

Abstract
Autophagy is a membrane-mediated intracellular degradation system. The serine/threonine kinase Atg1 plays an essential role in autophagosome formation. However, the role of the mammalian Atg1 homologues UNC-51–like kinase (ULK) 1 and 2 are not yet well understood. We found that murine ULK1 and 2 localized to autophagic isolation membrane under starvation conditions. Kinase-dead alleles of ULK1 and 2 exerted a dominant-negative effect on autophagosome formation, suggesting that ULK kinase activity is important for autophagy. We next screened for ULK binding proteins and identified the focal adhesion kinase family interacting protein of 200 kD (FIP200), which regulates diverse cellular functions such as cell size, proliferation, and migration. We found that FIP200 was redistributed from the cytoplasm to the isolation membrane under starvation conditions. In FIP200-deficient cells, autophagy induction by various treatments was abolished, and both stability and phosphorylation of ULK1 were impaired. These results suggest that FIP200 is a novel mammalian autophagy factor that functions together with ULKs.
Introduction
Autophagy is a primary route by which cytoplasmic contents are directed to the lysosome to be degraded (Cuervo, 2004; Levine and Klionsky, 2004; Rubinsztein, 2006; Mizushima, 2007; Mizushima et al., 2008). There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. Among them, only macroautophagy (referred to as autophagy hereafter) is mediated by the autophagosome. Upon induction of autophagy, a membrane cisterna called the isolation membrane (also termed the phagophore) enwraps a portion of cytoplasm to generate an autophagosome. The autophagosome then fuses with an endosome and, finally, with the lysosome, leading to degradation of cytoplasm-derived materials sequestered inside the autophagosome. Although autophagy occurs at low levels under normal conditions (Hara et al., 2006; Komatsu et al., 2006), autophagy is extensively activated under starvation conditions (Mizushima and Klionsky, 2007).
The molecular mechanism of autophagy has been revealed by genetic analyses performed in yeast (Klionsky, 2005; Suzuki and Ohsumi, 2007), in which 31 autophagy-related ATG genes have been identified so far. Among these genes, Atg1–10, 12–14, 16–18, 29, and 31 (collectively called AP-Atg) are required for autophagosome formation. In yeast, autophagosomes are generated at a special site near the vacuolar membrane, called the preautophagosomal structure (PAS), where most AP-Atg proteins are recruited (Kim et al., 2001a; Suzuki et al., 2001; Suzuki and Ohsumi, 2007). Although autophagy requires only these AP-Atg proteins, an autophagy-related pathway called cytoplasm-to-vacuole targeting (Cvt) pathway, which delivers two vacuolar enzymes, aminopeptidase 1 and α-mannosidase 1, from the cytoplasm to the vacuole, requires almost all Atg proteins except Atg17, 29, and 31.
AP-Atg proteins are classified into six functional groups: the Atg1 protein kinase complex; the Atg2–Atg18 complex; the Atg8 conjugation system; the Atg12 conjugation system; the Atg14–phosphatidylinositol 3-kinase complex; and Atg9 (Suzuki et al., 2007). Among these functional units, the Atg1 complex has a unique feature: it apparently receives the starvation signals. Atg1 is a serine/threonine protein kinase, and its kinase activity can be up-regulated after autophagy-inducible treatments such as nutrient starvation or rapamycin treatment (Kamada et al., 2000). The kinase activity of Atg1 is believed to be required for autophagy, although there have been debates (Kamada et al., 2000; Abeliovich et al., 2003; Kabeya et al., 2005; Cheong et al., 2008). The Atg1 complex includes Atg13, Atg17 (Kamada et al., 2000), Atg29 (Kawamata et al., 2008), Atg31/Cis1 (Kabeya et al., 2007), Atg11/Cvt9 (Kim et al., 2001b), Atg24/Cvt13 (Nice et al., 2002), Atg20/Cvt20 (Nice et al., 2002), and Vac8 (Scott et al., 2000). Atg17 (Kamada et al., 2000), 29 (Kawamata et al., 2005), and 31 (Kabeya et al., 2007) are specifically involved in autophagy, whereas Atg11, Atg20, Atg24, and Vac8 are specifically required for the Cvt pathway. Atg13 and 1 are involved in both pathways. A recent systematic analysis revealed that Atg17 and 11 are essential for PAS organization, and Atg17 has been suggested to behave as a scaffold protein (Suzuki et al., 2007). The Atg1–Atg17 interaction largely depends on Atg13 (Cheong et al., 2005; Kabeya et al., 2005), but a yeast two-hybrid analysis suggested that Atg1 and 17 can also directly interact with each other (Cheong et al., 2005). Interactions between Atg13, 1, and 17 are enhanced by starvation treatment, and both Atg13 and 17 are important for proper regulation of Atg1 kinase activity (Kamada et al., 2000; Kabeya et al., 2005).
The function of the Atg17–Atg13–Atg1 complex has yet to be fully understood. Although Atg1 and 13 sense nutrient conditions, this complex does not appear to function as a simple transducer of starvation signaling. For example, Atg1 is required for a late step of micropexophagy in Pichia pastoris (Mukaiyama et al., 2002) and for the Cvt pathway, which is a constitutive biosynthetic pathway proceeding under nutrient-rich conditions (Kamada et al., 2000; Abeliovich et al., 2003). Furthermore, Atg1 is known to be important for retrieval of Atg9 and 23 from PAS (Reggiori et al., 2004).
Homologues of Atg1 have been found in other species such as Dictyostelium discoideum (Otto et al., 2004), Caenorhabditis elegans (Ogura et al., 1994; Melendez et al., 2003), Drosophila melanogaster (Scott et al., 2004), Arabidopsis thaliana (Hanaoka et al., 2002), and mammals (Yan et al., 1998, 1999). Mutants of Atg1 in the species examined thus far indeed exhibit autophagy-defective phenotypes (Melendez et al., 2003; Otto et al., 2004; Scott et al., 2004; Chan et al., 2007).
In metazoa, however, the role of Atg1 seems not to be limited to macroautophagy. C. elegans Atg1 is known as UNC (uncoordinated movement)-51. The unc-51 mutants show neurological abnormalities such as paralysis and defects in axonal elongation (Ogura et al., 1994). In mammals, two Atg1 homologues have been reported: UNC-51–like kinase (ULK) 1 (also known as Unc51.1; Yan et al., 1998; Tomoda et al., 1999) and ULK2 (also known as Unc51.2; Tomoda et al., 1999; Yan et al., 1999). RNAi-mediated suppression of ULK1 expression alone is sufficient to inhibit autophagy (Chan et al., 2007) and redistribution of mAtg9 from the TGN to endosomes (Young et al., 2006). In addition to the autophagy phenotype, a dominant-negative form of ULK1, K46R, suppresses neurite extension of cerebellar granular neurons (Tomoda et al., 1999), which is consistent with the neurological phenotype of the unc-51 worm.
The observations that Atg1 is a multifunctional protein suggest that Atg1 should function in concert with several distinct partners. Indeed, C. elegans UNC-51 interacts with UNC-14, a protein involved in coordinated movement (Ogura et al., 1997). In mammals, ULK1 interacts with SynGAP, a negative regulator of Ras, and Syntenin, a Rab5-interacting protein (Tomoda et al., 2004). These interactions have been suggested to be important for axon guidance/elongation. Another recent paper also suggested that ULK1 is recruited to the TrkA–NGF receptor complex by p62 and regulates non–clathrin-coated endocytosis in growth cones, filopodia extension, and branching (Zhou et al., 2007). However, homologues of the yeast Atg1-interacting autophagy proteins have not been reported in higher eukaryotes (except for plant Atg13; Hanaoka et al., 2002).
To better understand the role of the Atg1/ULK family, we screened for ULK-interacting proteins and identified the FAK family–interacting protein of 200 kD (FIP200), which is also called RB1CC1 (retinoblastoma 1–inducible coiled-coil 1). FIP200 was reported to interact with multiple proteins, including FAK (Abbi et al., 2002), Pyk2 (Ueda et al., 2000), TSC1 (Gan et al., 2005), p53 (Melkoumian et al., 2005), ASK1, and TRAF2 (Gan et al., 2006), thereby regulating a variety of cellular functions such as cell migration, proliferation, cell size, and apoptosis. FIP200 was also independently identified as a novel inducer of RB1. We found that ULK1, ULK2, and FIP200 are present on autophagic isolation membrane. Using FIP200−/− mouse embryonic fibroblasts (MEFs), we revealed that FIP200 is a novel mammalian autophagy factor that functions together with ULKs.
