

Abstract
Amyotrophic lateral sclerosis (ALS), the most common motor neuron disease, is caused by a selective loss of motor neurons in the CNS. Mutations in the ALS2 gene have been linked to one form of autosomal recessive juvenile onset ALS (ALS2). To investigate the pathogenic mechanisms of ALS2, we generated ALS2 knock-out (ALS2-/-) mice. Although ALS2-/- mice lacked obvious developmental abnormalities, they exhibited age-dependent deficits in motor coordination and motor learning. Moreover, ALS2-/- mice showed a higher anxiety response in the open-field and elevated plus-maze tasks. Although they failed to recapitulate clinical or neuropathological phenotypes consistent with motor neuron disease by 20 months of age, ALS2-/- mice or primary cultured neurons derived from these mice were more susceptible to oxidative stress compared with wild-type controls. These observations suggest that loss of ALS2 function is insufficient to cause major motor deficits or motor neuron degeneration in a mouse model but predisposes neurons to oxidative stress.
Keywords: ALS2, knock-out mouse, motor neuron, motor coordination, motor learning, oxidative stress
Introduction
Amyotrophic lateral sclerosis (ALS), the most common motor neuron disease, is caused by a selective loss of lower and upper motor neurons in the CNS (Cleveland and Rothstein, 2001). Approximately 10% of ALS cases are familial ALS (FALS). Mutations in copper/zinc superoxide dismutase (SOD1) were first identified as a cause of autosomal dominant FALS (Rosen, 1993). Recently, mutations in a second ALS-related gene (ALS2) were identified to cause a rare recessive form of juvenile onset ALS (ALS2) (Hadano et al., 2001; Yang et al., 2001). One ALS2 mutation, the Tunisian mutation, is a single nucleotide deletion in exon 3 resulting in a premature stop codon and likely abrogates all the potential functions of alsin (the protein encoded by ALS2), including activities from its guanine-nucleotide-exchange factor (GEF) domains (Hadano et al., 2001; Yang et al., 2001). The other mutations in ALS2 also lead to a premature stop codon and result in a series of C-terminal truncated alsin proteins, which are unstable and have shorter half-life compared with full-length alsin (Yamanaka et al., 2003). All mutations in ALS2 affect motor neurons in the motor cortex (Devon et al., 2003). The Tunisian mutation in ALS2, however, is associated with both cortical and spinal motor neuron defects (Ben Hamida et al., 1990).
Interestingly, alsin may contain three different subfamilies of GEF-like domains: an N-terminal RCC1 (regulator of chromatin condensation)-like domain resembling GEF for Ran GTPase; a middle Dbl homology (DH)- and pleckstrin homology (PH)-like domain similar to GEF of Rho GTPase; and a C-terminal vacuolar protein sorting 9 (VPS9)-like domain homologous to GEF of Rab GTPase. Previous studies have shown that the VPS9 domain of alsin, in conjunction with its upstream membrane occupation and recognition nexus (MORN) motifs, can specifically activate the Rab5 GTPase subfamily (Otomo et al., 2003; Topp et al., 2004). The DH/PH motif of alsin is believed to facilitate the activation of members of Rho GTPase subfamily (Topp et al., 2004; Kanekura et al., 2005). Moreover, this alsin Rho GTPase activity may be important in protecting cells against mutant SOD1-mediated toxicity through a Rac1/phosphatidylinositol-3 kinase/Akt3 pathway (Kanekura et al., 2005). However, the precise pathologic mechanism underlying the selective motor neuron dysfunction caused by mutations in ALS2 is not known.
To study the physiological role of alsin and to reveal the pathogenic mechanism of ALS2-linked disease, we generated ALS2 knock-out (ALS2-/-) mice. In contrast to human cases of ALS2 (Ben Hamida et al., 1990), we show that ALS2-/- mice only developed a moderate age-dependent impairment in motor coordination and motor learning. Additionally, these mice exhibited higher levels of anxiety response and increased body weight compared with control littermates, which are alterations not documented in individuals carrying ALS2 mutations. Significantly, we found that mice and neurons lacking ALS2 were more susceptible to paraquat (PQ)-induced oxidative stress. These observations suggest that loss of ALS2 function is not sufficient to trigger motor neuron degeneration in mice but predisposes neurons to oxidative stress.
Materials and Methods
Gene targeting vector and embryonic stem cells. DNA fragments containing exons 3, 4, and 5 of ALS2 were isolated from a phage mouse genomic library (Stratagene, La Jolla, CA). To construct the targeting vector, initially an 8.04 kb XbaI/XbaI fragment carrying exon 3 of ALS2 was subcloned into the SpeI site of a modified pBluescript vector lacking the XbaI site; then, a 4.2 kb DNA fragment containing LacZ was used to replace the 39 bp of a BamHI fragment in exon3 of ALS2; finally, a 3.4 kb XbaI fragment containing the neomycin-resistance gene flanked with LoxP sites was inserted into the XbaI site at the 3′ untranslated region of the LacZ gene (see Fig. 1 A). The targeting vector was linearized at a unique NotI site and transfected into 129/SvJ embryonic stem (ES) cells, which were later subjected to G418 selection for 7 d. The G418-resistant ES clones were picked and screened by Southern blot analysis for the correctly targeted clones. Two targeted ES clones were expanded and injected into blastocysts. The resulting chimeras were bred with the C57BL/6J female mice to obtain the ALS2+/- mice. Finally, the ALS2+/- mice were intercrossed to obtain the ALS2-/- animals and identified by Southern blot (see Fig. 1 B). All ALS2 mice are a hybrid of 129/SvJ and C57BL/6J strain backgrounds. Genotypes were determined by PCR amplification (KOalsin5′, 5′gtagctcaacagaggcagaag; KOalsin3′, 5′ttcagtcagaagaactccatg; and KOlacZ3′, 5′taggttacgttggtgtagatg) of tail DNA and confirmed by Southern blot.
