

Abstract
The extracellular calcium (Ca2+o)-sensing receptor (CaSR) enables the parathyroid glands and other CaSR-expressing cells involved in calcium homeostasis, such as the kidney and bone, to sense alterations in the level of Ca2+o and to respond with changes in function that are directed at normalizing the blood calcium concentration. Several disorders of Ca2+o sensing arise from inherited or acquired abnormalities that ‘reset’ the serum calcium concentration upwards or downwards. Heterozygous inactivating mutations of the CaSR produce a benign form of hypercalcaemia, termed ‘familial hypocalciuric hypercalcaemia’, while homozygous mutations produce a much more severe hypercalcaemic disorder resulting from marked hyperparathyroidism, called ‘neonatal severe hyperparathyroidism’. Activating mutations cause a hypocalcaemic syndrome of varying severity, termed ‘autosomal-dominant hypocalcaemia or hypoparathyroidism’ as well as Bartter’s syndrome type V. Calcimimetic CaSR activators and calcilytic CaSR antagonists have also been developed with potential for use in the treatment of these disorders.
Keywords: seven transmembrane receptor, mutations, polymorphisms, calcium-sensing receptor, calcium homeostasis, calcimimetic, calcilytic, familial hypocalciuric hypercalcaemia, autosomal-dominant hypoparathyroidism, acquired hypoparathyroidism, osteoporosis, hyperparathyroidism, Bartter’s syndrome
The calcium-sensing receptor (CaSR) plays key roles in the maintenance of a narrow range (1.1–1.3 mM) of the extracellular ionized calcium concentration (Ca2+o), primarily by modulating the function of chief cells of the parathyroid gland. Here it regulates the synthesis and secretion of parathyroid hormone (PTH) as well as parathyroid cellular proliferation, inhibiting all three when Ca2+o is high and stimulating them when Ca2+o is low.1 Both very high and very low levels of Ca2+o can lead to serious clinical sequellae and, in some instances, can be life-threatening. The CaSR serves as a ‘calciostat’, informing the parathyroid glands and other tissues of the precise level of Ca2+o.
The CaSR (also known as CaSR1 and GPRC2A) was cloned using the expression-cloning technique in Xenopus laevis oocytes.2 It is a member of Family C of the superfamily of seven transmembrane (7TM), G-protein-coupled receptors (GPCRs). Other members of this family are so-called metabotropic receptors for glutamate, receptors for gamma-aminobutyric acid, and GPCRs for sensing pheromones, taste and odorants (in fish). Recently, another member of Family C, GPRC6A, has been found to share several pharmacological properties with the CaSR.3,4 Like the CaSR, GPRC6A is sensitive towards certain L-amino acids, but is most responsive to basic amino acids and less so to calcium,3,5 implicating GPRC6A as a second calcium-sensing receptor or at least a calcium-modulated amino acid sensor.
The physiological relevance of the CaSR in humans was proven by the identification of inherited disorders caused by mutations in the receptor leading to either loss or gain of function.6 Heterozygous gain-of-function mutations (CaSR activating) or loss-of-function mutations (CaSR inactivating) are the cause of a growing number of disorders of calcium metabolism, which typically manifest as asymptomatic hypo- or hypercalcaemia, respectively, with relative or absolute hyper- or hypocalciuria. When present in the homozygous or compound heterozygous state, in contrast, inactivating CaSR mutations produce neonatal severe primary hyperparathyroidism (NSHPT), a severe and sometimes lethal disease if it is left untreated.
CaSR expression is greatest in the parathyroid glands, calcitonin-secreting C-cells of the thyroid gland, and kidney. The CaSR is also found in the two other key organs that participate in calcium homeostasis: gut and bone (Figure 1).7,8 This review will briefly discuss the structure, function and normal physiology of the CaSR, and discuss in more depth the mutations and polymorphisms of the CaSR, and its role in various disorders of Ca2+o sensing that are the result of mutations in the CaSR.
Figure 1.
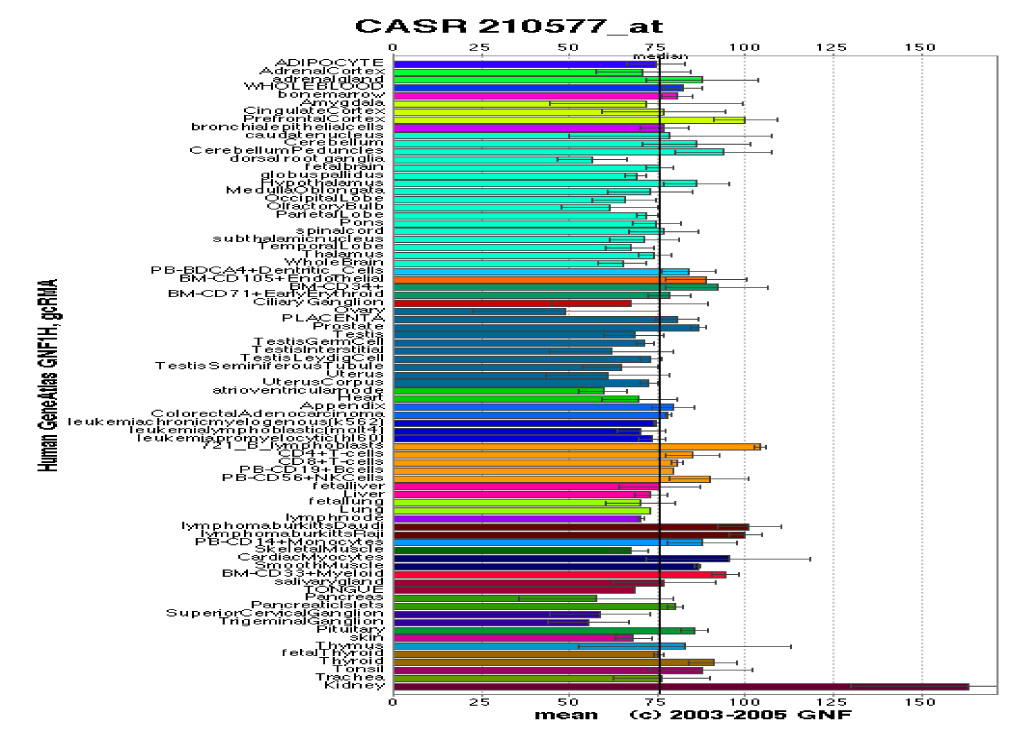
Tissue distribution and density of expression of the calcium-sensing receptor. Adopted with permission from the Novartis Research Foundation Gene Atlas Database.7
PHYSIOLOGY OF THE CASR
PTH, calcitonin and 1,25-dihydroxyvitamin D [1,25(OH)2D3] are the three most important Ca2+o-regulating hormones.9 As noted earlier, there is a functionally critical inverse relationship between Ca2+o and PTH, a calcium-elevating hormone. In contrast, high Ca2+o stimulates the secretion of calcitonin, a Ca2+o-lowering hormone; an action that is likewise mediated by the CaSR.10 Available data have demonstrated that the CaSR is expressed not only in the organs that secrete calcium-regulating hormones (e.g. the parathyroid glands and C-cells of the thyroid glands), but also in target tissues for these hormones. These latter tissues regulate Ca2+o by translocating calcium ions into or out of the bodily fluids, and include the kidney, which expresses the CaSR at robust levels in certain nephron segments, and bone and intestine, which express the receptor at lower levels (Figure 1).7 By acting on both hormone-secreting and hormone-responsive tissues through its own cell surface receptor, Ca2+o acts, in effect, as another Ca2+o-regulating ‘hormone’ (in this case, Ca2+o lowering) or ‘first messenger’. Elevations in Ca2+o stimulate the CaSR and lower Ca2+o by enhancing calcitonin secretion, promoting urinary calcium excretion and suppressing PTH release. The remainder of this section will provide a brief discussion of the CaSR’s known and putative roles in parathyroid gland, kidney and bone; three of the four main organs involved in calcium homeostasis. A recent review discusses the CaSR’s localization and potential roles along the gastrointestinal tract.11
Parathyroid glands
The CaSR is normally expressed at high levels on the surface of the parathyroid chief cells, where it mediates its most important function in minute-to-minute Ca2+o homeostasis by CaSR-regulated inhibition of PTH secretion. The steep inverse sigmoidal curve relating Ca2+o and PTH release was described long before the CaSR was cloned.12
The principal functions of the CaSR in the parathyroid gland are shown in Table 1. Relatively little is known about the control of its expression on the cell surface, but of note, CaSR expression in rat parathyroid and kidney was increased by 1,25(OH)2D3, while Ca2+o had no effect.13 In more recent studies, however, raising Ca2+o increased CaSR expression in avian parathyroid gland,14 and a calcimimetic elevated CaSR expression in pathological parathyroid glands.15 The upregulation of CaSR following its activation could clearly serve as a positive feedback loop contributing to CaSR-mediated actions in the parathyroid gland. Of note in this regard, Ca2+o also regulates the expression of the vitamin D receptor,16 which could potentiate the action of vitamin D on the level of CaSR expression and also enhance the biological actions of the CaSR.