Results
ULK1 and ULK2 localize on the isolation membrane (phagophore)
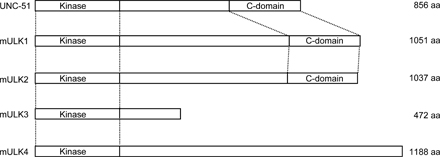
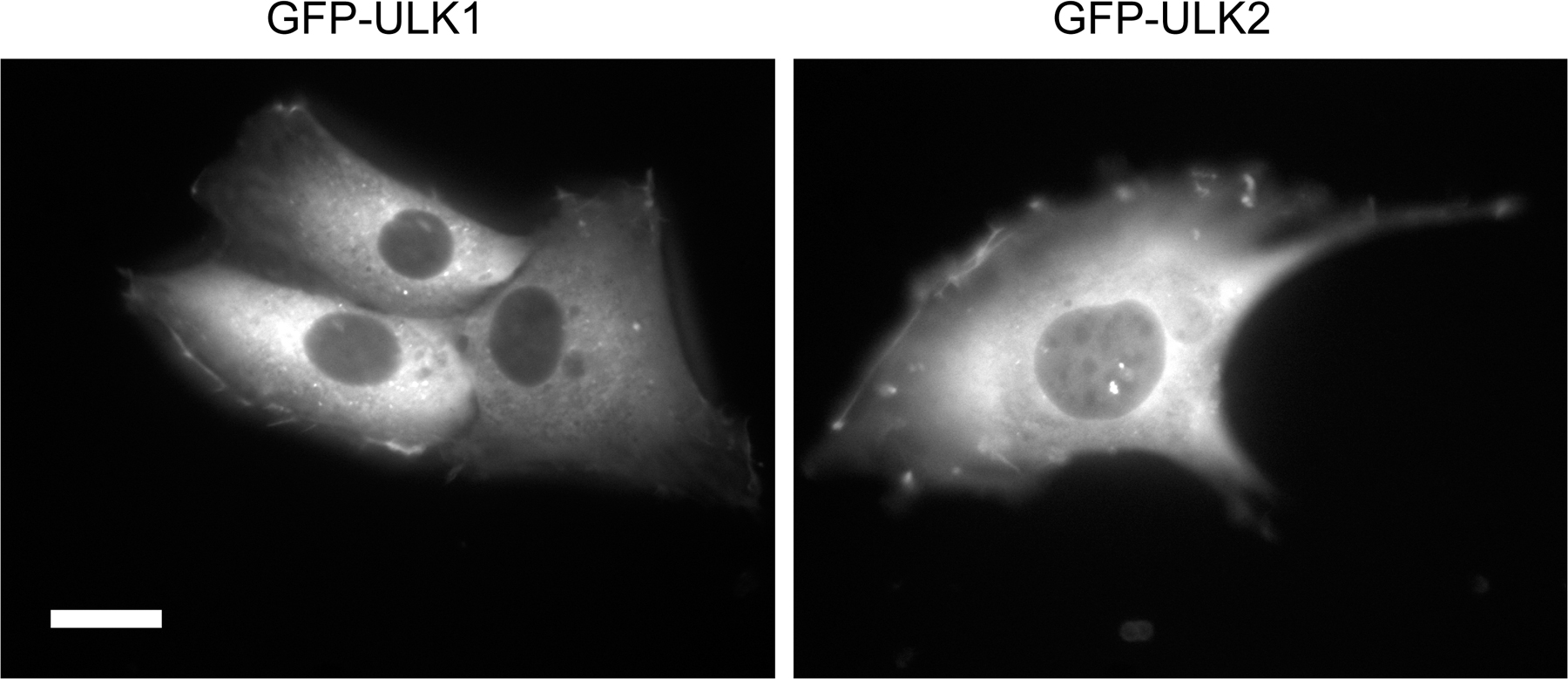
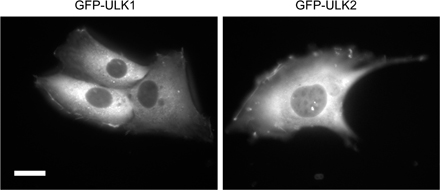
We found four ULK homologues in the mouse database. Although ULK1 and 2 are closely related to C. elegans UNC51, ULK3 and 4 show similarity to UNC51 only in the kinase domain (Fig. S1, available at http://jcb.org/cgi/content/full/jcb.200712064/DC1). We therefore analyzed the role of ULK1 and 2 in autophagosome formation. We first determined the subcellular distribution of ULK1 and 2 using NIH3T3 cells stably expressing ULKs fused with GFP at the N terminus (GFP-ULK1 and GFP-ULK2). Under nutrient-rich conditions, GFP-ULK1 and GFP-ULK2 were mostly found to distribute evenly throughout the cytoplasm, with few punctuate dots (Fig. 1, A and B, complete). We occasionally observed GFP-ULK1 and -ULK2 at the ruffled membrane in some cells (Fig. S2). After amino acid and serum starvation, GFP-ULK1 and -ULK2 localized to punctuate structures (Fig. 1, A and B, starvation). These punctuate structures immediately disappeared after replenishment with nutrient medium (Fig. 1, A and B, starvation→complete). As previously reported (Chan et al., 2007), these data suggest that ULK1 and 2 are targeted to autophagy-related structures. We then examined the localization of GFP-ULK in more detail and found that both ULK1 and 2 colocalized with endogenous Atg16L1 almost completely (Fig. 1, C and D). Because Atg16L1, together with the Atg12–Atg5 conjugate, specifically localizes to elongating isolation membrane (also called the phagophore; Mizushima et al., 2001), these data suggest that both ULK1 and 2 are targeted to the isolation membrane.
Figure 1.

ULK1 and 2 localize to the isolation membrane (phagophore) under starvation conditions. (A and B) NIH3T3 cells stably expressing GFP-ULK1 (A) and -ULK2 (B) were cultured in complete or starvation medium for 30 min. They were then cultured in fresh complete medium for an additional 30 min (starvation→complete). (C and D) NIH3T3 cells stably expressing GFP-ULK1 (C) and -ULK2 (D) were cultured in starvation medium for 120 min. The cells were fixed, permeabilized, and subjected to immunofluorescence microscopy using anti-Atg16L1 antibody and Alexa Fluor 660–conjugated secondary antibody. More than 90% of GFP-ULKs dots were positive for Atg16L1. (E–H) Wild-type (E and G) and Atg5−/− (F and H) MEFs were transfected with retroviral vectors encoding GFP-ULK1 and -ULK2. MEFs stably expressing GFP-ULK1 (E and F) and -ULK2 (G and H) were cultured in complete or starvation medium for 120 min. The cells were fixed and examined by fluorescence microscopy. Bars, 20 μm.
In yeast, Atg1 localizes to the PAS independently of Atg5 (Suzuki et al., 2007). Although it is not clear whether mammalian cells have a similar PAS, we examined whether the puncta formation of ULKs was independent of Atg5. Wild-type and Atg5−/− MEFs were transfected with retroviral vectors encoding GFP-ULK1 (Fig. 1, E and F) and -ULK2 (Fig. 1, G and H) and observed after starvation. Although both GFP-ULK1 and -ULK2 puncta were formed in wild-type MEFs (Fig. 1, E and G), these dots were never observed in Atg5−/− MEFs (Fig. 1, F and H). These results suggest that ULK puncta formation is dependent on Atg5 in mammalian cells.
Kinase-dead ULK mutants inhibit autophagy
In yeast, Atg1 kinase activity is up-regulated during autophagy induced by nitrogen starvation or Tor inactivation (Kamada et al., 2000). However, it is not known whether this is the case in mammalian cells. We therefore determined ULK kinase activity in MEFs under nutrient-rich and starved conditions. Endogenous ULK1 was precipitated with anti-ULK1 antibody, and the resultant immunoprecipitate was analyzed by an in vitro kinase assay using myelin basic protein as a model substrate. The ULK1 kinase activity level under the starvation condition is increased 1.3-fold relative to the nutrient-rich condition (Fig. 2 A). Although this change was modest, these data suggest that the increase in ULK1 kinase activity might be important for the induction of autophagosome formation.
Figure 2.

The kinase-dead ULK mutants inhibit autophagy. (A) In vitro kinase assay of endogenous ULK1. MEFs were cultured in complete or starvation medium for 60 min. ULK1 kinase activity was determined as described in Materials and methods. Relative kinase activity is shown. Data are the mean ± SE of five independent experiments. (B and C) NIH3T3 cells were transiently transfected with HA-ULK1, HA-ULK2, or their kinase-dead mutants and subjected to immunofluoresence microscopy using monoclonal anti-HA and polyclonal anti-Atg16L1 antibodies for primary staining and Alexa Fluor 488–conjugated goat anti–mouse IgG and Alexa Fluor 568–conjugated goat anti–rabbit IgG antibodies for secondary antibodies. Transfected cells are indicated by arrows. Bars, 20 μm. (D) NIH3T3 cells stably expressing GFP-ULK1K46N and GFP-ULK2K39T were cultured in starvation medium for 120 min. The cells were subjected to immunofluorescence microscopy using anti-Atg16L1 antibody. Bar, 20 μm. (E) NIH 3T3 cells were transfected with the retroviral vectors encoding HA-ULK1K46N or with the corresponding empty retrovirus (mock). They were cultured in complete or starvation medium for 120 min. Cell lysates were then analyzed by immunoblot analysis with the indicated antibodies.