Figure 1.

Generation of ALS2-/- mice. A, Maps of the wild-type ALS2 locus, the targeting vector, and the disrupted ALS2 allele. The first coding exon (exon 3) of ALS2 is indicated by a black box. The targeting vector shows the replacement of the 39 bp BamHI fragment of the exon 3 by the LacZ and neomycin (neo) genes. The correctly targeted ES clones are detected by a probe located in exon 4 outside the targeting vector (black bar). B, Analysis of genomic DNA from ALS2+/- intercrosses by Southern blot. The EcoRI fragments detected for wild-type (6.6 kb) and targeted (3.6 kb) ALS2 alleles with the 3′ probe are indicated. C, Total protein extracts (30 μg) from the cerebellum of wild type (+/+), heterozygous (+/-), and homozygous ALS2 knock-out (-/-) were immunoblotted using rabbit polyclonal antisera specific against the C-terminal 12 amino acids of ALS2 and a monoclonal antibody against βIII-tubulin.
The mice were housed in a 12 h light/dark cycle and fed a regular diet ad libitum. All mouse work was approved by the Institutional Animal Care and Use Committees of The Johns Hopkins University (P.C.W.) and National Institute on Aging (H.C.).
Antisera preparation. ALS2 anti-peptide and anti-fusion protein antibodies were generated in rabbits. KLH-coupled synthesized peptides corresponding to residues 2-16 and 1645-1657 of mouse alsin were used to generate the alsin anti-peptide antibodies (Covance Research Products, Denver, PA). To generate the his-tagged alsin fusion protein, a DNA fragment corresponding to residues 1-152 of mouse alsin was subcloned into pTrcHisA (Invitrogen, San Diego, CA). The resulting alsin fusion protein purified by Talon Metal Affinity Resin chromatography (Clontech, Palo Alto, CA) was used as the antigen for making the anti-alsin fusion protein antibody (Covance Research Products).
Western blot. Mouse brains and other tissues were dissected and homogenized in 1× TBS (10 mm Tris-HCl, pH 8.0, and 150 mm NaCl) with 1% SDS in the presence of protease inhibitors (Roche, Indianapolis, IN). Homogenates were centrifuged at 2500 × g to remove nuclei and non-soluble tissues, and the supernatants were saved for Western blotting. Immunoblots were performed using antibodies specific for alsin (1:1000) and βIII-tubulin (1:5000; Chemicon, Temecula, CA).
SHIRPA test. The general phenotypes of mice were assessed by the SHIRPA (for SmithKline Beecham, Harwell, Imperial College, Royal London Hospital, phenotype assessment) protocol (Rogers et al., 1997) for primary screening as described previously.
Open-field test. The Flex-Field animal activity system (San Diego Instruments, San Diego, CA) was used to monitor the spontaneous locomotor activity. Each mouse was first put in the same corner of the 16 × 16 inch Plexiglas cubicle and allowed to stay for 30 min under the dim diffuse light emitted from a liquid crystal display screen. Flex-Field software was used to trace and quantify the movement of mouse in the unit of number of beam breaks every 15 s. The number of entries to the central zone of the open field was also recorded.
Elevated plus-maze task. A plus maze (Andreasson et al., 2001) was used to test anxiety. For this test, the maze was raised 70 cm above the ground, and testing was performed in low diffuse lighting. Each subject was placed in the center of the apparatus and allowed to explore freely for 5 min. An observer scored the number of arm entries made to the open and closed arms as well as time spent in each area.
Rotarod test. The mouse was placed onto a rotating rod with autoacceleration from 0 to 40 rpm in 4 min (Rotor-Rod system; San Diego Instruments). The length of time the mouse was able to stay on the rotating rod was recorded. For the motor learning test, four trials were run for each mouse in a 2 h interval per day for 3 sequential days.
Grip strength measurement. Mice were allowed to use their forepaws to pull a triangular bar attached to a digital force gauge, which was set up to record the maximal pulling force (San Diego Instruments). Six measurements were taken for each animal during each test.
Y-maze test. Testing was performed on a Y-shaped maze raised 100 cm above the ground. Mice were placed into the end of one arm and allowed to explore freely for 5 min (Lalonde, 2002). Timing started once the animal had begun to traverse the start arm. An observer noted which arm the animal entered throughout the session. The spontaneous alternation behavior was calculated by determining the percentage of the time the animal entered three different arms consecutively.