Table 1.
Key roles of the calcium-sensing receptor in the parathyroid gland and kidney.
| • | Parathyroid gland |
| (1) Inhibit PTH secretion | |
| (2) Inhibit PTH gene expression | |
| (3) Inhibit parathyroid cellular proliferation | |
| • | Kidney |
| (1) Proximal tubule – blunt PTH-induced phosphaturia | |
| (2) MTAL – inhibit NaCl re-absorption | |
| (3) CTAL – inhibit re-absorption of Ca2+ and Mg2+ | |
| (4) IMCD – inhibit vasopressin-elicited water re-absorption |
PTH, parathyroid hormone; MTAL, medullary thick ascending limb; CTAL, cortical thick ascending limb; IMCD, inner medullary collecting duct.
Expression, function and regulation of the CaSR in the kidney
The kidney plays several critical roles in calcium homeostasis. The CaSR is widely expressed along essentially the whole nephron. The cellular localization and putative function(s) of the CaSR in the kidney seem to depend upon the region of the nephron in which the receptor resides.17 The CaSR’s cellular localization and expression along the nephron has been studied using in-situ hybridization, reverse transcriptase-polymerase chain reaction of micro-dissected nephron segments18 and immunofluorescence.19 One outcome of these studies was the recognition that the polarity of CaSR protein varies along the nephron. In the proximal tubule, the receptor is present on the apical surface of the proximal tubular epithelial cells. On the contrary, in the cells of the cortical thick ascending limb (CTAL), the receptor is localized in the basolateral membrane. Similarly, basolateral staining for the CaSR was observed in the medullary thick ascending limb, macula densa and the distal convoluted tubule. In the cortical collecting duct, immunostaining for the CaSR is located on some intercalated cells, while in the inner medullary collecting duct (IMCD), the receptor has primarily an apical distribution.
Available data support the following roles of the CaSR along the nephron: (1) diminishing the inhibitory effect of PTH on renal phosphate re-absorption in the proximal tubule;20 (2) inhibiting renal tubular re-absorption of calcium in the CTAL;21 and (3) reducing urinary concentrating ability in the IMCD by antagonizing the action of vasopressin.22
There has been relatively little work characterizing the factors that regulate CaSR expression in the kidney. A recent report demonstrated that in rat kidney, C-cell and parathyroid in vivo, as well as in a human proximal tubule cell line in vitro, transcription of the CaSR gene was significantly increased following 8 and 12 h of treatment with 1,25(OH)2D3,23 acting via vitamin D response elements upstream of the CaSR gene. Riccardi et al demonstrated in vivo in rats that a low phosphate diet as well as treatment with PTH downregulated CaSR protein in the proximal tubule.24 Thus, CaSR expression in the proximal tubule of the rat kidney is modulated by 1,25(OH)2D3, PTH and, perhaps, dietary phosphate.
Bone
Abundant data indicate that Ca2+o inhibits the formation and activity of osteoclasts and stimulates the activity of osteoblasts. The first evidence for the existence of a G-protein-coupled, cation-sensing mechanism in osteoblasts was presented shortly after the cloning of the CaSR.25 Since then, some, but not all, studies have found that the CaSR is expressed in various osteoblastic cell lines and primary osteoblasts.26,27 An interesting study demonstrated that osteoblasts from CaSR knockout mice still had a promitogenic response to Ca2+o, supporting the presence of a calcium-sensing mechanism other than the full-length CaSR. This mechanism could potentially be represented by the newly cloned GPRC6A, although the latter is responsive to calcimimetics, and the actions of Ca2+o on the CaSR knockout osteoblasts were not.5 The CaSR is also present in articular and hypertrophic chondrocytes.26 Utilizing a type II CaSR agonist in organ culture (fetal rat metatarsal bones) to study the possible role of the CasR in bone growth, Wu et al28 demonstrated that the receptor modulates chondrogenesis in the growth plate and enhances longitudinal bone growth.
The CaSR is expressed by some osteoclasts29 and by monocytes, which are of the same lineage as osteoclast precursors.30 In addition to inhibiting the formation and activity of osteoclasts, high Ca2+o has been shown to promote osteoclast apoptosis.31 However, the calcimimetic, AMG 073, produced none of the actions of elevated Ca2+o on osteoblast proliferation or osteoclast formation and resorption in one study.32 One possible mechanism that has been suggested to mediate calcium sensing in osteoclasts is a plasma membrane, ryanodine-like receptor that couples to increases in the intracellular calcium concentration.33 Therefore, although the CaSR and other calcium-sensing mechanisms may participate in the regulation of bone cell and cartilage function, further studies are clearly required to clarify the divergent results observed in studies to date.
MOLECULAR BIOLOGY OF THE CASR
This section briefly introduces key aspects of the structure and function of the CaSR to provide sufficient background information to understand the molecular basis for both normal mineral ion homeostasis and diseases from mutations of the CaSR.
Structure and signalling pathways of the CaSR
The 5.3-kb clone of the CaSR isolated by expression cloning, when expressed in oocytes, exhibited the same pharmacological properties as the Ca2+o-sensing mechanism previously characterized in dispersed bovine parathyroid cells, the prototypical calcium-sensing cell.2 The use of nucleic-acid-hybridization-based cloning enabled the CaSR to be cloned from humans34 and several other animals. The nucleic acid sequences of the mammalian receptors are at least 85% identical to that of the original bovine parathyroid CaSR. The amino acid sequences show even greater similarity (>90% identity using http://www.ncbi.nlm.nih.gov/BLAST/). Therefore, only a limited amount of divergence from a putative primordial calcium-sensing receptor has taken place throughout evolution, and the functionally important structural features have presumably been retained.
The CaSR is a member of Family C II of the superfamily of 7TM receptors, also called ‘GPCRs’,8 which is by far the largest group of cell surface receptors. They are critical in clinical medicine, since the 7TM receptors represent the targets of about 50% of currently available drugs. The human CaSR comprises 1078 amino acid residues and has three structural domains, as do all 7TM receptors (Figure 2). It has a large extracellular domain (ECD) (612 residues), a transmembrane domain (TMD) of 250 amino acids containing the seven membrane-spanning helices, and an intracellular, C-terminal domain (ICD) of 216 amino acids. The receptor exhibits substantial N-linked glycosylation, which is important for normal cell membrane expression of the receptor, but does not appear to modify the function of the receptor per se.35 The functional cell surface form of the CaSR is a dimer, and the two monomers within the dimeric CaSR are linked by disulphide bonds involving cysteine residues 129 and 131 within each monomer.36 The ECD of each CaSR monomer probably contains more than one binding site for Ca2+o because the Hill coefficient for the activation of the receptor by Ca2+o is 3–4, consistent with the presence of positive cooperativity amongst at least this number of binding sites within the dimeric CaSR.37,38 The TMD also appears to be involved in Ca2+o sensing, since a mutant CaSR lacking the ECD also responds to Ca2+o and other polyvalent cations.39
Figure 2.
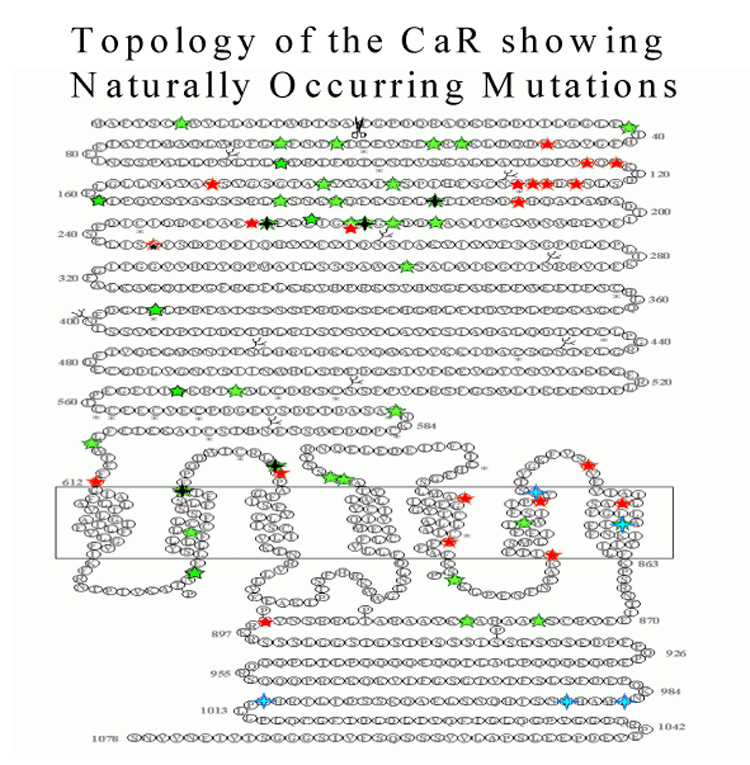
Topology of the calcium-sensing receptor showing some of the naturally occurring mutations.