We next examined the importance of the ULK kinase activity using ULK kinase-dead mutants. Overexpression of kinase-dead Atg1 mutants inhibits autophagy in D. discoideum (Tekinay et al., 2006) and D. melanogaster (Scott et al., 2007), whereas overexpression of wild-type Atg1 accelerates autophagy in D. melanogaster (Scott et al., 2007). However, in mammalian cells, both wild-type and kinase-dead ULK1 suppress autophagy, as judged by the LC3 conversion assay (Chan et al., 2007). We therefore carefully examined the effect of wild-type and kinase-dead mutants of ULK1 and 2 on Atg16L1 puncta formation. In transient transfection experiments, moderate expression of kinase-dead HA-ULK1K46N (Yan et al., 1998) and HA-ULK2K39T (Yan et al., 1999) efficiently suppress the starvation-induced Atg16L1 puncta formation (Fig. 2 C), whereas wild-type ULK1 and 2 showed almost no effect (Fig. 2 B; note that HA-ULK dots are not as clear as those in stable transformants [Fig. 1] because of high cytoplasmic signals caused by overexpression). In contrast, when overexpressed in higher levels, both wild-type and kinase-dead ULKs suppressed Atg16L1 puncta formation (unpublished data). The shape of wild-type ULK-overexpressing cells was abnormal (Fig. S3, available at http://jcb.org/cgi/content/full/jcb.200712064/DC1), as previously demonstrated by Chan et al. (2007). The cells generated protrusions and, finally, detached from the culture dish, which is consistent with the previous results in D. melanogaster that Atg1 overexpression caused apoptotic cell death (Scott et al., 2007). However, these abnormalities were not observed in cells overexpressing kinase-dead ULK1 and 2 (Fig. S3). Therefore, the effects of overexpression of wild-type and kinase-dead ULKs are different. The kinase-dead mutants indeed act as dominant-negative mutants, whereas wild-type overexpression may cause cytotoxicity by some other mechanism. Collectively, these data suggest that the kinase activity is important for the involvement of ULKs in autophagy.
We also generated NIH3T3 cells stably expressing GFP-ULK1K46N and GFP-ULK2K39T. Although both Atg16L1 and GFP-ULKs formed puncta in wild-type NIH3T3 cells after starvation, these puncta were only very rarely observed in NIH3T3 cells stably expressing GFP-ULK1K46N or GFP-ULK2K39T (Fig. 2 D), confirming that kinase-dead ULKs function as a dominant-negative mutant in autophagosome formation.
We next measured this effect by the LC3 conversion assay. Conversion of cytosolic LC3 (LC3-I) to membrane-bound phosphatidylethanolamine (PE)–conjugated LC3 (LC3-II) occurs during autophagy, and the amount of LC3-II is correlated with the number of autophagosomes (Kabeya et al., 2000). This LC3 conversion during starvation was markedly suppressed in NIH3T3 cells stably expressing ULK1K46N (Fig. 2 E), as recently reported by Chan et al. (2007). Another assay was conducted to monitor autophagy flux. Because p62 (SQSTM1/sequestosome 1) can bind LC3, p62 is preferentially incorporated into autophagosomes and degraded by autophagy (Bjørkøy et al., 2005; Mizushima and Yoshimori, 2007). The amount of p62 can therefore serve as a good indicator of autophagic activity. The base level of p62 was up-regulated in ULK1K46N-transfected cells compared with mock-transfected cells. Although the level of p62 decreased during starvation in mock-transfected cells, the decrease was modestly suppressed in ULK1K46N-transfected cells. These data suggest that autophagic flux was attenuated by expression of the kinase-dead ULKs.
Identification of FIP200 as a ULK-interacting protein
In yeast, Atg1 forms a complex with multiple proteins including Atg13 and 17. However, a different set of ULK-interacting proteins has been reported in mammals that includes GABARAP, GATE-16 (Okazaki et al., 2000), SynGAP, and Syntenin (Tomoda et al., 2004) but not other Atg homologues. To better understand the role of ULK, we searched for additional ULK-interacting proteins. Mouse ULK1-FLAG and FLAG-ULK2 were expressed in HEK293 cells and immunoprecipitated with an anti-FLAG antibody. We analyzed the immunoprecipitates by highly sensitive direct nanoflow liquid chromatography/tandem mass spectrometry (Natsume et al., 2002) and identified FIP200, which is also called RB1CC1, in both the ULK1 and 2 precipitates. We confirmed the interaction of FIP200 with ULK1 and 2 by immunoprecipitation analysis using HEK293T cells coexpressing FLAG-FIP200 and either HA-ULK1 or -ULK2 (Fig. 3 A). Furthermore, we generated antibody against FIP200 and detected the interaction between endogenous ULK1 and FIP200 in wild-type MEFs (Fig. 3 B). This coprecipitation was not observed if we performed the same experiments using FIP200−/− MEFs. The interaction between ULK1 and FIP200 was not affected by nutrient conditions, suggesting that ULK1 and FIP200 physically interact with each other under both nutrient-rich and starvation conditions (Fig. 3 B).
Figure 3.

ULK1 interacts with FIP200. (A) HEK293T cells were cotransfected with FLAG-FIP200 and HA-ULKs. Cell lysates were subjected to immunoprecipitation (IP) using antibodies against FLAG. The resulting precipitates were examined by immunoblot (IB) analysis with the indicated antibodies. The asterisk indicates nonspecific band. (B) Lysates of MEFs were immunoprecipitated with anti-ULK1 or anti-FIP200 antibody or preimmune rabbit serum, and the resulting precipitates were subjected to immunoblot analysis with antibodies against ULK1 and FIP200. (C) Schematic representation of ULK1 mutants used in D. (D) HEK293T cells were cotransfected with FLAG-FIP200 and various ULK1 mutants and analyzed as in A using anti-HA and anti-FLAG antibodies. (E) NIH3T3 cells stably expressing GFP-ULK1 (left) and GFP-ULK1ΔC (right) were cultured in starvation medium for 120 min. Bar, 20 μm.
We next determined which region of ULK1 is required for the interaction with FIP200, using several ULK1 mutants (Fig. 3 C). Although kinase-dead ULK1K46N could interact with FIP200, the C-terminal deletion mutants (ULK11–427 and ULK11–828) could not (Fig. 3 D). The C-terminal deletion mutant ULK11–828 also failed to accumulate to punctuate dots (Fig. 3 E). Thus, the C-terminal region (829–1051 aa) of ULK1 is required for both ULK-FIP200 interaction and puncta formation.
FIP200 localizes to the isolation membrane
FIP200, a ubiquitously expressed protein (Bamba et al., 2004), was originally identified as a Pyk2 (proline-rich tyrosine kinase 2)-interacting protein (Ueda et al., 2000). FIP200 binding inhibits Pyk2 kinase activity, thereby inhibiting Pyk2-induced apoptosis. FIP200 also associates with FAK as a negative regulator (Abbi et al., 2002). Additionally, FIP200 interacts with multiple proteins such as TSC1 (Gan et al., 2005), p53 (Melkoumian et al., 2005), ASK1, and TRAF2 (Gan et al., 2006). FIP200 also induces RB1 expression (Chano et al., 2002a). Therefore, FIP200 is a multifunctional protein that is involved in cell migration, proliferation, cell size regulation, cell death, and tumor suppression. However, its involvement in autophagy or membrane trafficking has not been reported.
To investigate the functional relevance of FIP200 in autophagosome formation, we first examined the subcellular localization of FIP200. FIP200 has been previously reported to localize to the nucleus (Chano et al., 2002a), cytoplasm (Ueda et al., 2000), and focal contacts in the cell periphery (Abbi et al., 2002). When we observed NIH3T3 cells transfected with a retrovirus vector encoding GFP-fused FIP200 under nutrient-rich conditions, most GFP signals were detected diffusely in the cytoplasm, with only a few punctate dots (Fig. 4 A). After amino acid and serum starvation, however, the number of these dots increased. These GFP-FIP200–positive dots were almost completely colocalized with Atg16L1 (Fig. 4 B). Furthermore, we observed almost complete colocalization between GFP-ULK1 and endogenous FIP200 (Fig. 4 C). All these data suggest that FIP200 localizes to elongating isolation membrane together with ULKs.
Figure 4.

FIP200 localizes to phagophore after starvation treatment. (A) NIH3T3 cells stably expressing GFP-FIP200 were cultured in complete or starvation medium for 120 min and the GFP signal was observed. (B) NIH3T3 cells stably expressing GFP-FIP200 were cultured in starvation medium for 120 min and then subjected to immunofluorescence microscopy using anti-Atg16L1 antibody and Alexa Fluor 568–conjugated secondary antibody. Bars, 20 μm. More than 90% of GFP-FIP200 dots were positive for Atg16L1. (C) NIH3T3 cells stably expressing GFP-ULK1 were starved for 120 min and then subjected to immunofluorescence microscopy using anti-FIP200 antibody and Alexa Fluor 568–conjugated secondary antibody. Black squares indicate the enlarged areas shown in insets. Bar, 20 μm.
FIP200 is required for autophagy
The isolation membrane localization of FIP200 prompted us to further examine its role in autophagy. FIP200 is known to be essential for embryonic development. FIP200−/− mice show embryonic lethality between embryonic day (E) 13.5 and 16.5 because of defective heart and liver development (Gan et al., 2006). We therefore examined the autophagic activity of MEFs derived from FIP200−/− embryos. In wild-type MEFs, 1 h of amino acid and serum starvation induced LC3 conversion, which was restored by an additional 1-h incubation in complete DME supplemented with 10% FCS (Fig. 5 A). In contrast, the starvation-induced LC3 conversion was almost completely abolished in FIP200−/− MEFs. Furthermore, p62 accumulated in FIP200−/− MEFs, suggesting that autophagy is profoundly suppressed in the absence of FIP200. It should be noted, however, that a small amount of LC3-II was detected in FIP200−/− MEFs irrespective of nutrient conditions. This phenotype was quite different from that of Atg5−/− MEFs, in which LC3-II was never detected (Fig. 5 A). These results suggest either that low-level autophagy constitutively occurs in FIP200−/− MEFs or that LC3 conversion occurs independently of autophagy.