Electrophysiology. Animals were anesthetized by inhalation anesthesia using VetEquip anesthesia setup. Compound motor action potentials (CMAPs) in the tibial nerve innervated foot muscles were measured and tail sensory nerve conduction studies were completed using a PowerLab signal acquisition system (ADInstruments, Castle Hill, Australia) as described previously (Heine et al., 2004). Briefly, CMAP measurements were performed by stimulating the sciatic nerve at the sciatic notch and recording from the intrinsic foot muscles. Orthodromic tail sensory nerve conduction studies were done by placing the recording electrodes proximally at the base of the tail and stimulating 5 cm distally. Sensory nerve action potentials (SNAPs) were averaged from 20 stimulations. Conduction velocities were calculated based on distal latency of the onset of the SNAP. In addition, needle electromyographic examination for denervation potentials in the form of spontaneous activity (fibrillation and positive sharp wave potentials) was done in the gastrocnemius muscle.
Histology and immunohistochemical analysis. Mice were perfused via cardiac infusion with 4% paraformaldehyde in cold PBS. To obtain frozen sections, tissues were removed and submerged in 30% sucrose for 24 h and sectioned at 40 μm thickness by sliding microtome. For paraffin sections, tissues were embedded in paraffin and sectioned at 8 μm thickness by rotary microtome. Antibodies specific for GFAP, microtubule-associated protein 2 (MAP2), synaptophysin, ubiquitin (Sigma, St. Louis, MO), Mac1 (CD11b) (Serotec, Raleigh, NC), SMI31 and SMI32 (Sternberg Monoclonals, Lutherville, MD), and chicken ovalbumin upstream promoter transcription factor-interacting protein 2 (CTIP2) (a gift from Dr. M. Leid, Oregon State University, Corvallis, OR) were used as suggested by the manufacturer followed by counterstaining with hematoxylin and eosin (HE).
Paraquat treatment in vivo. Mice received 25 or 35 mg/kg body weight of paraquat (Sigma) by intraperitoneal injection and were subsequently observed over a period of 2 weeks. Lungs and livers were harvested from affected mice and evaluated histologically.
Primary neuronal cultures, paraquat treatment in vitro, and cell counting. Primary neurons were cultured from newborn (postnatal day 0) pups. The pups were decapitated, and the cortex or cerebellum was rapidly removed and placed in 35 mm Petri dishes containing sterile Hanks buffer supplemented with the following (in mm): 2.5 HEPES, pH 7.4, 35 glucose, 1 CaCl2, 1 MgSO4, and 4 NaHCO3. The meninges surrounding the tissues were removed, and the neurons were dissociated by papain (Sigma) digestion. Neurons were plated at a density of 106 cells per well in six-well plate coated with poly-d-lysine (Becton Dickinson, San Jose, CA) in basal medium Eagle (Sigma) supplemented with B27, N2, 1 mm l-glutamine, and penicillin/streptomycin (Invitrogen). Arabinosylcytosine or β-d-arabinofuranoside cytosine (Sigma) was used to inhibit the growth of glial cells. Cultures were incubated at 37°C with 5% CO2. Seventy-five percent of culture medium was changed every 3 d along with any pharmacological agents. Before paraquat treatment, the neurons were counted under phase-contrast microscope in premarked fields. The neuron was judged by its appearance: a typical neuron has spherical soma, which surrounded by a bright ring under the phase contrast microscopy, and multiple elongated smooth branches, which look like dark lines under the microscope. This definition of neurons correlates well with the staining of neuronal markers such as MAP2 and βIII-tubulin. After treatment for 17 and 24 h, the cells were counted again in the same premarked field. Neurons were considered damaged if they demonstrated neurite fragmentation, cell body vacuolization, condensation, and distortion. The cell survival rate was expressed as percentage of surviving neurons after the treatment compared with the number of neurons before treatment.
Lactate dehydrogenase assay. To determine cell viability, lactate dehydrogenase (LDH) released into the medium was measured as described by Koh and Choi (1987). The media were removed from triplicate wells of a six-well plate after 24 h treatment with paraquat.
Statistical analysis. The data were analyzed using repeated-measures or simple main effect two-tailed t tests, Spearman correlations, and linear regression models at a minimal level of significance of p < 0.05.
Results
Generation of ALS2 knock-out mice
To define the physiological roles of alsin and to determine the pathogenic mechanism of ALS2-linked disease, we generated ALS2 knock-out mice as described in Materials and Methods (Fig. 1A,B). The deletion of ALS2 was confirmed by Western blot analysis of lysates derived from the cerebella of ALS2 wild-type, heterozygous (ALS2+/-), and homozygous knock-out (ALS2-/-) animals using antibodies specifically recognizing either the C terminal (Fig. 1C) or N terminal (data not shown) of alsin. As expected, alsin (184 kDa band) was absent in cerebellum (Fig. 1C) or other brain regions (data not shown) of ALS2-/- mice. Meanwhile, the level of alsin was significantly reduced in ALS2+/- sample (Fig. 1C). In addition, there were no extra lower-molecular-weight bands revealed by this ALS2 C-terminal antibody in both ALS2+/- and ALS2-/- samples, suggesting that no truncated forms of alsin were generated. ALS2-/- mice were viable, fertile, and displayed no obvious developmental abnormalities. Offspring derived from heterozygous crosses were born in normal Mendelian ratios.