The CaSR is located in caveolin-1-rich membrane domains called ‘caveolae’ from which it initiates several signalling pathways upon activation in cooperation with several other signalling molecules such as filamin-A and caveolin-1. The CaSR, through Gαq/11 or Gαi/0, activates PLA2, PLD, PLC, PKC, PI3K and mitogen-activated protein kinases like ERK-1/2, but the critical pathway(s) through which the CaSR mediates its biological effects is(are) yet to be identified. A detailed description of CaSR-mediated signalling is beyond the scope of this review, but the reader is referred to a recent discussion of this topic.40
Agonists and antagonists of the CaSR
The CaSR behaves in a promiscuous manner, as there are a considerable number of ligands that modulate its function. CaSR agonists are described as type I or type II.41 Type I agonists are direct agonists listed here in order of potency Gd3+≥La3+≫Ca2+=Ba2+>Sr2+>Mg2+,42 while type II agonists serve as allosteric modulators, requiring the presence of calcium to stimulate the CaSR. Type II modulators left-shift the calcium dose–response curve by sensitizing the receptor to type I agonists and include small molecule drugs and amino acids. The best characterized type I organic polycationic CaSR agonists are gadolinium, neomycin, spermine and amyloid β-peptides.2,43 Newer, more specific approaches, such as the use of pharmacological activators or inhibitors of the receptor, dominant negative constructs or RNA silencing, offer better opportunities for specifying the functional activity of the receptor.
Drugs that allosterically stimulate the CaSR are called ‘calcimimetics’. NPS R-467, NPS R-568 and AMG 073 are calcimimetics that have been used in various experimental studies and clinical trials and, more recently, as treatments for secondary hyperparathyroidism. AMG 073 (also called cinacalcet hydrochloride, Sensipar or Mimpara) is currently the drug of choice, because NPS R-467 and NPS R-568 are degraded by a cytochrome P-450 enzyme, CYP2D6 (Amgen, unpublished data). Five to seven percent of the general population express CYP2D6, which has reduced enzymatic activity, thereby resulting in higher blood levels and delayed metabolic clearance in this segment of the population. Calcimimetics interact with the TMD of the CaSR and increase the affinity of the receptor for calcium. Some L-amino acids also act as type II agonists, in contrast to the respective D-amino acids which are several-fold less potent in activating the receptor.44 Finally, calcilytics represent another type of pharmacological agent acting on the CaSR. They antagonize the action of Ca2+o on the receptor and are being studied for use in the treatment of osteoporosis, as they stimulate a pulse of endogenous PTH secretion which has the potential to exert an anabolic action on bone, similar to that of once-daily injected PTH.45
DISORDERS OF CALCIUM SENSING THAT INVOLVE CASR MUTATIONS
The principal disorders of Ca2+o sensing are listed in Table 2.
Table 2.
Use of calcium-sensing receptor (CaSR)-based therapeutics.
| • | Calcimimetics |
| (1) Approved by FDA | |
| (a) Secondary hyperparathyroidism in dialysis patients | |
| (2) Not yet approved | |
| (a) Primary hyperparathyroidism | |
| (b) Possibly FHH/NSHPT or inactivating CaSR antibodies | |
| • | Calcilytics |
| (1) Not yet FDA approved | |
| (a) Osteoporosis | |
| (b) Possibly activating CaSR mutations or antibodies |
FDA, Food and Drugs Administration; FHH, familial hypocalciurichypercalcaemia; NSHPT, neonatal severe primary hyperparathyroidism.
Clinical and genetic features of familial hypocalciuric hypercalcaemia (OMIM 14598)
The characteristic clinical features of the relatively benign condition now known as familial hypocalciuric hypercalcaemia (FHH) (initially called ‘familial benign hypercalcaemia’) were first descrided in 1972. The diagnosis of FHH can be made in a patient with mild-to-moderate, PTH-dependent hypercalcaemia averaging approximately 2.75 mM (total calcium), an autosomal-dominant pattern of inheritance of a similar degree of hypercalcaemia on family screening, and an inappropriately reduced rate of urinary calcium excretion in the face of hypercalcaemia. Several families, however, have been identified with more marked hypercalcaemia, averaging 3 and 3.4 mM. Patients with FHH are frequently not diagnosed until a routine measurement of the blood calcium concentration shows an unexpectedly high value, or family screening is carried out owing to the birth of a child with NSHPT. Patients with FHH commonly have normal serum levels of PTH despite their hypercalcaemia, although in approximately 15–20% of cases, PTH levels are frankly elevated.46
The hypercalcaemia in FHH, with PTH levels inappropriately high for that serum calcium concentration, reflects the presence of a right-shifted set-point for Ca2+o-regulated PTH release.47 An additional important finding is the normal to frankly reduced urinary calcium excretion in spite of the coexistent hypercalcaemia.48 This alteration in renal calcium handling reflects ‘resistance’ of the kidney to the usual hypercalciuric action of hypercalcaemia, and is the equivalent in the kidney of the resistance of PTH secretion in the parathyroid gland to the normal inhibitory effect of high calcium. Of note, administration of a loop diuretic (e.g. ethacrynic acid) promotes renal excretion of calcium in hypoparathyroid subjects with FHH,49 and more recently has been observed in CaSR knockout mouse models (unpublished observations). These observations point towards a key role of the thick ascending limb (the principal site of action for these classes of diuretics) in the anomalous renal calcium handling in FHH.
Short of carrying out mutational analysis, the most useful means of distinguishing FHH from other forms of hypercalcaemia, particularly primary hyperparathyroidism, is to determine the ratio of the renal clearance of calcium to that of creatinine. A value less than 0.01 is found in approximately 80% of individuals with FHH, while a similar proportion of cases of primary hyperparathyroidism have levels higher than this.50 Another biochemical finding in patients with FHH is their capacity to concentrate their urine normally, in contrast to patients with primary hyperparathyroidism in whom maximal urinary concentration elicited by dehydration is reduced.51 While this finding is not used diagnostically, it reflects renal resistance of FHH patients to the hypercalcaemia-induced diminution in urinary concentrating ability that is observed in other forms of hypercalcaemia. Individuals with FHH usually manifest serum magnesium concentrations that are in the upper normal range or mildly elevated.
While differentiating FHH from primary hyperparathyroidism is usually straightforward, a study of the genetic basis for familial isolated hyperparathyroidism showed that four of 22 unrelated probands harboured inactivating mutations of the CaSR52 restricted to the parathyroid gland. Therefore, the clinician should bear in mind that FHH can be an underdiagnosed but important cause of familial isolated hyperparathyroidism.52,53 Moreover, this study points out that there can be overlap in the clinical presentations of FHH and primary hyperparathyroidism that are important treatment considerations, particularly in familial forms of the latter. This is aptly exemplified by an atypical presentation in an FHH family that exhibited hypercalcaemia, high PTH levels, hypercalciuria and even renal stone formation, but was ultimately proven to harbour an inactivating FHH mutation.54 Subtotal parathyroidectomy in most affected family members provided long-term remission of their biochemical abnormalities, demonstrating that parathyroid surgery, while typically ineffective in curing hypercalcaemia in FHH, may be appropriate in occasional kindreds. While FHH most commonly presents as an asymptomatic form of hypercalcaemia, a few kindreds exhibit more severe hypercalcaemia. Nevertheless, even in these cases, the natural history of the disorder is usually so benign that the great majority of these patients should be followed without intervention, with a few exceptions (see above). In the unusual individual with FHH and symptomatic hypercalcaemia, the new calcimimetics could potentially provide a useful form of treatment.