Figure 5.

FIP200 is required for autophagy. (A) FIP200+/+, FIP200−/−, Atg5+/+, and Atg5−/− MEFs were cultured in complete or starvation medium for 60 min. In the recovery experiments, starved MEFs were cultured in fresh complete medium for an additional 60 min (replenishment). The cell lysates were subjected to immunoblot analysis with the indicated antibodies. (B) Wild-type and FIP200−/− MEFs were cultured in the complete or starvation medium for indicated time with or without 100 nM bafilomycin A1. The cell lysates were subjected to immunoblot analysis with anti-LC3 antibody. (C) Wild-type and FIP200−/− MEFs were transfected with retroviral vectors encoding GFP-Atg5 or GFP-LC3. Resulting cells were cultured in the starvation medium for 120 min. The cells were fixed and examined by fluorescence microscopy. Bar, 20 μm. (D–G) Wild-type (D and E) and FIP200−/− (F and G) MEFs were cultured in complete (D and F) or starvation (E and G) medium for 120 min and then fixed and subjected to EM analysis. Autophagosome-like structures (open arrowheads), and autolysosomes (closed arrowheads) are indicated. Bar, 1 μm. (H) The ratio of total area of autophagosomes (AP) and autolysosomes (AL) to total cytoplasmic area in D–G was determined by morphometric analysis.
To monitor the autophagic ability of FIP200−/− MEFs more precisely, we determined the autophagy flux in these cells by the LC3 turnover assay (Tanida et al., 2005; Mizushima and Yoshimori, 2007). Because LC3-II is present on both outer and inner autophagosome membranes, LC3-II itself is degraded after autophagosome-lysosome fusion. If autophagosome-lysosome fusion is blocked with the vacuolar H+ ATPase inhibitor bafilomycin A1 (Yamamoto et al., 1998), autophagic degradation of LC3-II should be suppressed. This treatment indeed caused accumulation of LC3-II in 2-h and 4-h starved wild-type MEFs (Fig. 5 B). However, this effect of bafilomycin A1 was not observed in FIP200−/− MEFs even after 4-h starvation, suggesting that autophagic degradation is suppressed almost completely in these cells.
We also examined autophagy induction in wild-type and FIP200−/− MEFs stably expressing GFP-Atg5 (isolation membrane marker) or GFP-LC3 (autophagosome marker) by monitoring the redistribution of cytosolic GFP-Atg5 and GFP-LC3 to membrane structures. After a 2-h culture in amino acid- and serum-deprived medium, several GFP-Atg5 and GFP-LC3 dots were observed in wild-type MEFs (Fig. 5 C, left). In FIP200−/− cells, however, these GFP-Atg5–positive dots were completely absent (Fig. 5 C, right, top). The number of GFP-LC3 dots was greatly reduced, but several GFP-LC3–positive dots were still observed in FIP200−/− cells (Fig. 5 C, right, bottom). However, these dots were irregular in shape, and their number did not change after starvation treatment (unpublished data). These structures therefore likely represent some aberrant structures or aggregates of GFP-LC3 protein caused by FIP200 deficiency. To further confirm the effect of loss of FIP200 on autophagosome formation, we performed EM analysis. In wild-type MEFs after a 2-h starvation, we could detect numerous autophagic vacuoles, which occupied ∼3% of the total cytoplasmic volume (∼2% autophagosomes and ∼1% autolysosomes; Fig. 5, D, E, and H). In contrast, autophagosome-like structures were hardly observed in FIP200−/− MEFs even under amino acid and serum starvation conditions (Fig. 5, F, G, and H). These results demonstrate that FIP200 is required for autophagosome formation.
The role of FIP200 in autophagy is independent of FAK
FIP200 can bind FAK and inhibit FAK function. FAK is one of the focal adhesion components that include multiple proteins such as vinculin, paxillin, and talin (Carragher and Frame, 2004). In addition, while we were preparing this manuscript, it was reported that paxillin also acts as a regulator of autophagy (Chen et al., 2007). On that basis, we investigated the potential role of FAK in autophagy using FAK−/− MEFs (Ilic et al., 1995). As far as we tested, FAK−/− MEFs demonstrated no abnormalities related to autophagy. Starvation-induced LC3 conversion with and without bafilomycin A1 and starvation-induced p62 degradation in the lysosome were normal (Fig. 6 A). There was also no difference in starvation-induced GFP-LC3 dot formation between FAK+/− and FAK−/− MEFs (Fig. 6 B). Additionally, GFP-fused FAK localized to focal adhesions but not to the Atg16L1-positive autophagy-related structure (Fig. 6 C). These results suggest that FAK is not involved in autophagy and that the role of FIP200 in autophagy is independent of FAK.
Figure 6.

FAK is not required for autophagy. (A) FAK+/− and FAK−/− MEFs were cultured in the complete or starvation medium for the indicated times with or without 100 nM bafilomycin A1. The cell lysates were subjected to immunoblot analysis with the indicated antibodies. (B) FAK+/− and FAK−/− MEFs stably expressing GFP-LC3 were cultured in complete or starvation medium for 120 min. The cells were fixed and examined by fluorescence microscopy. (C) Wild-type MEFs stably expressing GFP-FAK were cultured in complete or starvation medium for 120 min. The cells were fixed and subjected to immunofluorescence microscopy using anti-Atg16 antibody. Black squares indicate the enlarged areas shown in insets. Bars, 20 μm.
FIP200 functions downstream of mammalian target of rapamycin (mTOR) in autophagosome formation
FIP200 interacts with TSC1 and inhibits its function (Gan et al., 2005). The TSC1-TSC2 heterodimer inhibits mTOR function through Rheb inactivation (Inoki and Guan, 2006; Wullschleger et al., 2006; Guertin and Sabatini, 2007). As autophagy is negatively regulated by mTOR signaling (Meijer and Codogno, 2004), we investigated whether the autophagy defect of FIP200−/− MEFs is a result of aberrant TSC-mTOR signaling. If this were the case, autophagy should be induced by treatment with the mTOR inhibitor rapamycin. In wild-type MEFs, rapamycin induced LC3 conversion both in the absence and presence of bafilomycin A1. However, the LC3 conversion induced by rapamycin was still impaired in FIP200−/− MEFs (Fig. 7 A). We also observed that rapamycin was able to induce GFP-Atg5 and GFP-LC3 dot formation in wild-type MEFs but not in FIP200−/− MEFs (Fig. 7 B). Furthermore, mTOR appeared to be normally suppressed in FIP200−/− MEFs after serum and amino acid starvation, as judged by 4E-BP1 dephosphorylation, despite the suppression of autophagy (Fig. 5 A). Collectively, these data suggest that the defect in autophagosome formation of FIP200−/− MEFs is caused by loss of FIP200 function downstream of mTOR and not by aberrant nutrient signaling including the TSC complex.
Figure 7.

FIP200 functions downstream of mTOR in autophagosome formation. (A) Wild-type and FIP200−/− MEFs were treated with 100 ng/ml rapamycin (rapa) or vehicle (DMSO) for 120 min in the presence or absence of 100 nM bafilomycin A1. The cell lysates were subjected to immunoblot analysis with anti-LC3 antibody. (B) Wild-type and FIP200−/− MEFs stably expressing GFP-Atg5 or GFP-LC3 were cultured in the presence of 100 ng/ml rapamycin for 120 min. The formation of GFP-Atg5 (top) and GFP-LC3 (bottom) puncta was examined by fluorescence microscopy. Bar, 20 μm. (C) Wild-type and FIP200−/− MEFs were treated with 10 mM lithium chloride for 24 h or 100 μM C2-ceramide for 2 h.
FIP200−/− MEFs were also resistant to various autophagy-inducing reagents such as lithium chloride (Sarkar et al., 2005) and ceramide (Fig. 7 C; Scarlatti et al., 2004). Because the effect of lithium is independent of mTOR (Sarkar et al., 2005), FIP200 should function in both mTOR-dependent and -independent autophagy.