Increased body weight and higher level of anxiety in ALS2-/- mice
Because muscle wasting and the resultant body weight loss are often associated with onset of motor neuron diseases (Wong et al., 1995), we closely monitored the body weights of a cohort of 36 male mice (11 wild-type, 13 ALS2+/-, 12 ALS2-/- mice) for >20 months. Although no significant differences among these three groups in their body weights were observed up to 9 months of age, ALS2-/- mice were 18% (p < 0.02) and 25% (p < 0.01) heavier than wild-type littermates at 12 and 20 months of ages, respectively (Fig. 2A). The body weights of ALS2+/- mice were between the ALS2-/- and wild-type animals (Fig. 2A, inset), suggesting an alsin-dosage-dependent effect on body weight control. Despite of this discrepancy in their body weight, we found there were no significant differences in forearm grip strength measurements between the wild-type and ALS2-/- animals at any of the ages tested: 9, 12, 18, and 20 months (data not shown). To identify potential neurological abnormalities associated with ALS2-/- mice, the primary screen of the SHIRPA test battery (Rogers et al., 1997) was applied to 12-month-old ALS2-/- mice and their wild-type littermates. ALS2-/- mice displayed normal gait, grip, muscle tone, and startle response. When compared initially to ALS2+/+ mice, there was a significant decrease in the locomotor activity for ALS2-/- mice, in which the mice were transferred to a novel arena and the number of squares they had crossed in 30 s was counted (ALS2+/+, 14.2 ± 2.0 squares; ALS2-/-, 6.2 ± 1.6 squares; p < 0.005). However, when this same cohort of mice was monitored in a Flex-Field apparatus, we failed to detect a similar reduction in spontaneous locomotor activities in these ALS2-/- mice at 12 and 20 months of age (ALS2+/+, 1689.6 ± 209.4 beam breaks; ALS2-/-, 1689.8 ± 179.2 beam breaks; p = 0.5).
Figure 2.

Increased body weight and higher anxiety response in ALS2-/- mice. A, Aged ALS2-/- mice were heavier than their wild-type littermates. The body weights of ALS2+/+ (n = 11), ALS2+/- (n = 13), and ALS2-/- (n = 12) mice were measured monthly. The ALS2-/- mice were significantly heavier at 12 months of age (#p < 0.02 and ##p < 0.01). B, ALS2-/- mice spent less time in the center part of the arena of the Flex-Field apparatus compared with littermate controls (n = 12 and 11, respectively; *p < 0.05). C, The ALS2-/- mice spent less time in the open arms of the elevated plus-maze test compared with littermate controls (n = 12 and 11, respectively; *p < 0.05). D, The ALS2-/- mice visited the open arms of the elevated plus-maze test less often compared with littermate controls (n = 12 and 11, respectively; *p < 0.05). WT, Wild type; HEZ, heterozygous; KO, knock-out. Error bars represent SEM.
The decrease of locomotor activity observed in SHIRPA test for ALS2-/- mice could be contributed by their higher anxiety response to a novel environment when compared with control littermates. To test this hypothesis, an open-field task was used to evaluate their anxiety-like behaviors (Crawley, 1999; Prut and Belzung, 2003). In such a situation, mice spontaneously prefer the periphery of the apparatus (thigmotaxis) because of an anxiety-induced inhibition of exploration in the adverse central parts of the open field (Crawley, 1999). Consistent with our initial hypothesis, we observed a significant decrease in the time the ALS2-/- animals spent in the central part of the Flex-Field arena within the first 5 min of the trial period (p < 0.05) (Fig. 2B), indicating a higher level of anxiety in these mice. To further examine the anxiety response of these mice, we subjected this same cohort of mice to the elevated plus-maze test, a well established emotion test paradigm (Handley and Mithani, 1984; Andreasson et al., 2001). We found that ALS2-/- mice spent less time in the open arms (p < 0.05) and made fewer visits to the open arms (p < 0.05) of the elevated plus maze compared with that of the wild-type littermates (Fig. 2C,D), confirming an increased anxiety response for these ALS2-/- mice.
Motor coordination and motor learning deficiencies in ALS2-/- mice
We next examined the motor coordination and balance of ALS2+/+, ALS2+/-, and ALS2-/- mice at various ages (ranging from 9 to 20 months of age; n = 12, 13, and 11, respectively) on an accelerating (0-40 rpm in 4 min) rotarod test (Fig. 3A). Deficits in the ALS2-/- mice were observed beginning from 12 months of age, in which they had a reduced (25%) latency to fall from the rotating rod (51.77 ± 5.12 s) compared with age-matched wild-type controls (69.17 ± 4.68 s; p < 0.02). By 20 months of age, this deficit was even more dramatic (47.6% reduction in latency to fall from the rotating rod) in the ALS2-/- mice (40.95 ± 2.26 s) when compared with their wild-type littermates (78.13 ± 6.68 s; p < 0.0002) (Fig. 3A).
Figure 3.

Motor coordination and motor learning deficiencies in ALS2-/- mice. A, The latency to fall of ALS2-/- mice from the accelerating rotatory rod was shorter after 12 months of age compared with the control mice (n = 12 and 11, respectively; #p < 0.01; ##p < 0.005; **p < 0.0002). B, The ALS2-/- and ALS2+/- mice exhibited a reduced rate of motor learning in the rotarod test over a 3 d test period at 20 months of age compared with the wild-type animals (n = 12, 13, and 11, respectively; *p < 0.05; ##p < 0.005; and **p < 0.0002). WT, Wild type; HEZ, heterozygous; KO, knock-out. Error bars represent SEM.