Finally, studies have described the presence of single and multiple parathyroid ‘adenomas’ in several patients with FHH, although the parathyroid glands in FHH are typically of normal size and histology, or in some cases exhibit mild chief cell or lipohyperplasia with one or more enlarged glands resembling adenomas.54,55
Two hundred and thirteen mutations have been described for the CaSR (188 mis-sense, 17 non-sense, six insertion and/or deletion, one silent and one splice mutation) in the CaSR mutation database (http://www.casrdb.mcgill.ca) related to FHH, NSHPT or ADH families or as de-novo disease. Of these mutations, most are inactivating with the most frequently reported for FHH being 59 mis-sense mutations, six non-sense mutations, six insertions and/or deletions including an Alu element insertion56 and one splice mutation.57 A number of dominant negative mutations (S137P, R185Q, R227L, R795W and F881L) have also been described that appear to inhibit the wild-type partner in mutant wild-type heterodimers, thereby increasing the degree of hypercalcaemia. Linkage analysis showed that the predominant locus of the FHH disease gene (e.g. the CaSR gene) resided on the long arm of chromosome 3 (band q21–24).58 However, FHH is not always linked to chromosome 3q. Notably, two families with clinical features similar to FHH showed linkage to the short and long arms of chromosome 19, respectively; one of these was called the ‘Oklahoma variant’ and exhibited a tendency for the biochemical abnormalities to progress with time.59 FHH that is linked to these latter two loci may be present in a minority of the ~30% of FHH cases without an identifiable mutation in the CaSR gene. The remaining cases of FHH without an identifiable mutation presumably harbour mutations in regulatory regions of the CaSR gene that control its expression, but this remains to be shown directly. Three cases of de-novo NSHPT have been described that were heterozygous for mis-sense mutations located in the ECD, and no mutation was found in the parents.60,61 One individual with de-novo NSHPT was heterozygous for a previously described mutation in an FHH family.60
Clinical and genetic features of neonatal severe primary hyperparathyroidism (OMIM 239200)
There appears to be a gene dosage effect in many inactivating mutations of the CaSR, with heterozygous inactivating mutations leading to FHH with mild hypercalcaemia, and homozygous mutations resulting in NSHPT, a more severe phenotype that manifests very early in life with severe hypercalcaemia, bone demineralization and failure to thrive.
In most cases, NSHPT presents within the first 6 months of life. Affected infants have severe, symptomatic, PTH-dependent hypercalcaemia, along with the bony changes of severe hyperparathyroidism. Infants with NSHPT can exhibit polyuria, dehydration, hypotonia and failure to thrive.46,62,63 A prominent feature of the disease is the associated hyperparathyroid bone disease, which can be associated with multiple fractures. Rib fractures can, in some cases, produce a ‘flail chest’ syndrome that causes respiratory difficulties, owing to a decreased capacity of the affected infant to expand its chest wall and generate the negative intrathoracic pressure needed for normal respiration.64
The mass of the parathyroid glands in NSHPT is generally increased several fold, and they exhibit prominent chief cell hyperplasia. Biochemical evaluation shows hypercalcaemia, hyperparathyroidism and relative hypocalciuria.65 Total serum calcium concentrations range from moderately elevated (e.g. 3–3.25 mM) to levels as high as 7.7 mM in the most severely affected cases.46,66 PTH levels are often 10-fold higher than the upper limit of normal. Early diagnosis is critical as untreated NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy to alleviate the hyperparathyroidism and hypercalcaemia.65 As noted earlier, the most severe cases of NSHPT develop ribcage deformities, as well as rachitic changes, skeletal undermineralization, and fractures of the long bones and other skeletal sites.64,67
More recently, a broader clinical spectrum for NSHPT has become apparent, particularly given the availability of genetic testing of the CaSR gene. As a result, a number of studies have shown that some infants have milder hyperparathyroidism and a substantially milder clinical presentation and natural history.46,61 This latter form of the disease might be better termed ‘neonatal hyperparathyroidism’ to emphasize this milder phenotype in these infants, most of whom harbour heterozygous inactivating CaSR mutations. In these latter cases, the condition can revert with time to a phenotype resembling FHH with medical management alone.46 Therefore, at the moment, parathyroidectomy should be reserved for the most severely affected infants in whom intensive medical therapy (e.g. with aggressive hydration and, if appropriate, bisphosphonates) has failed to stabilize the patient, and there is concern for the infant’s survival.
Three cases of de-novo NSHTP reported in the literature are heterozygous for mis-sense mutations located in the ECD, with only one mutated allele and no mutation found in the parents.60,61 One individual with de-novo NSHPT was heterozygous for a previously described mutation in a FHH family,60 and it is thought that the mutant receptors with these mutations in some cases exert a dominant negative action on the wild-type partner in mutant wild-type heterodimers. Recent reports have described patients with homozygous mutations in the CaSR gene who escaped detection until adulthood, at which time they did not have the usual symptoms and signs of hypercalcaemia and were only identified serendipitously by routine biochemical screening. One such patient, a 35-year-old woman, had two copies of the mis-sense mutation pro39ala from related parents. She was asymptomatic, despite a serum calcium concentration of 3.75–4.25 mM.68 Another such patient, who was homozygous for a distinct inactivating CaSR mutation, was also not diagnosed until adulthood.69 Both mutations produced relatively mild defects of their function when expressed heterologously, perhaps enabling sufficient control of PTH release by calcium to be compatible with a relatively normal life, despite quite marked hypercalcaemia. Indeed, the seeming lack of hypercalcaemic symptoms in the face of moderate to severe hypercalcaemia supports the notion that at least some of these symptoms are mediated by the CaSR. That is, these patients seem to be resistant not only to the effects of calcium on parathyroid gland and kidney, but also to the development of hypercalcaemic symptoms. In these patients, a calcimimetic might represent a means of lowering the serum calcium concentration, assuming the mutant CaSRs were responsive to the drug, thereby providing not only a diagnostic test to determine whether the patient obtained any symptomatic benefit from parathyroidectomy but also, potentially, an effective long-term medical therapy.
NSHPT is most commonly an autosomal-recessive condition; that is, the CaSR genes from both of the parents are mutated (e.g. homozygous FHH). Pollak et al studied 11 kindreds with FHH in whom consanguineous unions engendered four infants with NSHPT.70 It should be recognized, however, that NSHPT is quite uncommon in FHH families considered as a whole. In one case of NSHPT, two distinct mutations, one a mutation in exon 7 from the mother and the other a mutation in exon 4 from the father, caused the disease as a result of the compound heterozygosity in the proband, who thereby lacked any normal CaSRs.71 In theory, NSHPT can result from: (1) homozygosity from a consanguineous FHH union; (2) two mutant alleles of the CaSR gene arising from two distinct FHH kindreds; or (3) from a de-novo mutational event, with or without an inherited, mutant parental allele.61 In addition, an investigation of a girl with phenotypic NSHPT and her family revealed a single mutant allele (present in exon 6, Gly552Arg) in her CaSR gene, while her sister, despite having the same genotype, had phenotypic FHH.72 Thus, factors leading to this degree of phenotypic variation are still only partly understood.
Clinical and genetic features of autosomal-dominant hypoparathyroidism (OMIM 601298)
Patients with this inherited form of hypocalcaemia/hypoparathyroidism are commonly asymptomatic. Some patients, especially children during febrile episodes, can exhibit neuromuscular irritability, seizures and basal ganglia calcification. Patients generally exhibit mild to moderate hypocalcaemia, with serum PTH levels that are inappropriately low given the hypocalcaemia, i.e. within the lower half of the normal range or frankly subnormal.73 Affected individuals often exhibit relative or absolute hypercalciuria, with normal or frankly elevated urinary calcium excretion, respectively, in spite of their low serum calcium concentration. Some, but not all, studies have shown that renal calcium excretion in ADH is higher than that in typical hypoparathyroidism. It is important to prevent renal complications, including nephrocalcinosis, impaired renal function and nephrolithiasis, during treatment of ADH patients with calcium and vitamin D. Treatment with calcium supplements and vitamin D should be reserved for those patients with symptomatic ADH; the goal should be to increase the serum calcium concentration only to a level sufficient to render the patient asymptomatic and not necessarily to a normocalcaemic level. Renal excretion of calcium requires monitoring, and it may be necessary to co-administer a hypocalciuric agent, such as a thiazide diuretic or injectable PTH,74 that can lower urinary calcium excretion at any given level of serum calcium.
ADH, although rare, in index cases may comprise a sizeable fraction of cases of idiopathic hypoparathyroidism, perhaps representing as many as one-third of such cases.75 Patients with this condition harbour an activating mutation of the CaSR gene that resets the set-point of Ca2+o-regulated PTH secretion to the left and lowers renal calcium re-absorption (Figure 2). Soon after the cloning of the CaSR, investigators76 showed linkage of ADH to a locus on chromosome 3 q13; the same locus containing the gene for the CaSR. Shortly afterwards, a heterozygous mis-sense mutation, Q127A, was shown to be the cause of ADH in an unrelated family.73 Since these first reports, 40 mutations have been characterized causing ADH. The majority are mis-sense mutations within the CaSR’s ECD and TMD. In addition, two deletion mutations have been described. Most ADH patients are heterozygous for the activating mutation. In one family, a homozygous mutation was described but it was not associated with a more severe phenotype,77 and although there is a spectrum of phenotypic severity for a given genotype, the symptoms present in affected members of the same family tend to be similar.