FIP200 is required for ULK puncta formation and is important for the stability and proper phosphorylation of ULK
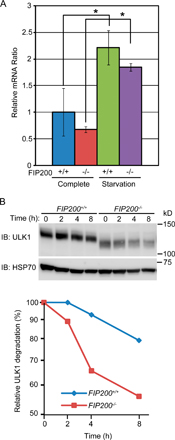
Given that FIP200 interacts with ULK1 and 2, we postulated that ULKs and FIP200 function together. We first examined the membrane targeting of ULKs in wild-type and FIP200−/− MEFs. As we demonstrated in Fig. 1, GFP-ULK puncta were formed in wild-type MEFs during starvation. These puncta represent the isolation membrane (Fig. 8 A, left). However, these dots were never observed in FIP200−/− MEFs, suggesting that puncta formation of ULK1 and 2 depends on FIP200 (Fig. 8 A, right). However, this observation may simply reflect impairment of isolation membrane formation in the absence of FIP200, because even GFP-Atg5 dot formation was suppressed in FIP200−/− MEFs (Fig. 5 C). To explore more direct functional connections between ULK and FIP200, we next examined the expression status of ULK1 in wild-type and FIP200−/− MEFs. The expression level of ULK1 in FIP200−/− MEFs was much lower than in wild-type MEFs under both nutrient rich (Fig. 8 B) and starvation (not depicted) conditions. The ULK1 mRNA expression was enhanced after starvation, but there was no difference between wild-type and FIP200−/− cells (Fig. S4 A, available at http://jcb.org/cgi/content/full/jcb.200712064/DC1). However, we observed faster decay of ULK1 protein in FIP200−/− cells after cycloheximide treatment, suggesting that ULK1 is destabilized in the absence of FIP200 (Fig. S4 B). Additionally, we found that ULK1 was detected as a smeared band with faster mobility in FIP200−/− MEFs. As previously reported (Yan et al., 1998, 1999), we observed that mobility of ULK1K46N is faster than that of wild-type ULK1, suggesting that ULK1 is autophosphorylated (Fig. 3 D). Consistent with this, when we treated ULK1 immunoprecipitated from wild-type cells with λ phosphatase, ULK1 migrated to a lower position that was suppressed in the presence of phosphatase inhibitors, confirming that ULK1 is phosphorylated (Fig. 8 C). Likewise, the phosphatase treatment of ULK1 from FIP200−/− cell lysates produced a band with the same mobility (Fig. 8 C, compare lanes 2 and 5), indicating that the faster migration of endogenous ULK1 in FIP200−/− MEFs represents reduced phosphorylation rather than other modifications such as protein processing. Therefore, FIP200 is likely important for maintenance of the ULK1 kinase activity. All these results suggest that FIP200 and ULK1 are functionally connected and that this complex plays an important role in autophagosome formation.
Figure 8.

FIP200 is required for membrane targeting and proper function of ULK. (A) FIP200+/− and FIP200−/− MEFs stably expressing GFP-ULK1 or -ULK2 were cultured in starvation medium for 120 min. The cells were fixed and examined by fluorescence microscopy. Bar, 20 μm. (B) FIP200+/+ and FIP200−/− MEFs were cultured in complete medium. The cell lysates were subjected to immunoblot analysis with antibodies against ULK1 or HSP90 (loading control). (C) Phosphatase sensitivity of ULK1. FIP200+/+ and FIP200−/− MEFs were cultured in complete medium. ULK1 was immunoprecipitated from cell lysates and treated with λ phosphatase for 30 min at 30°C in the absence or presence of phosphatase inhibitors (1 mM Na2VO4, 50 mM KF, 15 mM Na4P2O7, and 1 mM EGTA).
Discussion
We have identified a novel ULK-interacting protein, FIP200, and demonstrated that ULK1, ULK2, and FIP200 localize to the isolation membrane. Because FIP200-deficient cells displayed virtually no autophagosomes, we propose that the ULK–FIP200 complex is essential for an early step, not a later or completion step, of autophagosome formation.
Comparison between yeast Atg1 and mammalian ULK in the autophagy pathway
Although yeast Atg1 and mammalian ULK possess several common features, there are some differences. One difference is that although both proteins target to autophagic structures, precise localization may not be the same. Yeast Atg1 is known to localize to the PAS (Suzuki et al., 2007). However, because Atg1 is delivered to the vacuole (Suzuki et al., 2001), it should be present on the inner membrane of autophagosomes or captured inside autophagosomes. In contrast, we detected ULK1 and 2 only on the isolation membrane, which suggests that ULK1 and 2 associate primarily with the outer membrane of autophagosomes like the Atg12–Atg5 conjugate (Mizushima et al., 2001). Accordingly, both ULK1 and FIP200 are not degraded in lysosome because expression levels of these proteins did not change after treatments of bafilomycin A1 (Fig. S5, available at http://jcb.org/cgi/content/full/jcb.200712064/DC1).
Another difference is the Atg5 dependency of Atg1/ULK dot formation. Although PAS targeting of yeast Atg1 does not depend on Atg5 (Suzuki et al., 2007), puncta formation of ULK1 and 2 depends on Atg5 in mammalian cells (Fig. 1). One possible explanation of this apparent discrepancy between yeast and mammalian Atg1/ULK would be that the PAS-equivalent structure is not visible in mammalian cells, and localization of Atg1/ULK to isolation membrane should be dependent on Atg5 both in yeast and mammals. Dissection of PAS and the isolation membrane in yeast would clarify this issue.
We did not observe any difference between ULK1 and 2 in the present study, although ULK2 has been reported to be dispensable for autophagy (Young et al., 2006; Chan et al., 2007). The function of ULK2 thus remains unclear. Examination of endogenous ULK2 will be required.
FIP200, a possible counterpart of yeast Atg17?
It was previously shown that FIP200 interacted with TSC1, and phosphorylation of S6 kinase after amino acid stimulation was impaired in cells treated with FIP200 siRNA (Gan et al., 2005). However, this effect was observed only moderately in FIP200−/− MEFs (Gan et al., 2005). We also failed to detect clear defect in this pathway as we tested the phosphorylation levels of 4E-BP1 (Fig. 5 A). There might be adaptation to the FIP200-deficient conditions in permanently knocked out cells. However, our present study does not exclude the possibility that FIP200 functions with the TSC complex. FIP200 may function both upstream and downstream of mTOR, but the downstream effect can be dominant when we analyze autophagy.
As FIP200 interacts with ULK1 and 2 and is essential for autophagy, one might speculate that this protein is a counterpart of yeast Atg13 or 17. Indeed, although FIP200 (1594 aa) is much larger than Atg17 (417 aa), FIP200 shares several features with yeast Atg17. First, both Atg17 and FIP200 have multiple coiled-coil domains. Second, just as Atg17 is required for Atg1 kinase activity (Kamada et al., 2000; Kabeya et al., 2005), the phosphorylation status of ULK1 is reduced in FIP200-deficient cells, which may indicate diminished ULK1 activity (Fig. 8). It is therefore likely that Atg17 and FIP200 can support the function of Atg1 and ULKs, respectively. We also found that ULK1 kinase activity per cell was reduced in FIP200−/− cells (unpublished data). However, as ULK1 expression level itself was also lower in FIP200−/− cells than in wild-type cells (Fig. 8), we could not conclude how much specific activity of ULK1 reduced in the absence of FIP200. Third, the C-terminal region of ULK1 is essential for both FIP200 interaction and ULK1 puncta formation (Fig. 3; Chan et al., 2007), suggesting that interaction with FIP200 is important for membrane targeting. This is consistent with the previous finding that yeast Atg17 is required for PAS targeting of Atg1, which is particularly apparent in the atg11 mutant (Suzuki et al., 2007; Cheong et al., 2008; Kawamata et al., 2008). Fourth, FIP200 has multiple binding partners other than ULK1 and 2. Similarly, systematic analyses of yeast protein interactions by two-hybrid analysis and mass spectrometry identified many potential Atg17-interacting proteins, some of which are unrelated to autophagy (Ho et al., 2002; Gavin et al., 2006; Krogan et al., 2006). Therefore, both Atg17 and FIP200 may function as a scaffold for multiple proteins. Finally, homologues of Atg17 are present in Kluyveromyces lactis and Eremothecium gossypii, where FIP200 homologue is missing. On the other hand, FIP200 homologues are found in Homo sapiens, Mus musculus, Gallus gallus, and D. melanogaster, where Atg17 homologues are absent, but not in Saccharomyces cerevisiae. This mutual exclusiveness suggests that these two groups of proteins may serve similar functions for which one, but not both, proteins are required. Identification and analysis of a functional homologue of mammalian Atg13, a critical factor of this complex, will further clarify the functional relationship between yeast and mammalian Atg1 complex.
LC3 conversion in FIP200−/− cells
One notable finding in this study is that significant amounts of LC3-II, which is the most widely used autophagy indicator, are detectable in FIP200-deficient cells. This may indicate that a very low level of autophagy occurs even in the absence of FIP200, which is consistent with the finding that small autophagosomes are occasionally generated in yeast atg17 mutant cells (Cheong et al., 2005; Kabeya et al., 2005). However, this leaky phenotype is likely mediated by Cvt-specific Atg11, which is absent in mammalian cells (Suzuki et al., 2007; Cheong et al., 2008; Kawamata et al., 2008). Indeed, the autophagy flux analysis of FIP200−/− cells suggested that autophagic degradation is nearly completely abolished (Fig. 5 B). Thus, the small amount of LC3-II detected in FIP200−/− cells may be generated in an autophagy-independent manner. In yeast, Atg8–PE is generated even in atg17 mutant cells as well as in several other autophagy mutants such as atg1, 2, 6, 9, 13, 14, and 16 mutants (Suzuki et al., 2001). Atg8–PE conjugation is severely affected only in atg5, 10, and 12 mutant cells and is completely blocked in atg3, 4, and 7 mutant cells (Suzuki et al., 2001). The Atg12–Atg5 conjugate was recently reported to have an E3-like enzyme activity for the Atg8–PE conjugation reaction (Hanada et al., 2007). Therefore, the Atg8–PE conjugation can take place in an autophagy-independent manner. The Atg knockout mammalian cells reported so far are Atg5−/− and Atg7−/−. Although LC3-II formation is absent in these cells, this is probably caused by deficiency of Atg12 and 8 conjugation and not simply autophagy deficiency. As observed in FIP200−/− cells, Beclin 1 silencing blocks autophagosome formation but does not cause complete suppression of the LC3 conversion (Zeng et al., 2006; Matsui et al., 2007). Therefore, it is not contradictory that FIP200−/− cells show some LC3-II, even if the autophagic activity is virtually suppressed. Rather, these data suggest that we should measure the autophagy flux, not the absolute amount of LC3-II, to monitor autophagy (Tanida et al., 2005; Mizushima and Yoshimori, 2007).