Because alsin highly accumulates in the cerebellum (Hadano et al., 2001; Yamanaka et al., 2003; our unpublished observations), we examined whether loss of alsin may affect any cerebellum-associated motor learning ability in the ALS2-/- mice. The ability of cerebellum-mediated motor learning can be measured by numbers of trials needed for these animals to reach the maximal latency to fall on the accelerating rotarod (Lalonde et al., 1995). Four trials were performed daily for 3 consecutive days for ALS2+/+, ALS2+/-, and ALS2-/- mice at 9 and 20 months of age (n = 11, 13, and 12, respectively). All groups had similar improvements as determined by latency to fall during the 12 trials at 9 months of age, indicating that motor learning is intact in ALS2-/- animals at this age (data not shown). As expected, wild-type mice showed a significant increase (p < 0.02) in latency to fall from the rotating rod between 9 and 20 months of age, reflecting an active motor learning and memory processes in these animals through this period of time. In contrast, no significant change (p = 0.4) in the latency to fall was observed in ALS2-/- mice between 9 and 20 months of age (Fig. 3A), suggesting a possible deficit in motor learning when these mice aged. To examine whether motor learning is impaired in aged ALS2-/-, we ran the same 3 d 12 trial motor learning task as described above on 20-month-old mice. Both ALS2+/- (50.30 ± 6.59 s) and ALS2-/- (46.86 ± 5.55 s) mice performed poorly compared with wild-type littermates (79.42 ± 5.52 s) on the first trial of the first day (ALS2+/-, p < 0.002; ALS2-/-, p < 0.0005), but the rate of improvement for all three groups were similar during the subsequent trials (Fig. 3B). On the second day, whereas the performance of ALS2+/- mice on the first trial improved over the last trial of the first day and approached to the level of wild-type mice, the performance of ALS2-/- mice on the first two trials was worse than the last trial of the first day (Fig. 3B). On the third day, whereas the performance of ALS2+/- mice was indistinguishable from the wild-type littermates, ALS2-/- mice still exhibited significantly shorter latency to fall in the first two trials compared with either ALS2+/+ or ALS2+/- mice (Fig. 3B). Finally, all three groups of animals performed at a similar level in the last two trials (Fig. 3B). Thus, our finding that wild-type and ALS2+/- mice performed significantly better than ALS2-/- mice on repeated rotarod test indicates that aged ALS2-/- mice are impaired in motor learning. Furthermore, this motor learning deficit appears to be restricted to the motor system, because no differences were observed between ALS2+/+ and ALS2-/- mice in the Y-maze test, a task designed for assessing spatial learning and memory in mice (data not shown) (Lalonde, 2002).
No effect of body weight on the rotarod test in ALS2-/- mice
To ascertain that heavier body weight observed in the ALS2-/- mice (Fig. 2A) do not contribute to their poor performance on the rotarod test, we determined that there was no correlation between the animals' body weight and the latency to fall from the rotating rod within each group of ALS2-/- or wild-type animals by using a Spearman correlation test (ALS2-/-, p = 0.93; ALS2+/+, p = 0.096). Moreover, its body weight was not significant (at p = 0.05 level) for predicting the time an animal was able to stay on the rod using a linear regression approach within the groups. Therefore, the observed reduction of latency to fall from the rotating rod in ALS2-/- mice was not related to their increased body weight.
No alteration of peripheral nerve conduction or electromyography in ALS2-/- mice
We assessed the electrophysiology of periphery nerves of 21-month-old ALS2-/- mice and their wild-type littermates. As summarized in Table 1, there were no significant differences in the CMAP amplitudes, SNAP amplitudes, or sensory nerve conduction velocities (SNAPCVs) between ALS2+/+ and ALS2-/- mice. Needle electromyography did not show any significant degree of denervation potentials (data not shown).
Table 1.
The electrophysiological properties of the periphery nerves of ALS2−/− mice
|
|
SNAP (μV) |
SNAPCV (m/s) |
CMAP (mV) |
|---|---|---|---|
| ALS2+/+ (n = 8) | 24.3 ± 4.6 | 38.6 ± 1.8 | 3.5 ± 0.4 |
|
ALS2−/− (n = 9) |
18.5 ± 4.2 |
39.0 ± 0.8 |
3.9 ± 0.4 |
There was no significant difference between ALS2−/− and their littermate controls in CMAPs of the tibial nerve innervated foot muscles (p = 0.52). There were no significant differences between ALS2−/− and their littermate controls in SNAPs and SNAPCVs (p = 0.36 and p = 0.82, respectively).
Lack of obvious neuropathological abnormalities in ALS2-/- mice
To determine whether there are significant changes in the number and morphology of spinal motor neurons in ALS2-/- mice, we examined a series of coronal sections of lumbar spinal cords derived from six pairs of 20-month-old ALS2 wild-type (Fig. 4A,C) and ALS2-/- (Fig. 4B,D) animals. We found no evidence of chromatolysis in spinal motor neurons of ALS2-/- mice and no significant differences in the numbers of motor neurons between ALS2-/- and control mice (ALS2+/+, 38.0 ± 1.9 per section; ALS2-/-, 37.5 ± 1.3 per section; p = 0.79). In addition, we found no obvious changes in the number and morphology of motor neurons in layer V of the motor cortices of ALS2-/- animals (Fig. 4F) compared with control littermates (Fig. 4E) using a monoclonal antibody against CTIP2, which is specifically expressed by the corticospinal motor neurons (Arlotta et al., 2005).