When expressed in heterologous systems, these mutations cause a left-shift in the activation of the CaSR by Ca2+o; they only rarely induce constitutive activation of the receptor.58,73,78–80 A recent report identified a family with a large deletion of 181 amino acids within the C-terminus of the CaSR, which increased the sensitivity of the receptor for Ca2+o.77 This family contained the only individual to date known to be homozygous for an activating mutation, but this individual exhibited a phenotype very similar to that of the heterozygous family members. Thus, one mutated allele may be enough to induce a maximal shift in the set-point of Ca2+o-regulated PTH secretion, and the presence of the second mutated allele does not alter the biochemical properties of the receptor dimers any further, perhaps due to a ‘dominant positive’ effect of the mutant receptor on its wild-type partner within the heterodimeric CaSR. Another activating mutation of the CaSR changed a cysteine at amino acid 129 to a serine (Cys129Ser).58,81 As this cysteine participates in dimerization of the CaSR, this result suggests that this cysteine constrains the receptor in its inactive state.
Clinical and genetic features of Bartter’s syndrome subtype V (OMIM 601199.0035)
The Bartter-Gitelman spectrum of disorders is a heterogeneous syndrome of abnormal renal tubular transport characterized by defects in sodium and chloride re-absorption in the thick ascending limb of the loop of Henle (mimicking a furosemide-like effect). One key transporter involved in this spectrum of disorders is the apical Na-2Cl-K transporter that transports sodium and potassium down an electrochemical gradient. The efficiency of this transporter and the maintenance of a transepithelial voltage gradient is enhanced by potassium recycling across the apical renal outer medullary potassium channel (ROMK) that also drives the paracellular absorption of calcium and magnesium.
The overall feature of this class of disorders includes: renal salt wasting; hypokalaemic metabolic alkalosis; elevated renin and aldosterone levels; and normal to low blood pressure. In a small proportion of individuals with Bartter’s syndrome subtype V, additional features of hypercalciuria, hypocalcaemia and hypomagnesaemia may be present and represent gain-of-function mutations in the CaSR that further implicate the CaSR not only in calcium and magnesium homeostasis but also in salt and water homeostasis.82 The CaSR-related cases of Bartter’s syndrome identified to date have been inherited in an autosomal-dominant manner, unlike other subtypes that are inherited as autosomal-recessive traits.
In a 2002 case report, investigators found activating mutations of the CaSR gene in three patients involving L125P, C131W and A843E, which inhibited the activity of the ROMK channel that is mutated in Bartter's syndrome subtype II.83,84 This observation provides the missing link that explains why some activating mutations of CaSR can cause the Bartter's syndrome phenotype. Another recent case report of this syndrome in monozygotic twins involving the K29E mutation in the ECD of the CaSR described mild hypokalaemia, minimal aldosterone and renin production, absent alkalosis but notable hypocalcaemia.85 The K29E mutation has been reported to be a significant activating mutation of the CaSR,86 and buttresses previous observations that the phenotype of Bartters syndrome is variable and not directly related to the in-vitro potency of the known genetic changes associated with this syndrome.87
The clinical presentation of Bartter’s syndrome can occur in the perinatal period with polyhydramnios and premature delivery, or in the first few years of life with polyuria, polydypsia, isosthenuria or hyposthenuria, failure to thrive and frequent episodes of dehydration. The mainstay of treatment at the present time is non-specific and includes fluid and electrolyte replacement. Chronic hypokalaemia worsens the condition and can be ameliorated with amiloride or aldosterone antagonists. Magnesium repletion is frequently required.
POLYMORPHISMS OF THE CASR
In addition to clinically relevant activating and inactivating mutations, single nucleotide polymorphisms (SNPs) have been identified in the general population or in families with FHH and ADH in which the base pair change is present in affected and unaffected persons and does not segregate with known diseases of divalent ion metabolism. Six SNPs have been found in the CaSR gene; one in intron 5 just before exon 6 (IVS 5-88 t/c) and the remaining five in exon 7 in the coding region [one in the sixth TM (A826T), one in the seventh TM (C851S) and three in the ICD (A986S, R990G and Q1011E)]. The polymorphism in intron 5, IVS 5-88 t/c, is very common,88 but no correlation has been found between this mutation and the incidence of parathyroid adenoma or diabetes.89 The A826T mutation has been seen in 16% of 50 normal subjects' samples.90 The C851S mutation was found in an ADH family in both affected and unaffected members,79 but the investigators also found another mutation in this family (A116T) which segregates with the disease, and concluded that C851S was a rare polymorphism. The frequency of three common polymorphisms in the cytoplasmic tail varies in different populations. In one investigation of 377 unrelated DNA samples in a normal Caucasian (Italian) population, the relative frequencies for CaSR SNPs 986S, 990G and 1011E minor alleles were 24%, 4% and 3%, respectively.91
Interestingly, a study analysing serum calcium levels in samples from a normal population found that the homozygous polymorphism A986S was associated with higher serum calcium levels compared with the heterozygous form, while the heterozygous A986S had the lowest calcium levels, suggesting a spectrum of severity in phenotypic features and variable penetrance associated with various mutations.91,92 Further analysis of tri-locus haplotypes for the A986, R990 and Q1011 alleles in the same study91 showed that the highest ionized blood calcium levels were in subjects with the SRQ/ARE genotype compared with the ARQ/ARQ (wild-type) genotype, and suggests that tri-locus haplotyping may be more informative in studies of association between variation in CaSR and disease. At the present time, there is little evidence that the SNPs are disease causing, but there is the possibility that they may be associated with increased risk for disease. For example, on the basis of a CaSR polymorphism haplotype study in stone-forming patients, it is suggested that the 990G variant could influence renal CaSR activation and calcium excretion.93
CASR-BASED THERAPEUTICS
Specific gene-based therapies for disorders of mineral metabolism as a result of mutations of the CaSR are not yet available. Similarly, there are currently no pharmacological agents approved by the Food and Drug Administration (FDA) for treatment of these disorders. However, clinically symptomatic disease as a result of these mutations could potentially be addressed with the use of modulators of the CaSR. The development of allosteric activators (‘calcimimetics’)41 and antagonists (‘calcilytics’)45 of the CaSR has made possible CaSR-based therapy of disorders of Ca2+o homeostasis (Table 1). One foreseeable problem with the use of modulators of the CaSR is the heterogeneity of response to treatment as a result of uniqueness of mutations and variability of binding of drug-ligand with mutant receptors.
One agent that could potentially be used in the management of disorders involving inactivating mutations of the CaSR (i.e. FHH and NSHPT) is AMG073, known as cinacalcet hydrochloride. It has recently been approved by the FDA for use in treating secondary hyperparathyroidism in patients receiving dialysis therapy for end-stage kidney disease,94 as well as in parathyroid cancer. The drug also shows efficacy in mild primary hyperparathyroidism, as described below, but has not yet received FDA approval for this indication.
Calcium receptor antagonists, so-called calcilytics, have also been developed, and their clinical utility is being explored. Again, the FDA has not approved the use of these agents in the treatment of any disorders involving mutations of the CaSR. In the presence of a calcilytic, a higher than usual calcium concentration is needed to suppress PTH levels to a given extent. As a result, the calcium receptor reads normocalcaemia as hypocalcaemia and secretes a pulse of PTH. Similar adjustment in the sensing function of mutant ‘activated CaSR’ in the kidney or other CaSR-expressing tissues can be hypothesized to occur with use of calcylitics, as has been shown with wild-type CaSR in the parathyroid.45,95
SUMMARY AND FUTURE ISSUES
The CaSR is a membrane-bound 7TM receptor expressed in several tissues such as the parathyroid gland, kidney, gut and bone, and regulates Ca2+o homeostasis by acting as the body’s ‘calciostat’.
Patients who have loss-of-function mutations in the CaSR gene exhibit a form of hypercalcaemia that is accompanied by absolute calciuria or hypocalciuria. In the heterozygous form, it produces a benign hypercalcaemic condition, FHH. In the homozygous form (NSHPT), the hypercalcaemia may be lethal if it is not treated surgically.
Gain-of-function mutations produce a generally benign state of hypocalcaemia with relative or absolute hypercalciuria, as seen in ADH or Bartter’s subtype V syndromes.
Polymorphisms of the CaSR exist in the general population but, by definition, are not associated with clinically relevant disease.