ULK/Atg1 and focal adhesion components
We have demonstrated that FIP200 localizes to the autophagic isolation membrane and is essential for autophagy. We do not think that FIP200 is an autophagy-specific protein. The most striking evidence for this is that FIP200−/− mice die between E13.5 and 16.5 with defective heart and liver development (Gan et al., 2006), whereas autophagy-deficient Atg5−/− and Atg7−/− mice survive embryogenesis (although they die within one day of birth). Indeed, FIP200 involvement has been suggested in various cellular processes, such as inhibition of Pyk2-induced signaling (Ueda et al., 2000), inhibition of cell migration through FAK inhibition (Abbi et al., 2002), tumor suppression (Chano et al., 2002b), induction of RB expression (Chano et al., 2002a), cell size regulation through inhibition of the TSC complex (Gan et al., 2005; Chano et al., 2006), inhibition of p53-mediated G1-S progression (Melkoumian et al., 2005), and inhibition of TNF-induced apoptosis, probably through inhibition with ASK1 and TRAF2 (Gan et al., 2006). These data suggest that FIP200 is a multifunctional protein.
An important question is whether FIP200 alone or FIP200 and its interacting proteins are involved in autophagy. While we were preparing this manuscript, a D. melanogaster genetic analysis revealed a genetic interaction between Atg1 and paxillin, a cytoskeletal scaffolding protein (Chen et al., 2007). The study further demonstrates that paxillin and vinculin redistribute from focal adhesions to intracellular structures under starvation conditions and that paxillin-deficient MEFs are defective in autophagy. In contrast, our analysis showed that FAK is not required for autophagy, suggesting that the focal adhesion complex per se is not involved in autophagy. FAK-independent communication between FIP200 and paxillin will be the subject of future studies in both autophagy and focal adhesion.
Given that at least three of the focal adhesion components function in autophagy, it is possible that ULKs also plays a role in cell adhesion that is independent of its function in autophagy. Indeed, GFP-ULK1 and -ULK2 were occasionally detected at the cell periphery (Fig. S2), and overexpression of wild-type ULKs caused abnormal cell morphology such as cell rounding and protrusion (Fig. S3). So far, several studies have suggested autophagy-independent roles of Atg1/ULK in various species, such as axonal elongation and branching in C. elegans (Ogura et al., 1994) and mammals (Tomoda et al., 1999; Zhou et al., 2007). These functions may be related to the role of Atg1 in focal adhesion.
In conclusion, we have identified a novel ULK-interacting protein, FIP200, which is functionally similar to yeast Atg17. The ULK–FIP200 complex is essential for autophagy, but the precise role of this complex is unknown. Identification of physiologically relevant substrates of Atg1–ULK is one of the most straightforward approaches, although it has not been successful thus far in any species studied (Deminoff and Herman, 2007). Searching for additional interacting proteins will also facilitate understanding of the role of Atg1–ULK complex and the regulation of autophagy induction and autophagosome formation.
Materials and methods
Plasmids
IMAGE Consortium cDNA clones (GenBank accession no. BC017556) encoding human RB1CC1/FIP200 were obtained from Invitrogen. The cDNA encoding FIP200 was cloned into p3×FLAG CMV10 (Sigma-Aldrich). Expression constructs for wild-type and kinase-dead mouse ULK1 and 2 were gifts from N. Okazaki (Kazusa DNA Research Institute, Kisarazu, Japan) and M. Muramatsu (Tokyo Medical and Dental University, Tokyo, Japan). The kinase-dead ULK1 mutant (with conserved ATP binding Lys 46 replaced with Asn) and the kinase-dead mutant ULK2 (with Lys 39 to Thr replacement) were previously described (Yan et al., 1998, 1999). FAK cDNA constructs were provided by S. Hanks (Vanderbilt University, Nashville, TN).
Cell culture and transfection
Atg5−/− (Kuma et al., 2004), FIP200−/− (Gan et al., 2006), and FAK+/−, FAK−/− (gift from S. Aizawa, Institute of Physical and Chemical Research, Kobe, Japan; Ilic et al., 1995) MEFs were generated previously. MEFs and HEK293T cells were cultured in DME supplemented with 10% FBS and 50 μg/ml penicillin and streptomycin (complete medium) in a 5% CO2 incubator. Bovine calf serum was used instead of FBS for NIH3T3 cells. For starvation, cells were washed with PBS and incubated in amino acid–free DME without FBS (starvation medium). Fugene 6 reagent (Roche) and lipofectamine 2000 reagent (Invitrogen) were used for transfection.
Antibodies and reagents
Polyclonal antibodies against FIP200 or ULK1 were generated in rabbits by standard procedures with fragments of recombinant human FIP200 (residues 200–413 and 1–633) or mouse ULK1 (residues 738–1052) as antigens. Polyclonal anti-LC3 (Hosokawa et al., 2006) and anti-Atg16L1 antibodies (Mizushima et al., 2003) were described previously. Polyclonal anti-ULK1 antibody (A7481) and monoclonal anti-FLAG (M2) and anti–α-tubulin (DM1A) were purchased from Sigma-Aldrich. Another polyclonal anti-ULK1 antibody was purchased from Santa Cruz Biotechnology, Inc. A monoclonal anti-HA antibody (HA11) was purchased from Covance. Monoclonal anti-HSP90 and anti-HSP70 antibodies were purchased from BD Biosciences. Polyclonal antibodies to 4E-BP1 were purchased from Cell Signaling Technology. Polyclonal antibodies to p62 were purchased from American Research Products, Inc. Alexa Fluor 488–, 568–, and 660–conjugated goat anti–rabbit IgG (H + L) antibodies (Invitrogen) were used for immunochemistry. Rapamycin and lithium chloride were purchased from Sigma-Aldrich. Bafilomycin A1 was purchased from Wako Pure Chemical Industries, Ltd. C2-ceramide was purchased from EMD.
EM
MEFs were fixed in 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4, for 2 h. The cells were washed three times in phosphate buffer containing 1 mM glycine and were postfixed in 1% OsO4 in 0.1 M phosphate buffer, pH 7.4, for 1 h. The cells were further dehydrated with a graded series of ethanol and were embedded in epoxy resin. Ultrathin sections were doubly stained with uranyl acetate and lead citrate and observed using an electron microscope (7100; Hitachi). For morphometric analysis, at least 20 sections of each sample were analyzed using MetaMorph image analysis software (version 6.2; MDS Analytical Technologies).
Retroviral expression system
cDNAs encoding human FIP200, wild-type mouse ULK1, ULK1K46N, ULK1ΔC, ULK2, ULK2K39T, Atg5, and FAK, and rat LC3 were N-terminally fused to the GFP fragment. A cDNA encoding mouse ULK1K46N was N-terminally tagged with the HA epitope. These cDNAs were subcloned into pMXs-puro or pMXs-IP (provided by T. Kitamura, University of Tokyo, Tokyo, Japan). The resulting vectors were used to transfect Plat E cells and thereby generate recombinant retroviruses. MEFs and NIH 3T3 cells were infected with the recombinant retroviruses and selected in medium containing 1 μg/ml puromycin. Cells stably expressing the recombinant proteins were pooled for experiments (Kamura et al., 2004).
Immunoprecipitation and immunoblotting
Cell lysates were prepared in a lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor cocktail [Complete EDTA-free protease inhibitor; Roche]). The lysates were clarified by centrifugation at 15,000 rpm for 15 min and were subjected to immunoprecipitation using specific antibodies in combination with protein G–Sepharose (GE Healthcare). Precipitated immunocomplexes were washed five times in lysis buffer and boiled in sample buffer. Samples were subsequently separated by SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore). Immunoblot analysis was performed with the indicated antibodies and visualized with SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific). The signal intensities were analyzed using an imaging analyzer (LAS-3000mini; Fujifilm) and Multi Gauge software (version 3.0; Fujifilm). Contrast and brightness adjustment was applied to the whole images using Photoshop 7.0.1 (Adobe).
Fluorescence microscopy
MEF or NIH3T3 cells expressing protein fused to GFP were directly observed with a fluorescence microscope (IX81; Olympus) equipped with a charge-coupled device camera (ORCA ER; Hamamatsu Photonics). A 60× PlanAPO oil immersion lens (1.42 NA; Olympus) was used. Images were acquired using MetaMorph image analysis software version. For examination by immunofluorescence microscopy, cells grown on gelatinized coverslips were fixed and stained with an anti-Atg16L1, anti-FIP200, or anti-HA antibody as previously described (Mizushima et al., 2001).