Figure 4.
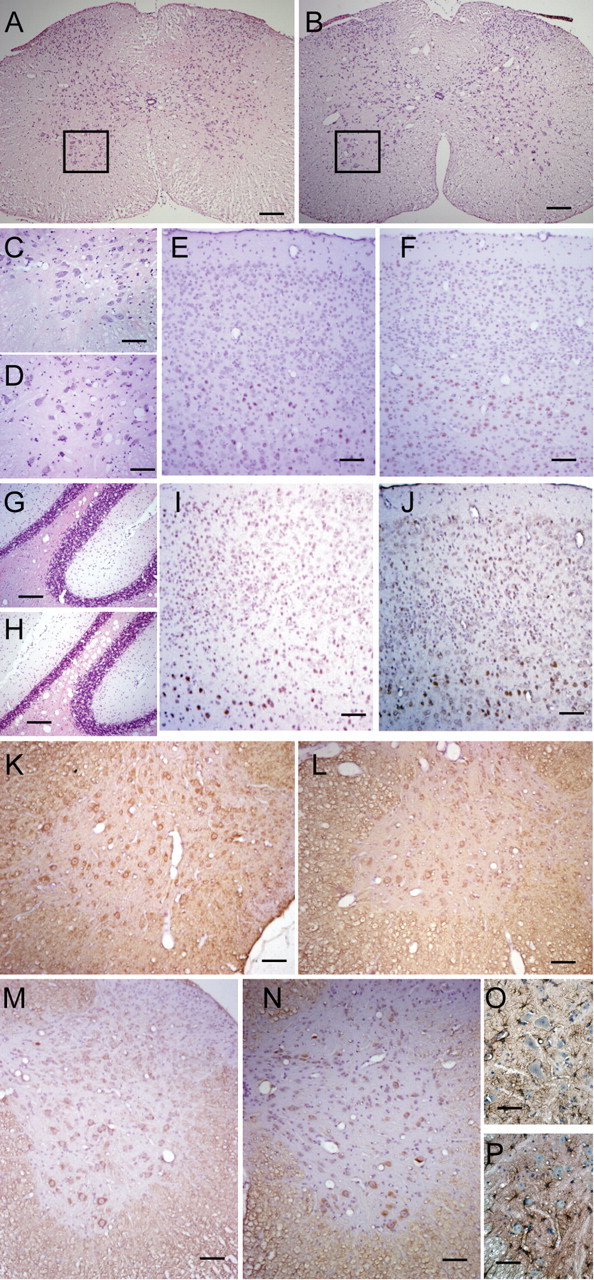
Histological analysis of the nervous system of 20-month-old ALS2-/- mice.A-D, The coronal sections of lumbar spinal cord were stained with HE, and no obvious differences were observed between the wild-type (n = 6; A, C) and ALS2-/- (n = 6; B, D) tissues. The ventral spinal cords and motor neurons (C, D) were revealed under higher magnification (the boxed areas in A, B). E, F, The cortical spinal motor neurons were labeled with CTIP2 antibody (brown) and counterstained with HE (blue and purple) in the motor cortices of wild-type (E; n = 6) and ALS2-/- (F; n = 6) mice. G, H, The cerebella of wild-type (G) and ALS2-/- (H) mice were stained with HE. I, J, The motor cortices of wild-type (E) and ALS2-/- (F) mice were stained with SMI31 (brown) and counterstained with HE. K, L, The coronal sections of lumbar spinal cords from wild-type (K) and ALS2-/- (L) animals were immunostained with SMI31 (brown). M, N, The coronal sections of lumbar spinal cords from wild-type (M) and ALS2-/- (N) animals were immunostained with SMI32 (brown) and counterstained with HE. O, P, The coronal sections of lumbar spinal cords from wild-type (O) and ALS2-/- (P) animals were immunostained with GFAP (brown) and counterstained with HE. Scale bars: A, B, 200 μm; C, D, 50 μm; E-N, 100 μm; O, P, 25 μm.
Because alsin is most abundant in the cerebellum (Hadano et al., 2001; Yamanaka et al., 2003) and the deficiency of ALS2-/- mice in motor learning test, we assessed the gross anatomy of the cerebella from these animals. We found no significant morphological or cell number change in ALS2-/- mice compared with wild-type ones (Fig. 4G,H). Similarly, because cortico-striatal system is also involved in the motor learning process, we examined the gross anatomy of the striatum of ALS2-/- mice and did not detected any obvious morphological and structure changes in this region either (data not shown). In addition, we examined the brain sections containing amygdala, a region related to anxiety response, and did not find any significant morphological alterations in this area (data not shown).
To further reveal any potential morphological and structure changes in the brain, we stained the brain sections with a series of antibodies. Immunocytochemical analysis of spinal cord and brain sections using antibodies against phosphorylated neurofilament H (SMI31) revealed only moderate or weak staining in ALS2-/- animals (Fig. 4L,J), which is comparable to the staining in the control tissues (Fig. 4K,I). To examine potential alterations of the cytoskeleton, particularly the pattern of neurofilaments, in ALS2-/- motor neurons, we also stained the spinal cord and cortical sections with SMI32, an antibody recognizing the nonphosphorylated neurofilament H, and did not detect any obvious changes (Fig. 4M,N). In addition, we examined the expression of synaptophysin, a presynaptic marker, and MAP2, a dendritic marker, in brain and spinal cord sections from the ALS2-/- animals and detected no significant change (data not shown).