Disease-causing mutations of the CaSR can be associated with a spectrum of clinical phenotypes within and between kindreds.
At the present time, treatment for disorders of hyper- and hypocalcaemia due to mutations of the CaSR involve generally supportive measures.
Mutant CaSR is a target for therapeutic manipulation using gene-based therapy, which is currently unavailable. There is potential for use of pharmaceutical agents (calcimimetics and calcylitics). However, this approach is yet to be approved by drug-regulating bodies. One foreseeable problem with the use of modulators of the CaSR is the heterogeneity of response to treatment as a result of uniqueness of mutations and variability of binding of drug-ligand with mutant receptors.
It will be of interest in future studies to collect detailed clinical information in large kindreds to study the possible implications of the CaSR’s altered sensitivity to calcium in these patients, and the resulting alterations in the levels of serum calcium. This could be even more relevant than previously considered, since the CaSR is expressed in numerous organs, such as the breast, brain, intestine and cardiovascular system, which are not thought to be involved in systemic calcium homeostasis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ogo I. Egbuna, Division of Endocrinology, Diabetes and Hypertension, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, and Division of Nephrology, Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA.
Edward M. Brown, Division of Endocrinology, Diabetes and Hypertension, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA.
REFERENCES
- *1.Tfelt-Hansen J, Brown EM. The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci. 2005;42:35–70. doi: 10.1080/10408360590886606. [DOI] [PubMed] [Google Scholar]
- 2.Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 3.Wellendorph P, Hansen KB, Balsgaard A, Greenwood JR, Egebjerg J, Brauner-Osborne H. Deorphanization of GPRC6A: a promiscuous L-alpha-amino acid receptor with preference for basic amino acids. Mol Pharmacol. 2005;67:589–597. doi: 10.1124/mol.104.007559. [DOI] [PubMed] [Google Scholar]
- 4.Wellendorph P, Brauner-Osborne H. Molecular cloning, expression, and sequence analysis of GPRC6A, a novel family C G-protein-coupled receptor. Gene. 2004;335:37–46. doi: 10.1016/j.gene.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Pi M, Faber P, Ekema G, et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J Biol Chem. 2005;280:40201–40209. doi: 10.1074/jbc.M505186200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *6.Hendy GN, D'Souza-Li L, Yang B, Canaff L, Cole DE. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2000;16:281–296. doi: 10.1002/1098-1004(200010)16:4<281::AID-HUMU1>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- *7.Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 9.Bringhurst FR, Demay MB, Kronenberg HM. Hormones and disorders of mineral metabolism. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams Textbook of Endocrinology. 9th edn. Philadelphia: W.B. Saunders; 1998. pp. 1155–1209. [Google Scholar]
- 10.Fudge NJ, Kovacs CS. Physiological studies in heterozygous calcium sensing receptor (CaSR) gene-ablated mice confirm that the CaSR regulates calcitonin release in vivo. BMC Physiol. 2004;4:5. doi: 10.1186/1472-6793-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebert SC, Cheng S, Geibel J. Functions and roles of the extracellular Ca2+-sensing receptor in the gastrointestinal tract. Cell Calcium. 2004;35:239–247. doi: 10.1016/j.ceca.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Brown EM. Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. J Clin Endocrinol Metab. 1983;56:572–581. doi: 10.1210/jcem-56-3-572. [DOI] [PubMed] [Google Scholar]
- 13.Brown AJ, Zhong M, Finch J, et al. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am J Physiol. 1996;270:F454–F460. doi: 10.1152/ajprenal.1996.270.3.F454. [DOI] [PubMed] [Google Scholar]
- 14.Yarden N, Lavelin I, Genina O, et al. Expression of calcium-sensing receptor gene by avian parathyroid gland in vivo: relationship to plasma calcium. Gen Comp Endocrinol. 2000;117:173–181. doi: 10.1006/gcen.1999.7405. [DOI] [PubMed] [Google Scholar]
- 15.Mizobuchi M, Hatamura I, Ogata H, et al. Calcimimetic compound upregulates decreased calcium-sensing receptor expression level in parathyroid glands of rats with chronic renal insufficiency. J Am Soc Nephrol. 2004;15:2579–2587. doi: 10.1097/01.ASN.0000141016.20133.33. [DOI] [PubMed] [Google Scholar]
- 16.Bajwa A, Horst RL, Beckman MJ. Gene profiling the effects of calcium deficiency versus 1,25-dihydroxyvitamin D induced hypercalcemia in rat kidney cortex. Arch Biochem Biophys. 2005;438:182–194. doi: 10.1016/j.abb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Ward DT, Riccardi D. Renal physiology of the extracellular calcium-sensing receptor. Pflugers Arch. 2002;445:169–176. doi: 10.1007/s00424-002-0914-x. [DOI] [PubMed] [Google Scholar]
- 18.Riccardi D, Lee WS, Lee K, Segre GV, Brown EM, Hebert SC. Localization of the extracellular Ca(2+)-sensing receptor and PTH/PTHrP receptor in rat kidney. Am J Physiol. 1996;271:F951–F956. doi: 10.1152/ajprenal.1996.271.4.F951. [DOI] [PubMed] [Google Scholar]
- *19.Riccardi D, Hall AE, Chattopadhyay N, Xu JZ, Brown EM, Hebert SC. Localization of the extracellular Ca2+/polyvalent cation-sensing protein in rat kidney. Am J Physiol. 1998;274:F611–F622. doi: 10.1152/ajprenal.1998.274.3.F611. [DOI] [PubMed] [Google Scholar]
- 20.Ba J, Brown D, Friedman PA. Calcium-sensing receptor regulation of PTH-inhibitable proximal tubule phosphate transport. Am J Physiol Renal Physiol. 2003;285:F1233–F1243. doi: 10.1152/ajprenal.00249.2003. [DOI] [PubMed] [Google Scholar]
- 21.Motoyama HI, Friedman PA. Calcium-sensing receptor regulation of PTH-dependent calcium absorption by mouse cortical ascending limbs. Am J Physiol Renal Physiol. 2002;283:F399–F406. doi: 10.1152/ajprenal.00346.2001. [DOI] [PubMed] [Google Scholar]
- 22.Sands JM, Naruse M, Baum M, et al. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest. 1997;99:1399–1405. doi: 10.1172/JCI119299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canaff L, Hendy GN. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25- dihydroxyvitamin D. J Biol Chem. 2002;277:30337–30350. doi: 10.1074/jbc.M201804200. [DOI] [PubMed] [Google Scholar]
- 24.Riccardi D, Traebert M, Ward DT, et al. Dietary phosphate and parathyroid hormone alter the expression of the calcium-sensing receptor (CaR) and the Na+-dependent Pi transporter (NaPi-2) in the rat proximal tubule. Pflugers Arch. 2000;441:379–387. doi: 10.1007/s004240000436. [DOI] [PubMed] [Google Scholar]
- 25.Quarles LD, Hartle JE, 2nd, Middleton JP, Zhang J, Arthur JM, Raymond JR. Aluminum-induced DNA synthesis in osteoblasts: mediation by a G-protein coupled cation sensing mechanism. J Cell Biochem. 1994;56:106–117. doi: 10.1002/jcb.240560115. [DOI] [PubMed] [Google Scholar]
- 26.Chang W, Tu C, Chen T-H, et al. Expression and signal transduction of calcium-sensing receptors in cartilage and bone. Endocrinology. 1999;140:5883–5893. doi: 10.1210/endo.140.12.7190. [DOI] [PubMed] [Google Scholar]
- 27.Chattopadhyay N, Yano S, Tfelt-Hansen J, et al. Mitogenic action of calcium-sensing receptor on rat calvarial osteoblasts. Endocrinology. 2004;145:3451–3462. doi: 10.1210/en.2003-1127. [DOI] [PubMed] [Google Scholar]
- 28.Wu S, Palese T, Mishra OP, Delivoria-Papadopoulos M, De Luca F. Effects of Ca2+ sensing receptor activation in the growth plate. Faseb J. 2004;18:143–145. doi: 10.1096/fj.03-0294fje. [DOI] [PubMed] [Google Scholar]
