogenesis to be able to accurately judge the risks associated with exposure to airborne PCBs.
The BigBlue In Vivo Mutation Assay
As mentioned before, DNA mutation is a prerequisite for initiation, the first step in the carcinogenic transformation of cells. All our experiments so far indicate that lower chlorinated biphenyls are activated to DNA damaging intermediates which cause preneoplastic changes in the liver of treated rats. There is, however, a gap in the evidence going from PCB congener to liver cancer, and that is the actual mutation induction. We have begun experiments with the BigBlue rat in vivo mutation assay from Stratagene. These transgenic rats carry multiple copies of an indicator gene that can be isolated, packed in phages, and selected on agar plates after transfection into bacteria. In the first experiment male rats were treated weekly for 4 weeks with i.p. injections of 3-methylcholantrene, PCB3, 4-HO-PCB3 or vehicle alone (corn oil) and killed after a period of additional 10 days. Liver tissue was collected, the target DNA isolated and packed into phages, transfected into bacteria, and mutated (blue) colonies identified. The mutant frequency per 100,000 pfu was 1.7 in the negative control, and 8.8 (3-MC), 4.8 (PCB3), and 4.0 (4-HO-PCB3) in the treatment groups (Table 1 and Lehmann et al., 2007).
Table 1.
Mutants and type of mutation
| Corn Oil | 3-MC | PCB3 | 4OH-PCB3 | |
|---|---|---|---|---|
| Mutants ±SD /106pfu | 17±4 | 88±15 | 48±4 | 40±12 |
| Level of significance | p=0.0037 | p=0.0014 | p=0.115 | |
| Mutation Types* (%) | ||||
| Transitions | 63 | 30 | 30 | 56 |
| Tranversions | 19 | 43 | 43 | 22 |
| Insertions/Deletions | 19 | 27 | 26 | 22 |
Number of Mutations sequenced: 16 (corn oil), 44 (3-MC), 23 (PCB3), 27 (4OH-PCB3)
Sequencing of the reporter gene revealed that 3-MC and PCB3 induced different types of mutations compared to those in the control group. Whereas the spontaneous mutations in the control group were mostly GC to AT transitions, a strong increase in the percent of GC to TA transversions and -1 frameshift mutations was seen in the 3-MC and PCB3 groups. The mutation types in the 4-HO-PCB3 group were somewhat similar to the control group, although the percentage of GC to TA transversions was more than doubled and insertions were nearly twice as frequent. GC to TA transversions are often produced by bulky PAH adducts with the DNA or by DNA oxidation. Thus neither epoxides, nor quinones or ROS can be excluded as ultimate mutagen of PCB3. These results clearly indicate that we are able to see mutation induction with PCB congeners and possibly with the metabolites.
Summary
In summary, these results clearly show that lower halogenated biphenyls are metabolized by microsomal enzymes to mono- and dihydroxy metabolites which may be further oxidized to quinones and that intermediates of this metabolic pathway, probably both, arene oxides and (semi)quinones, react with cellular macromolecules. Individual PCB congeners caused DNA adduct formation, mutations, and initiated foci in vivo. At least one metabolite, the 3,4-quinone, is a probable ultimate carcinogen. These data taken together strengthen our hypothesis that PCBs are metabolized to genotoxic species in vitro and in vivo, may act as initiating agents and induce liver cancer in rodents. The effects and mechanisms that we observed so far may also be a characteristic of PCBs like PCB 5/8 (coelute), 15/17 (coelute), 28, 33, 44, 52, or 95, all of which are prominent constituents of Chicago air. The fact that very little is known about the activity of these congeners warrants further studies.