To assess inflammatory responses in the ALS2-/- tissues, we stained the spinal cord and cortical sections with markers for microgliosis (CD11b) and astrocytosis (GFAP). There were only very weak signals for CD11b in the cortices and spinal cords of both wild-type and ALS2-/- animals (data not shown). The GFAP staining was comparable between ALS2-/- (Fig. 4P) and control spinal cord sections (Fig. 4O). Together, we found no evidence of neuropathological abnormalities occurring in ALS2-/- mice.
ALS2-/- mice and neurons were more vulnerable to paraquat-mediated oxidative stress
Our data showed that ALS2-/- mice developed a late-onset motor deficit in the absence of obvious neuropathological abnormalities. Recent studies suggest that alsin plays a protective role in vitro against mutant SOD1 or other toxic challenges (Kanekura et al., 2005). It has been documented that both SOD1- and copper chaperone for SOD1-(CCS) deficient mice are hypersensitive to PQ exposure (Wong et al., 2000). The herbicide PQ is highly toxic for cells through generation of superoxide anion, a precursor of reactive oxygen species (ROS) (Suntres, 2002). We examined the possibility that there is an increased sensitivity to PQ-mediated oxidative stress in ALS2-/- mice. Intraperitoneal administration of PQ (25 mg/kg body weight), which killed >90% of CCS-deficient mice (Wong et al., 2000), had limited effects on ALS2-/- mice (n = 15) (Fig. 4A). With a higher dosage of PQ (35 mg/kg body weight), in which no control mice (n = 15) were affected, 50% of ALS2-/- mice were killed (n = 15) (Fig. 5A). Examination of PQ-treated ALS2-/- mice revealed characteristic features of PQ toxicity: edema and hemorrhage in the lungs (Wong et al., 2000).
Figure 5.
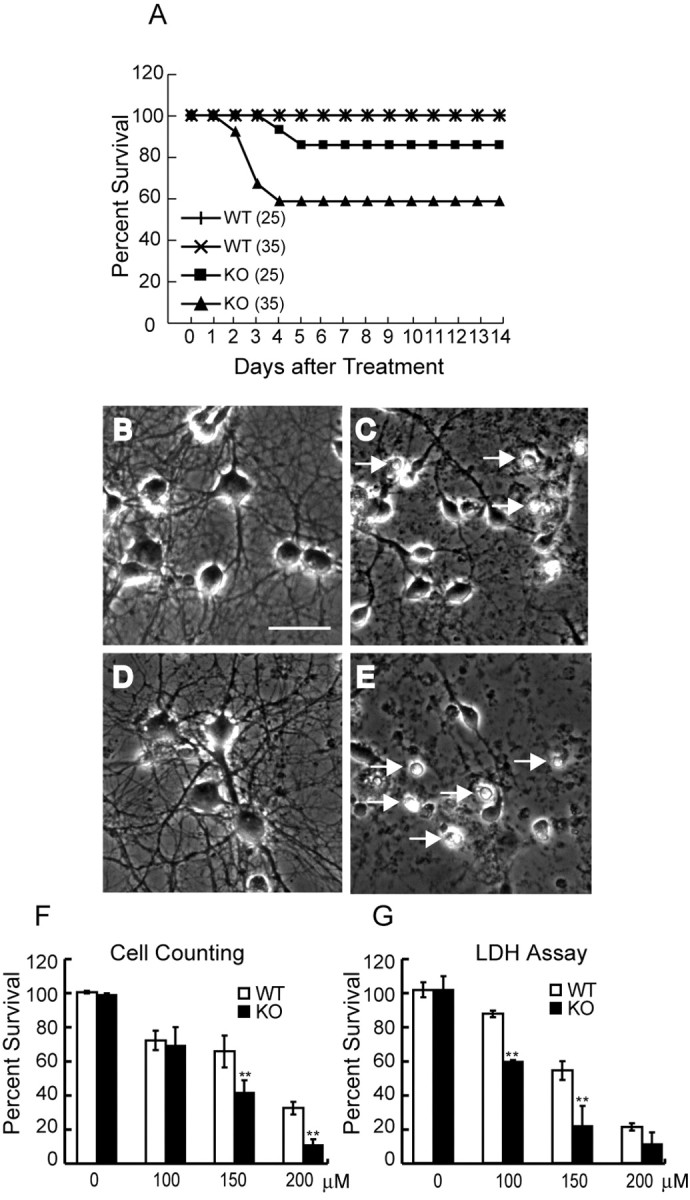
ALS2-deficient mice and neurons were more susceptible to paraquat-induced oxidative stress. A, Intraperitoneal administration of PQ (25 mg/kg body weight) had limited effects on the survival of 5-month-old female ALS2-/- knock-out [KO (25); n = 15] compared with wild-type [WT (25); n = 15] mice. With a higher dose of PQ (35 mg/kg body weight), at which none of control mice [WT (35); n = 15] were affected, 50% of ALS2-/- mice were killed [KO (35); n = 15]. B-D, Sample images of primary cultured cortical neurons derived from wild-type (B, C) and ALS2-/- (D, E) pups were shown under the phase-contrast microscopy before (B, D) and 17 h after (C, E) treatment with 150 μm PQ. The increase of condensed cell bodies of dying cells (marked by arrows in C and E) was observed after PQ treatment. Scale bar, (in B) 50 μm. F, The ALS2-/- neurons were more sensitive to PQ-induced cell death revealed by cell counting 17 h after treatment (**p < 0.001). Each data point represented three independent experiments in which >2000 neurons were counted. G, The ALS2-/- neurons were more susceptible to PQ-mediated cell death revealed by LDH assay 24 h after the treatment (n = 6; **p < 0.001). Error bars represent SEM.