Kinase assay
Cells were washed with PBS and then lysed in an extraction buffer (20 mM Tris-HCl, pH7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 5 mM EGTA, 1 mM sodium pyrophosphate, 5 mM NaF, 1 mM Na3VO4, and 0.5% Triton X-100) supplemented with protease inhibitors (10 μg/ml each of pepstatin A, chymostatin, leupeptin, and E64 [Peptide Institute, Inc.]). ULK1 was immunoprecipitated with anti-ULK1 antibody, and an in vitro protein kinase assay was performed for 30 min at 30°C in the presence of γ-[32P]ATP (GE Healthcare) and myelin basic protein (Millipore). The reaction products were separated by SDS-PAGE and the intensities of the 32P-labeled myelin basic protein bands were visualized with a BAS image analyzer (Fujifilm). The signal intensities were quantified using Multi Gauge software. The amount of immunoprecipitated ULK1 was monitored by immunoblot analysis using ULK1 antibody (Santa Cruz Biotechnology, Inc.).
Real-time PCR
Real-time PCR was performed on a Thermal Cycler Dice (Takara) using SYBR premix EX Taq (Takara). The primer sets used were as follows: mouse ULK1 forward, 5′-TTACCAGCGCATCGAGCA-3′; and reverse, 5′-TGGGGAGAAGGTGTGTAGGG-3′; and mouse β-actin forward, 5′-CTGGGTATGGAATCCTGTGG-3′; and reverse, 5′-GTACTTGCGCTCAGGAGGAG-3′. Amplicon expression in each sample was normalized to its β-actin mRNA content.
Online supplemental material
Fig. S1 shows the structural comparison of murine ULK homologues (ULK1–4). Fig. S2 shows the peripheral localization pattern of ULK1 and 2. Fig. S3 shows the aberrant cell morphology of cells overexpressing wild-type ULK but not in kinase-dead ULK. Fig. S4 shows the ULK1 mRNA expression and protein turnover in wild-type and FIP200−/− MEFs. Fig. S5 shows endogenous expression of ULK1 and FIP200 in MEFs in the presence or absence of 100 nM bafilomycin A1. Online supplemental material is available at http://jcb.org/cgi/content/full/jcb.200712064/DC1.
Supplemental Material
Acknowledgments
We thank Dr. Shinichi Aizawa (Institute of Physical and Chemical Research) for providing FAK−/− MEFs, Dr. Steven K. Hanks (Vanderbilt University) for FAK cDNA, and Dr. Toshio Kitamura (The University of Tokyo) for the retroviral vectors and Plat E cells.
This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors also thank the Kato Memorial Bioscience Foundation and the Toray Science Foundation for financial support.
Abbreviations used in this paper: Cvt, cytoplasm-to-vacuole targeting; E, embryonic day; FIP200, FAK family–interacting protein of 200 kD; MEF, mouse embryonic fibroblast; mTOR, mammalian target of rapamycin; PAS, preautophagosomal structure; PE, phosphatidylethanolamine; ULK, UNC-51–like kinase.
References
- Abbi, S., H. Ueda, C. Zheng, L.A. Cooper, J. Zhao, R. Christopher, and J.L. Guan. 2002. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell. 13:3178–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeliovich, H., C. Zhang, W.A. Dunn Jr., K.M. Shokat, and D.J. Klionsky. 2003. Chemical genetic analysis of Apg1 reveals a non-kinase role in the induction of autophagy. Mol. Biol. Cell. 14:477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamba, N., T. Chano, T. Taga, S. Ohta, Y. Takeuchi, and H. Okabe. 2004. Expression and regulation of RB1CC1 in developing murine and human tissues. Int. J. Mol. Med. 14:583–587. [PubMed] [Google Scholar]
- Bjørkøy, G., T. Lamark, A. Brech, H. Outzen, M. Perander, A. Øvervatn, H. Stenmark, and T. Johansen. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher, N.O., and M.C. Frame. 2004. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14:241–249. [DOI] [PubMed] [Google Scholar]
- Chan, E.Y., S. Kir, and S.A. Tooze. 2007. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 282:25464–25474. [DOI] [PubMed] [Google Scholar]
- Chano, T., S. Ikegawa, K. Kontani, H. Okabe, N. Baldini, and Y. Saeki. 2002. a. Identification of RB1CC1, a novel human gene that can induce RB1 in various human cells. Oncogene. 21:1295–1298. [DOI] [PubMed] [Google Scholar]
- Chano, T., K. Kontani, K. Teramoto, H. Okabe, and S. Ikegawa. 2002. b. Truncating mutations of RB1CC1 in human breast cancer. Nat. Genet. 31:285–288. [DOI] [PubMed] [Google Scholar]
- Chano, T., M. Saji, H. Inoue, K. Minami, T. Kobayashi, O. Hino, and H. Okabe. 2006. Neuromuscular abundance of RB1CC1 contributes to the non-proliferating enlarged cell phenotype through both RB1 maintenance and TSC1 degradation. Int. J. Mol. Med. 18:425–432. [PubMed] [Google Scholar]
- Chen, G.C., J.Y. Lee, H.W. Tang, J. Debnath, S.M. Thomas, and J. Settleman. 2007. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 4:37–45. [DOI] [PubMed] [Google Scholar]
- Cheong, H., T. Yorimitsu, F. Reggiori, J.E. Legakis, C.W. Wang, and D.J. Klionsky. 2005. Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell. 16:3438–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong, H., U. Nair, J. Geng, and D.J. Klionsky. 2008. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae.Mol. Biol. Cell. 19:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo, A.M. 2004. Autophagy: in sickness and in health. Trends Cell Biol. 14:70–77. [DOI] [PubMed] [Google Scholar]
- Deminoff, S.J., and P.K. Herman. 2007. Identifying atg1 substrates: four means to an end. Autophagy. 3:667–673. [DOI] [PubMed] [Google Scholar]
- Gan, B., Z.K. Melkoumian, X. Wu, K.L. Guan, and J.L. Guan. 2005. Identification of FIP200 interaction with the TSC1–TSC2 complex and its role in regulation of cell size control. J. Cell Biol. 170:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan, B., X. Peng, T. Nagy, A. Alcaraz, H. Gu, and J.L. Guan. 2006. Role of FIP200 in cardiac and liver development and its regulation of TNFα and TSC–mTOR signaling pathways. J. Cell Biol. 175:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin, A.C., P. Aloy, P. Grandi, R. Krause, M. Boesche, M. Marzioch, C. Rau, L.J. Jensen, S. Bastuck, B. Dumpelfeld, et al. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature. 440:631–636. [DOI] [PubMed] [Google Scholar]
- Guertin, D.A., and D.M. Sabatini. 2007. Defining the role of mTOR in cancer. Cancer Cell. 12:9–22. [DOI] [PubMed] [Google Scholar]
- Hanada, T., N.N. Noda, Y. Satomi, Y. Ichimura, Y. Fujioka, T. Takao, F. Inagaki, and Y. Ohusmi. 2007. The ATG12-ATG5 conjugate has a novel e3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282:37298–37302. [DOI] [PubMed] [Google Scholar]
- Hanaoka, H., T. Noda, Y. Shirano, T. Kato, H. Hayashi, D. Shibata, S. Tabata, and Y. Ohsumi. 2002. Leaf senescence and starvation-induced chlorosis are accelerated by the disruption of an Arabidopsis autophagy gene. Plant Physiol. 129:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, T., K. Nakamura, M. Matsui, A. Yamamoto, Y. Nakahara, R. Suzuki-Migishima, M. Yokoyama, K. Mishima, I. Saito, H. Okano, and N. Mizushima. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889. [DOI] [PubMed] [Google Scholar]
- Ho, Y., A. Gruhler, A. Heilbut, G.D. Bader, L. Moore, S.L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 415:180–183. [DOI] [PubMed] [Google Scholar]
- Hosokawa, N., Y. Hara, and N. Mizushima. 2006. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 580:2623–2629. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, T. Yamamoto, and S. Aizawa. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- Inoki, K., and K.L. Guan. 2006. Complexity of the TOR signaling network. Trends Cell Biol. 16:206–212. [DOI] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., Y. Kamada, M. Baba, H. Takikawa, M. Sasaki, and Y. Ohsumi. 2005. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell. 16:2544–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., T. Kawamata, K. Suzuki, and Y. Ohsumi. 2007. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae.Biochem. Biophys. Res. Commun. 356:405–410. [DOI] [PubMed] [Google Scholar]
- Kamada, Y., T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi, and Y. Ohsumi. 2000. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150:1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura, T., T. Hara, M. Matsumoto, N. Ishida, F. Okumura, S. Hatakeyama, M. Yoshida, K. Nakayama, and K.I. Nakayama. 2004. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 6:1229–1235. [DOI] [PubMed] [Google Scholar]
- Kawamata, T., Y. Kamada, K. Suzuki, N. Kuboshima, H. Akimatsu, S. Ota, M. Ohsumi, and Y. Ohsumi. 2005. Characterization of a novel autophagy-specific gene, ATG29. Biochem. Biophys. Res. Commun. 338:1884–1889. [DOI] [PubMed] [Google Scholar]
- Kawamata, T., Y. Kamada, Y. Kabeya, T. Sekito, and Y. Ohsumi. 2008. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell. DOI: 10.1091/mbc.E07-10-1048. [DOI] [PMC free article] [PubMed]