Acknowledgments
These results are based on cooperation with many scientists of whom we wish to specifically mention our colleagues, i.e., Dr R. Gupta (U. of Louisville) and Dr. H.P. Glauert (U. of Kentucky), and former and current students, i.e., Dr. P. Espandiari (FDA), Dr. N. Tampal (FDA), Dr. D. Pereg (U. of Laval, Canada), J. Jacobus, and C. Maddox (both U. of Iowa). (Supported by NIEHS (P42 ES07380 and P42 ES013661), DOD (DAMD17-02-1-0241), and EPA (R-82902102-0).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaro AR, Oakley GG, Bauer U, Spielmann HP, Robertson LW. Metabolic activation of PCBs to quinones: reactivity toward nitrogen and sulfur nucleophiles and influence of superoxide dismutase. Chem Res Toxicol. 1996;9(3):623–9. doi: 10.1021/tx950117e. [DOI] [PubMed] [Google Scholar]
- Angulo R, Martinez P, Jodral ML. PCB congeners transferred by human milk, with an estimate of their daily intake. Food Chem Toxicol. 1999;37(11):1081–8. doi: 10.1016/s0278-6915(99)00101-5. [DOI] [PubMed] [Google Scholar]
- Anonymous. PCB contamination of ceiling tiles - Madison Wisconsin. Morb Mortal Wkly Rep. 1988;37(2):17–19. [PubMed] [Google Scholar]
- Balfanz E, Fuchs J, Kieper H. Sampling and analysis of polychlorinated biphenyls (PCB) in indoor air due to permanently elastic sealants. Chemosphere. 1993;26(5):871–880. [Google Scholar]
- Bender RP, Ham AJ, Osheroff N. Quinone-induced enhancement of DNA cleavage by human topoisomerase IIalpha: adduction of cysteine residues 392 and 405. Biochemistry. 2007;46(10):2856–64. doi: 10.1021/bi062017l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman A, Klasson-Wehler E, Kuroki H. Selective retention of hydroxylated PCB metabolites in blood. Environ Health Perspect. 1994;102(5):464–9. doi: 10.1289/ehp.94102464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B, Beach J, Wade M, Klein AK, Hoch K. Risk Assessment of Polychlorinated Biphenyls (PCBs) in Indoor Air. The Toxicologist, Supplement to Toxicological Sciences. 2002;66(106):516. [Google Scholar]
- Deml E, Oesterle D, Wiebel FJ, Wolff T. Correlation between promotion of enzyme-deficient islands and induction of monooxygenase activities by halogenated hydrocarbons in rat liver. Food Chem Toxicol. 1985;23:880. [Google Scholar]
- Digernes V, Astrup EG. Are datascreen terminals a source of increased PCB-concentrations in the working atmosphere? Int Arch Occup Environ Health. 1982;49(34):193–7. doi: 10.1007/BF00377928. [DOI] [PubMed] [Google Scholar]
- Espandiari P, Glauert HP, Lehmler HJ, Lee EY, Srinivasan C, Robertson LW. Polychlorinated biphenyls as initiators in liver carcinogenesis: resistant hepatocyte model. Toxicol Appl Pharmacol. 2003;186(1):55–62. doi: 10.1016/s0041-008x(02)00018-2. [DOI] [PubMed] [Google Scholar]
- Espandiari P, Glauert HP, Lehmler HJ, Lee EY, Srinivasan C, Robertson LW. Initiating activity of 4-chlorobiphenyl metabolites in the resistant hepatocyte model. Toxicol Sci. 2004;79(1):41–6. doi: 10.1093/toxsci/kfh097. [DOI] [PubMed] [Google Scholar]
- Farber E, Solt D, Cameron R, Laishes B, Ogawa K, Medline A. Newer insights into the pathogenesis of liver cancer. Am J Pathol. 1977;89(2):477–82. [PMC free article] [PubMed] [Google Scholar]
- Glauert HP, Robertson LW, Silberhorn EM. PCBs and tumor promotion. In: Robertson LW, Hansen LG, editors. PCBs: Recent Advances in Environmental Toxicology and Health Effects Lexington. The University Press of Kentucky; 2001. pp. 355–371. [Google Scholar]
- Harrad S, Hazrati S, Ibarra C. Concentrations of polychlorinated biphenyls in indoor air and polybrominated diphenyl ethers in indoor air and dust in Birmingham, United Kingdom: implications for human exposure. Environ Sci Technol. 2006;40(15):4633–8. doi: 10.1021/es0609147. [DOI] [PubMed] [Google Scholar]
- Harris MG. PCB exposure from fluorescent lights. Am J Public Health. 1985;75(8):892. doi: 10.2105/ajph.75.8.892-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Noma Y, Yamamoto T, Mori Y, Sakai S. PCB decomposition and formation in thermal treatment plant equipment. Chemosphere. 2007;67(7):1383–93. doi: 10.1016/j.chemosphere.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Jensen S. A new chemical hazard. New Sci. 1966;32:612. [Google Scholar]
- Kohler M, Zennegg M, Waeber R. Coplanar polychlorinated biphenyls (PCB) in indoor air. Environ Sci Technol. 2002;36(22):4735–40. doi: 10.1021/es025622u. [DOI] [PubMed] [Google Scholar]
- Lehmann L, H LE, P AK, L WR, Ludewig G. 4-monochlorobiphenyl (PCB3) induces mutations in the livers of transgenic Fisher 344 rats. Carcinogenesis. 2007;28(2):471–8. doi: 10.1093/carcin/bgl157. [DOI] [PubMed] [Google Scholar]