- 29.Kanatani M, Sugimoto T, Kanzawa M, Yano S, Chihara K. High extracellular calcium inhibits osteoclast-like cell formation by directly acting on the calcium-sensing receptor existing in osteoclast precursor cells. Biochem Biophys Res Commun. 1999;261:144–148. doi: 10.1006/bbrc.1999.0932. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi T, Ye C, Chattopadhyay N, Sanders JL, Vassilev PM, Brown EM. Enhanced expression of extracellular calcium sensing receptor in monocyte-differentiated versus undifferentiated HL-60 cells: potential role in regulation of a nonselective cation channel. Calcif Tissue Int. 2000;66:375–382. doi: 10.1007/s002230010076. [DOI] [PubMed] [Google Scholar]
- 31.Lorget F, Kamel S, Mentaverri R, et al. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem Biophys Res Commun. 2000;268:899–903. doi: 10.1006/bbrc.2000.2229. [DOI] [PubMed] [Google Scholar]
- 32.Shalhoub V, Grisanti M, Padagas J, et al. In vitro studies with the calcimimetic, cinacalcet HCl, on normal human adult osteoblastic and osteoclastic cells. Crit Rev Eukaryot Gene Expr. 2003;13:89–106. doi: 10.1615/critreveukaryotgeneexpr.v13.i24.30. [DOI] [PubMed] [Google Scholar]
- 33.Zaidi M, Adebanjo OA, Moonga BS, Sun L, Huang CL. Emerging insights into the role of calcium ions in osteoclast regulation. J Bone Miner Res. 1999;14:669–674. doi: 10.1359/jbmr.1999.14.5.669. [DOI] [PubMed] [Google Scholar]
- 34.Garrett JE, Capuano IV, Hammerland LG, et al. Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J Biol Chem. 1995;270:12919–12925. doi: 10.1074/jbc.270.21.12919. [DOI] [PubMed] [Google Scholar]
- 35.Ray K, Clapp P, Goldsmith PK, Spiegel AM. Identification of the sites of N-linked glycosylation on the human calcium receptor and assessment of their role in cell surface expression and signal transduction. J Biol Chem. 1998;273:34558–34567. doi: 10.1074/jbc.273.51.34558. [DOI] [PubMed] [Google Scholar]
- 36.Bai M, Trivedi S, Brown EM. Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem. 1998;273:23605–23610. doi: 10.1074/jbc.273.36.23605. [DOI] [PubMed] [Google Scholar]
- *37.Bai M, Quinn S, Trivedi S, et al. Expression and characterization of inactivating and activating mutations in the human Ca2+o-sensing receptor. J Biol Chem. 1996;271:19537–19545. doi: 10.1074/jbc.271.32.19537. [DOI] [PubMed] [Google Scholar]
- 38.Sanders JL, Chattopadhyay N, Kifor O, Yamaguchi T, Brown EM. Extracellular calcium-sensing receptor (CaR) expression and its potential role in parathyroid hormone-related peptide (PTHrP) secretion in the H-500 rat Leydig cell model of humoral hypercalcemia of malignancy. Biochem Biophys Res Commun. 2000;269:427–432. doi: 10.1006/bbrc.2000.2157. [DOI] [PubMed] [Google Scholar]
- 39.Hu J, McLarnon SJ, Mora S, et al. A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ J Biol Chem. 2005;280:5113–5120. doi: 10.1074/jbc.M413403200. [DOI] [PubMed] [Google Scholar]
- 40.Brown EM. Calcium sensing by endocrine cells. Endocr Pathol. 2004;15:187–220. doi: 10.1385/ep:15:3:187. [DOI] [PubMed] [Google Scholar]
- 41.Nemeth EF, Steffey ME, Hammerland LG, et al. Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA. 1998;95:4040–4045. doi: 10.1073/pnas.95.7.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown EM, Fuleihan Ge-H, Chen CJ, Kifor O. A comparison of the effects of divalent and trivalent cations on parathyroid hormone release, 3',5'-cyclic-adenosine monophosphate accumulation, and the levels of inositol phosphates in bovine parathyroid cells. Endocrinology. 1990;127:1064–1071. doi: 10.1210/endo-127-3-1064. [DOI] [PubMed] [Google Scholar]
- 43.Ye C, Ho-Pao CL, Kanazirska M, et al. Amyloid-beta proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J Neurosci Res. 1997;47:547–554. doi: 10.1002/(sici)1097-4547(19970301)47:5<547::aid-jnr10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 44.Conigrave AD, Quinn SJ, Brown EM. L-amino acid sensing by the extracellular Ca2+- sensing receptor. Proc Natl Acad Sci USA. 2000;97:4814–4819. doi: 10.1073/pnas.97.9.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gowen M, Stroup GB, Dodds RA, et al. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest. 2000;105:1595–1604. doi: 10.1172/JCI9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heath D. Familial benign hypercalcemia. Trends Endocrinol Metab. 1989;1:6–9. doi: 10.1016/1043-2760(89)90022-2. [DOI] [PubMed] [Google Scholar]
- 47.Auwerx J, Demedts M, Bouillon R. Altered parathyroid set point to calcium in familial hypocalciuric hypercalcaemia. Acta Endocrinol (Copenh) 1984;106:215–218. doi: 10.1530/acta.0.1060215. [DOI] [PubMed] [Google Scholar]
- 48.Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW, Jr, Lasker RD. The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Medicine (Baltimore) 1981;60:397–412. doi: 10.1097/00005792-198111000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Attie MF, Gill J, Jr, Stock JL, et al. Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest. 1983;72:667–676. doi: 10.1172/JCI111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuleihan Gel H. Familial benign hypocalciuric hypercalcemia. J Bone Miner Res. 2002;17 Suppl 2:N51–N56. [PubMed] [Google Scholar]
- *51.Marx SJ, Attie MF, Stock JL, Spiegel AM, Levine MA. Maximal urine-concentrating ability: familial hypocalciuric hypercalcemia versus typical primary hyperparathyroidism. J Clin Endocrinol Metab. 1981;52:736–740. doi: 10.1210/jcem-52-4-736. [DOI] [PubMed] [Google Scholar]
- 52.Warner J, Epstein M, Sweet A, et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet. 2004;41:155–160. doi: 10.1136/jmg.2003.016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simonds WF, James-Newton LA, Agarwal SK, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 2002;81:1–26. doi: 10.1097/00005792-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Carling T, Szabo E, Bai M, et al. Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab. 2000;85:2042–2047. doi: 10.1210/jcem.85.5.6477. [DOI] [PubMed] [Google Scholar]
- 55.Burski K, Torjussen B, Paulsen AQ, Boman H, Bollerslev J. Parathyroid adenoma in a subject with familial hypocalciuric hypercalcemia: coincidence or causality? J Clin Endocrinol Metab. 2002;87:1015–1016. doi: 10.1210/jcem.87.3.8304. [DOI] [PubMed] [Google Scholar]
- 56.Janicic N, Pausova Z, Cole DE, Hendy GN. Insertion of an Alu sequence in the Ca(2+)- sensing receptor gene in familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Am J Hum Genet. 1995;56:880–886. [PMC free article] [PubMed] [Google Scholar]
- 57.D'Souza-Li L, Canaff L, Janicic N, Cole DEC, Hendy GN. An acceptor splice site mutation in the calcium-sensing receptor gene in familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Hum Mutat. 2001;18:411–421. doi: 10.1002/humu.1212. [DOI] [PubMed] [Google Scholar]
- 58.Hauache OM. Extracellular calcium-sensing receptor: structural and functional features and association with diseases. Braz J Med Biol Res. 2001;34:577–584. doi: 10.1590/s0100-879x2001000500004. [DOI] [PubMed] [Google Scholar]
- 59.Lloyd SE, Pannett AA, Dixon PH, Whyte MP, Thakker RV. Localization of familial benign hypercalcemia, Oklahoma variant (FBHOk), to chromosome 19q13. Am J Hum Genet. 1999;64:189–195. doi: 10.1086/302202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai M, Pearce SH, Kifor O, et al. In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2+- sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest. 1997;99:88–96. doi: 10.1172/JCI119137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pearce SH, Trump D, Wooding C, et al. Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest. 1995;96:2683–2692. doi: 10.1172/JCI118335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marx SJ, Fraser D, Rapoport A. Familial hypocalciuric hypercalcemia. Mild expression of the gene in heterozygotes and severe expression in homozygotes. Am J Med. 1985;78:15–22. doi: 10.1016/0002-9343(85)90455-3. [DOI] [PubMed] [Google Scholar]