- Kim, J., W.-P. Huang, and D.J. Klionsky. 2001. a. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J. Cell Biol. 152:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., Y. Kamada, P.E. Stromhaug, J. Guan, A. Hefner-Gravink, M. Baba, S.V. Scott, Y. Ohsumi, W.A. Dunn Jr., and D.J. Klionsky. 2001. b. Cvt9/Gsa9 functions in sequestering selective cytosolic cargo destined for the vacuole. J. Cell Biol. 153:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, M., S. Waguri, T. Chiba, S. Murata, J.I. Iwata, I. Tanida, T. Ueno, M. Koike, Y. Uchiyama, E. Kominami, and K. Tanaka. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 441:880–884. [DOI] [PubMed] [Google Scholar]
- Krogan, N.J., G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, S. Pu, N. Datta, A.P. Tikuisis, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 440:637–643. [DOI] [PubMed] [Google Scholar]
- Kuma, A., M. Hatano, M. Matsui, A. Yamamoto, H. Nakaya, T. Yoshimori, Y. Ohsumi, T. Tokuhisa, and N. Mizushima. 2004. The role of autophagy during the early neonatal starvation period. Nature. 432:1032–1036. [DOI] [PubMed] [Google Scholar]
- Levine, B., and D.J. Klionsky. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 6:463–477. [DOI] [PubMed] [Google Scholar]
- Matsui, Y., H. Takagi, X. Qu, M. Abdellatif, H. Sakoda, T. Asano, B. Levine, and J. Sadoshima. 2007. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100:914–922. [DOI] [PubMed] [Google Scholar]
- Meijer, A.J., and P. Codogno. 2004. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 36:2445–2462. [DOI] [PubMed] [Google Scholar]
- Melendez, A., Z. Tallóczy, M. Seaman, E.-L. Eskelinen, D.H. Hall, and B. Levine. 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans.Science. 301:1387–1391. [DOI] [PubMed] [Google Scholar]
- Melkoumian, Z.K., X. Peng, B. Gan, X. Wu, and J.L. Guan. 2005. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 65:6676–6684. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. 2007. Autophagy: process and function. Genes Dev. 21:2861–2873. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., and D.J. Klionsky. 2007. Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27:19–40. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., and T. Yoshimori. 2007. How to interpret LC3 immunoblotting. Autophagy. 3:542–545. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., A. Yamamoto, M. Hatano, Y. Kobayashi, Y. Kabeya, K. Suzuki, T. Tokuhisa, Y. Ohsumi, and T. Yoshimori. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N., A. Kuma, Y. Kobayashi, A. Yamamoto, M. Matsubae, T. Takao, T. Natsume, Y. Ohsumi, and T. Yoshimori. 2003. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116:1679–1688. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., B. Levine, A.M. Cuervo, and D.J. Klionsky. 2008. Autophagy fights disease through cellular self-digestion. Nature. 451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaiyama, H., M. Oku, M. Baba, T. Samizo, A.T. Hammond, B.S. Glick, N. Kato, and Y. Sakai. 2002. Paz2 and 13 other PAZ gene products regulate vacuolar engulfment of peroxisomes during micropexophagy. Genes Cells. 7:75–90. [DOI] [PubMed] [Google Scholar]
- Natsume, T., Y. Yamauchi, H. Nakayama, T. Shinkawa, M. Yanagida, N. Takahashi, and T. Isobe. 2002. A direct nanoflow liquid chromatography-tandem mass spectrometry system for interaction proteomics. Anal. Chem. 74:4725–4733. [DOI] [PubMed] [Google Scholar]
- Nice, D.C., T.K. Sato, P.E. Stromhaug, S.D. Emr, and D.J. Klionsky. 2002. Cooperative binding of the cytoplasm to vacuole targeting pathway proteins, Cvt13 and Cvt20, to phosphatidylinositol 3-phosphate at the pre-autophagosomal structure is required for selective autophagy. J. Biol. Chem. 277:30198–30207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, K., C. Wicky, L. Magnenat, H. Tobler, I. Mori, F. Muller, and Y. Ohshima. 1994. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 8:2389–2400. [DOI] [PubMed] [Google Scholar]
- Ogura, K., M. Shirakawa, T.M. Barnes, S. Hekimi, and Y. Ohshima. 1997. The UNC-14 protein required for axonal elongation and guidance in Caenorhabditis elegans interacts with the serine/threonine kinase UNC-51. Genes Dev. 11:1801–1811. [DOI] [PubMed] [Google Scholar]
- Okazaki, N., J. Yan, S. Yuasa, T. Ueno, E. Kominami, Y. Masuho, H. Koga, and M. Muramatsu. 2000. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res. Mol. Brain Res. 85:1–12. [DOI] [PubMed] [Google Scholar]
- Otto, G.P., M.Y. Wu, N. Kazgan, O.R. Anderson, and R.H. Kessin. 2004. Dictyostelium macroautophagy mutants vary in the severity of their developmental defects. J. Biol. Chem. 279:15621–15629. [DOI] [PubMed] [Google Scholar]
- Reggiori, F., K.A. Tucker, P.E. Stromhaug, and D.J. Klionsky. 2004. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell. 6:79–90. [DOI] [PubMed] [Google Scholar]
- Rubinsztein, D.C. 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 443:780–786. [DOI] [PubMed] [Google Scholar]
- Sarkar, S., R.A. Floto, Z. Berger, S. Imarisio, A. Cordenier, M. Pasco, L.J. Cook, and D.C. Rubinsztein. 2005. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlatti, F., C. Bauvy, A. Ventruti, G. Sala, F. Cluzeaud, A. Vandewalle, R. Ghidoni, and P. Codogno. 2004. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 279:18384–18391. [DOI] [PubMed] [Google Scholar]
- Scott, R.C., O. Schuldiner, and T.P. Neufeld. 2004. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 7:167–178. [DOI] [PubMed] [Google Scholar]
- Scott, R.C., G. Juhasz, and T.P. Neufeld. 2007. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, S.V., D.C. Nice III, J.J. Nau, L.S. Weisman, Y. Kamada, I. Keizer-Gunnink, T. Funakoshi, M. Veenhuis, Y. Ohsumi, and D.J. Klionsky. 2000. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J. Biol. Chem. 275:25840–25849. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., and Y. Ohsumi. 2007. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae.FEBS Lett. 581:2156–2161. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., T. Kirisako, Y. Kamada, N. Mizushima, T. Noda, and Y. Ohsumi. 2001. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20:5971–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K., Y. Kubota, T. Sekito, and Y. Ohsumi. 2007. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 12:209–218. [DOI] [PubMed] [Google Scholar]
- Tanida, I., N. Minematsu-Ikeguchi, T. Ueno, and E. Kominami. 2005. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. [DOI] [PubMed] [Google Scholar]
- Tekinay, T., M.Y. Wu, G.P. Otto, O.R. Anderson, and R.H. Kessin. 2006. Function of the Dictyostelium discoideum Atg1 kinase during autophagy and development. Eukaryot. Cell. 5:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda, T., R.S. Bhatt, H. Kuroyanagi, T. Shirasawa, and M.E. Hatten. 1999. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 24:833–846. [DOI] [PubMed] [Google Scholar]
- Tomoda, T., J.H. Kim, C. Zhan, and M.E. Hatten. 2004. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 18:541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, H., S. Abbi, C. Zheng, and J.L. Guan. 2000. Suppression of Pyk2 kinase and cellular activities by FIP200. J. Cell Biol. 149:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger, S., R. Loewith, and M.N. Hall. 2006. TOR signaling in growth and metabolism. Cell. 124:471–484. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., Y. Tagawa, T. Yoshimori, Y. Moriyama, R. Masaki, and Y. Tashiro. 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23:33–42. [DOI] [PubMed] [Google Scholar]
- Yan, J., H. Kuroyanagi, A. Kuroiwa, Y. Matsuda, H. Tokumitsu, T. Tomoda, T. Shirasawa, and M. Muramatsu. 1998. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem. Biophys. Res. Commun. 246:222–227. [DOI] [PubMed] [Google Scholar]
- Yan, J., H. Kuroyanagi, T. Tomemori, N. Okazaki, K. Asato, Y. Matsuda, Y. Suzuki, Y. Ohshima, S. Mitani, Y. Masuho, et al. 1999. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene. 18:5850–5859. [DOI] [PubMed] [Google Scholar]
- Young, A.R., E.Y. Chan, X.W. Hu, R. Kochl, S.G. Crawshaw, S. High, D.W. Hailey, J. Lippincott-Schwartz, and S.A. Tooze. 2006. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 119:3888–3900. [DOI] [PubMed] [Google Scholar]
- Zeng, X., J.H. Overmeyer, and W.A. Maltese. 2006. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 119:259–270. [DOI] [PubMed] [Google Scholar]
- Zhou, X., J.R. Babu, S. da Silva, Q. Shu, I.A. Graef, T. Oliver, T. Tomoda, T. Tani, M.W. Wooten, and F. Wang. 2007. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc. Natl. Acad. Sci. USA. 104:5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}