- Lin PH, Sangaiah R, Ranasinghe A, Upton PB, La DK, Gold A, Swenberg JA. Formation of quinonoid-derived protein adducts in the liver and brain of Sprague-Dawley rats treated with 2,2′,5, 5′-tetrachlorobiphenyl. Chem Res Toxicol. 2000;13(8):710–8. doi: 10.1021/tx000030f. [DOI] [PubMed] [Google Scholar]
- Mayes BA, McConnell EE, Neal BH, Brunner MJ, Hamilton SB, Sullivan TM, Peters AC, Ryan MJ, Toft JD, Singer AW, et al. Comparative carcinogenicity in Sprague-Dawley rats of the polychlorinated biphenyl mixtures Aroclors 1016, 1242, 1254, and 1260. Toxicol Sci. 1998;41(1):62–76. doi: 10.1093/toxsci/41.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean MR, Bauer U, Amaro AR, Robertson LW. Identification of catechol and hydroquinone metabolites of 4-monochlorobiphenyl. Chem Res Toxicol. 1996a;9(1):158–64. doi: 10.1021/tx950083a. [DOI] [PubMed] [Google Scholar]
- McLean MR, Robertson LW, Gupta RC. Detection of PCB adducts by the 32P-postlabeling technique. Chem Res Toxicol. 1996b;9(1):165–71. doi: 10.1021/tx9500843. [DOI] [PubMed] [Google Scholar]
- McLean MR, Twaroski TP, Robertson LW. Redox cycling of 2-(x'-mono, -di, -trichlorophenyl)- 1, 4-benzoquinones, oxidation products of polychlorinated biphenyls. Arch Biochem Biophys. 2000;376(2):449–55. doi: 10.1006/abbi.2000.1754. [DOI] [PubMed] [Google Scholar]
- Oakley GG, Devanaboyina U, Robertson LW, Gupta RC. Oxidative DNA damage induced by activation of polychlorinated biphenyls (PCBs): implications for PCB-induced oxidative stress in breast cancer. Chem Res Toxicol. 1996;9(8):1285–92. doi: 10.1021/tx960103o. [DOI] [PubMed] [Google Scholar]
- Parkinson A, Robertson L, Safe L, Safe S. Polychlorinated biphenyls as inducers of hepatic microsomal enzymes: structure-activity rules. Chem Biol Interact. 1980;30(3):271–85. doi: 10.1016/0009-2797(80)90050-2. [DOI] [PubMed] [Google Scholar]
- Pereg D, Tampal N, Espandiari P, Robertson LW. Distribution and macromolecular binding of benzo[a]pyrene and two polychlorinated biphenyl congeners in female mice. Chem Biol Interact. 2001;137(3):243–58. doi: 10.1016/s0009-2797(01)00256-3. [DOI] [PubMed] [Google Scholar]
- Safe S. Metabolism, uptake, storage and bioaccumulation. In: Kimbrough, Jensen, editors. Halogenated Biphenyls, Terphenyls, Naphthalens, Dibenzodioxins and Related Products. Elsevier; New York: 1989. [Google Scholar]
- Schneider AR, Stapleton HM, Cornwell J, Baker JE. Recent declines in PAH, PCB, and toxaphene levels in the northern Great Lakes as determined from high resolution sediment cores. Environmental Science & Technology. 2001;35:3809–3815. doi: 10.1021/es002044d. [DOI] [PubMed] [Google Scholar]
- Schwenk M, Gabrio T, Papke O, Wallenhorst T. Human biomonitoring of polychlorinated biphenyls and polychlorinated dibenzodioxins and dibenzofuranes in teachers working in a PCB-contaminated school. Chemosphere. 2002;47(2):229–33. doi: 10.1016/s0045-6535(01)00307-1. [DOI] [PubMed] [Google Scholar]
- Silberhorn EM, Glauert HP, Robertson LW. Carcinogenicity of polyhalogenated biphenyls: PCBs and PBBs. Crit Rev Toxicol. 1990;20(6):440–96. doi: 10.3109/10408449009029331. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Lehmler HJ, Robertson LW, Ludewig G. Production of DNA strand breaks in vitro and reactive oxygen species in vitro and in HL-60 cells by PCB metabolites. Toxicol Sci. 2001;60(1):92–102. doi: 10.1093/toxsci/60.1.92. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Robertson LW, Ludewig G. Sulfhydryl binding and topoisomerase inhibition by PCB metabolites. Chem Res Toxicol. 2002;15(4):497–505. doi: 10.1021/tx010128+. [DOI] [PubMed] [Google Scholar]
- Tampal N, Myers S, Robertson LW. Binding of polychlorinated biphenyls/metabolites to hemoglobin. Toxicol Lett. 2003;142(12):53–60. doi: 10.1016/s0378-4274(02)00484-8. [DOI] [PubMed] [Google Scholar]
- Ueno D, Darling C, Alaee M, Campbell L, Pacepavicius G, Teixeira C, Muir D. Detection of hydroxylated polychlorinated biphenyls (OH-PCBs) in the abiotic environment: surface water and precipitation from Ontario, Canada. Environ Sci Technol. 2007;41(6):1841–8. doi: 10.1021/es061539l. [DOI] [PubMed] [Google Scholar]
- Vorhees DJ, Cullen AC, Altshul LM. Exposure to polychlorinated biphenyls in residential indoor air and outdoor air near a Superfund site. Environ Sci Technol. 1997;31(12):3612–3618. [Google Scholar]
- Wallace JC, Basu I, Hites RA. Sampling and analysis artifacts caused by elevated indoor air polychlorinated biphenyl concentrations. Environ Sci Technol. 1996;30(9):2730–2734. [Google Scholar]
- WHO. Environmenal Health Criteria 2. WHO; Geneva, Switzerland: 1976. Polychlorinated Biphenyls and Terphenyls; p. 85. [Google Scholar]
- Wolff MS. Occupational exposure to polychlorinated biphenyls (PCBs) Environ Health Perspect. 1985;60:133–8. doi: 10.1289/ehp.8560133. [DOI] [PMC free article] [PubMed] [Google Scholar]