- 63.Brown EM, Bai M, Pollak MR. Familial benign hypocalciuric hypercalcemia and other syndromes of altered responsiveness to extracellular calcium. In: Krane S, Avioli LV, editors. Metabolic Bone Diseases and Clinically Related Disorders. 3rd edn. Academic Press: San Diego; 1997. pp. 479–499. [Google Scholar]
- 64.Grantmyre E. Roentgenographic features of ‘primary’ hyperparathyroidism in infancy. J Can Assoc Radiol. 1973;24:257–260. [PubMed] [Google Scholar]
- 65.Cole DE, Janicic N, Salisbury SR, Hendy GN. Neonatal severe hyperparathyroidism, secondary hyperparathyroidism, and familial hypocalciuric hypercalcemia: multiple different phenotypes associated with an inactivating Alu insertion mutation of the calcium-sensing receptor gene. Am J Med Genet. 1997;71:202–210. doi: 10.1002/(sici)1096-8628(19970808)71:2<202::aid-ajmg16>3.0.co;2-i. published erratum appears in Am J Med Genet 1997; 72: 251–252. [DOI] [PubMed] [Google Scholar]
- *66.Brown EM. Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin North Am. 2000;29:503–522. doi: 10.1016/s0889-8529(05)70148-1. [DOI] [PubMed] [Google Scholar]
- 67.Eftekhari F, Yousefzadeh D. Primary infantile hyperparathyroidism: clinical, laboratory, and radiographic features in 21 cases. Skeletal Radiol. 1982;8:201–208. doi: 10.1007/BF00355507. [DOI] [PubMed] [Google Scholar]
- 68.Aida K, Koishi S, Inoue M, Nakazato M, Tawata M, Onaya T. Familial hypocalciuric hypercalcemia associated with mutation in the human Ca(2+)-sensing receptor gene. J Clin Endocrinol Metab. 1995;80:2594–2598. doi: 10.1210/jcem.80.9.7673400. [DOI] [PubMed] [Google Scholar]
- 69.Fukumoto S, Chikatsu N, Okazaki R, et al. Inactivating mutations of calcium-sensing receptor results in parathyroid lipohyperplasia. Diagn Mol Pathol. 2001;10:242–247. doi: 10.1097/00019606-200112000-00006. [DOI] [PubMed] [Google Scholar]
- 70.Pollak MR, Chou YH, Marx SJ, et al. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest. 1994;93:1108–1112. doi: 10.1172/JCI117062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi M, Tanaka H, Tsuzuki K, et al. Two novel missense mutations in calcium-sensing receptor gene associated with neonatal severe hyperparathyroidism. J Clin Endocrinol Metab. 1997;82:2716–2719. doi: 10.1210/jcem.82.8.4135. [DOI] [PubMed] [Google Scholar]
- 72.Schwarz P, Larsen NE, Lonborg Friis IM, Lillquist K, Brown EM, Gammeltoft S. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism associated with mutations in the human Ca2+-sensing receptor gene in three Danish families. Scand J Clin Lab Invest. 2000;60:221–227. doi: 10.1080/003655100750044875. [DOI] [PubMed] [Google Scholar]
- 73.Pollak MR, Brown EM, Estep HL, et al. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet. 1994;8:303–307. doi: 10.1038/ng1194-303. [DOI] [PubMed] [Google Scholar]
- 74.Winer KK, Yanovski JA, Sarani B, Cutler GB., Jr. A randomized, cross-over trial of once-daily versus twice-daily parathyroid hormone 1-34 in treatment of hypoparathyroidism. J Clin Endocrinol Metab. 1998;83:3480–3486. doi: 10.1210/jcem.83.10.5185. [DOI] [PubMed] [Google Scholar]
- *75.Lienhardt A, Bai M, Lagarde JP, et al. Activating mutations of the calcium-sensing receptor: management of hypocalcemia. J Clin Endocrinol Metab. 2001;86:5313–5323. doi: 10.1210/jcem.86.11.8016. [DOI] [PubMed] [Google Scholar]
- 76.Finegold DN, Armitage MM, Galiani M, et al. Preliminary localization of a gene for autosomal dominant hypoparathyroidism to chromosome 3q13. Pediatr Res. 1994;36:414–417. doi: 10.1203/00006450-199409000-00024. [DOI] [PubMed] [Google Scholar]
- 77.Lienhardt A, Garabedian M, Bai M, et al. A large homozygous or heterozygous in-frame deletion within the calcium-sensing receptor's carboxylterminal cytoplasmic tail that causes autosomal dominant hypocalcemia. J Clin Endocrinol Metab. 2000;85:1695–1702. doi: 10.1210/jcem.85.4.6570. [DOI] [PubMed] [Google Scholar]
- 78.Pearce SH, Williamson C, Kifor O, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335:1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- 79.Baron J, Winer KK, Yanovski JA, et al. Mutations in the Ca(2+)-sensing receptor gene cause autosomal dominant and sporadic hypoparathyroidism. Hum Mol Genet. 1996;5:601–606. doi: 10.1093/hmg/5.5.601. [DOI] [PubMed] [Google Scholar]
- 80.D'Souza-Li L, Yang B, Canaff L, et al. Identification and functional characterization of novel calcium-sensing receptor mutations in familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia. J Clin Endocrinol Metab. 2002;87:1309–1318. doi: 10.1210/jcem.87.3.8280. [DOI] [PubMed] [Google Scholar]
- 81.Hirai H, Nakajima S, Miyauchi A, et al. A novel activating mutation (C129S) in the calcium-sensing receptor gene in a Japanese family with autosomal dominant hypocalcemia. J Hum Genet. 2001;46:41–44. doi: 10.1007/s100380170124. [DOI] [PubMed] [Google Scholar]
- 82.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology. 2005;146:5063–5070. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- *83.Watanabe S, Fukumoto S, Chang H, et al. Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet. 2002;360:692–694. doi: 10.1016/S0140-6736(02)09842-2. [DOI] [PubMed] [Google Scholar]
- 84.Vargas-Poussou R, Huang C, Hulin P, et al. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol. 2002;13:2259–2266. doi: 10.1097/01.asn.0000025781.16723.68. [DOI] [PubMed] [Google Scholar]
- *85.Vezzoli G, Arcidiacono T, Paloschi V, et al. Autosomal dominant hypocalcemia with mild type 5 Bartter syndrome. J Nephrol. 2006;19:525–528. [PubMed] [Google Scholar]
- 86.Hu J, Mora S, Weber G, Zamproni I, Proverbio MC, Spiegel AM. Autosomal dominant hypocalcemia in monozygotic twins caused by a de novo germline mutation near the amino-terminus of the human calcium receptor. J Bone Miner Res. 2004;19:578–586. doi: 10.1359/JBMR.040106. [DOI] [PubMed] [Google Scholar]
- 87.Bettinelli A, Vezzoli G, Colussi G, Bianchetti MG, Sereni F, Casari G. Genotypephenotype correlations in normotensive patients with primary renal tubular hypokalemic metabolic alkalosis. J Nephrol. 1998;11:61–69. [PubMed] [Google Scholar]
- 88.Lovlie R, Eiken HG, Sorheim JI, Boman H. The Ca(2+)-sensing receptor gene (PCAR1) mutation T151M in isolated autosomal dominant hypoparathyroidism. Hum Genet. 1996;98:129–133. doi: 10.1007/s004390050174. [DOI] [PubMed] [Google Scholar]
- 89.Koishi S, Aida K, Tawata M, Onaya T. Polymorphism of the human Ca(2+)-sensing receptor gene in Japanese individuals: no relation to non-insulin dependent diabetes mellitus. Horm Metab Res. 1996;28:541–544. doi: 10.1055/s-2007-979848. [DOI] [PubMed] [Google Scholar]
- 90.Cetani F, Pinchera A, Pardi E, et al. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Miner Res. 1999;14:878–882. doi: 10.1359/jbmr.1999.14.6.878. [DOI] [PubMed] [Google Scholar]
- 91.Scillitani A, Guarnieri V, De Geronimo S, et al. Blood ionized calcium is associated with clustered polymorphisms in the carboxyl-terminal tail of the calcium-sensing receptor. J Clin Endocrinol Metab. 2004;89:5634–5638. doi: 10.1210/jc.2004-0129. [DOI] [PubMed] [Google Scholar]
- 92.Cole DE, Peltekova VD, Rubin LA, et al. A986S polymorphism of the calcium-sensing receptor and circulating calcium concentrations. Lancet. 1999;353:112–115. doi: 10.1016/s0140-6736(98)06434-4. [DOI] [PubMed] [Google Scholar]
- 93.Vezzoli G, Tanini A, Ferrucci L, et al. Influence of the calcium-sensing receptor gene on urinary calium excretion in stone forming patients. J Am Soc Nephrol. 2002;13:2517–2523. doi: 10.1097/01.asn.0000030077.72157.d2. [DOI] [PubMed] [Google Scholar]
- 94.Block GA, Martin KJ, de Francisco AL, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350:1516–1525. doi: 10.1056/NEJMoa031633. [DOI] [PubMed] [Google Scholar]
- 95.Nemeth EF, Fox J, Delmar EG, et al. Stimulation of parathyroid hormone secretion by a small molecule antagonist of the calcium receptor. J Bone Miner Res. 1998;23:S156. abstract 1030. [Google Scholar]