Because ALS2 is enriched in neuronal tissues, we tested a possible neuroprotective role of ALS2 from PQ-mediated toxicity. Primary cultured cortical neurons derived from newborn ALS2 wild-type and ALS2-/- pups were used for cytotoxicity studies. PQ treatment caused neuron death in both wild-type and ALS2-/- cultures manifested as condensation of soma and nuclei and fragmentation of neurites (Fig. 5C,E). Neurons with normal morphology were counted in the marked fields before and after PQ treatment. After exposure to 150 μm PQ for 17 h, only 40.5% of ALS2-/- neurons survived compared with 64.7% observed in wild-type neurons (p < 0.0002) (Fig. 5F). At a higher dosage (200 μm), only 10.6% of ALS2-/- neurons remained viable compared with 31.8% of wild-type neurons (p < 0.001) (Fig. 5F). In parallel, the LDH assay was used to assess the survival rate of neurons after treatment with PQ, showing a similar reduction in survival rate of ALS2-/- cells after treatment with 100 and 150 μm PQ (p < 0.001) (Fig. 5G). Together, our data indicates that absence of alsin predisposes neurons and animals to PQ-induced toxicity.
Discussion
The onset of ALS2 occurs in the first or second decade and progresses slowly for 10-15 years (Ben Hamida et al., 1990). Three clinical subtypes of ALS2 based on the initial involvement of either limbs or muscles are recognized. The ALS2 Tunisian family, which carries a complete loss-of-function mutation, is characterized as the type 3 ALS2 as a result of predominance of progressive facial and limb muscles spasticity (Hentati et al., 1994). In an attempt to model this type of ALS2, we generated ALS2 knock-out mice (Fig. 1). In contrast to the clinical and neurological abnormalities observed in affected individuals, ALS2-/- mice did not exhibit any evidence consistent with motor neuron disease up to 20 months of age (Figs. 2, 3, 4). Instead, a higher level of anxiety response was observed in these ALS2-/- mice (Fig. 2B-D), a trait not reported in clinical studies of individuals with mutant ALS2. In addition to the alteration of anxiety response, we also found mice deficient in ALS2 perform poorly in the rotarod test for motor coordination at 12 months of age compared with their wild-type littermates (Fig. 3A).
Whereas wild-type mice significantly improved their performance in the rotarod test from 9 to 20 months, there was no significant change for the performance of ALS2-/- mice (Fig. 3A). In conjunction with observations that alsin is most abundant in the cerebellum (Hadano et al., 2001; Yamanaka et al., 2003), we speculated that alsin may participate in cerebellum-dependent motor learning. To test this hypothesis, we used repeated testing on the accelerating rotarod for ALS2+/+, ALS2+/-, and ALS2-/- mice at 9 and 20 months of age to evaluate cerebellum-mediated motor learning in these animals. ALS2-/- mice at 9 months of age improved at a similar rate as both the wild-type and ALS2+/- animals in repeated rotarod tests over the 3 d trials, indicating that motor learning was not affected at this age. However, both ALS2+/- and ALS2-/- mice displayed motor learning deficits at 20 months of age in the same series of rotarod tests, in which ALS2-/- mice displayed more severe motor learning disability compared with the ALS2+/- littermates (Fig. 3B). Supporting this notion, we did not find any evidences consistent with motor neuron disease (Fig. 4), including muscle weakness or alteration of periphery nerve transduction in these aged ALS2-/- mice. Together, the deficits in motor coordination and motor learning observed in ALS2-/- are likely caused by the absence of ALS2 in cerebellum. A selective deletion of ALS2 in the cerebellum will be useful to directly test this hypothesis.
The lack of overt motor phenotypes in mice deficient in ALS2 may reflect the differences in the organizations of motor systems between humans and mice. For example, upper motor neurons located in the human primary motor cortex, which descend through the lateral cortical spinal track, can directly innervate lower motor neurons in the spinal cord to control movement. In rodents, there is no direct synaptic connection between the lower and upper motor neurons, because upper motor neurons send their axons mostly through the ventral medial cortical spinal tract and make synapses with only the interneurons in the spinal cord (Yang and Lemon, 2003). As revealed by previous genetic and clinical studies, most mutations in ALS2 preferentially affect the upper motor neurons. The only exception is the Tunisian mutation in which a lower motor neuron involvement has been suggested (Ben Hamida et al., 1990; Hentati et al., 1994; Yang et al., 2001; Eymard-Pierre et al., 2002; Devon et al., 2003). Conceivably, the anatomical and potential physiological discrepancy between the human and mouse upper motor neurons may explain the relatively normal motor phenotype of the aged ALS2-/- mice.
The absence of obvious motor phenotypes in mice deficient in ALS2 may also be related to gene redundancy, where genes with similar function can compensate for the loss of function of ALS2. Recently, an ALS2-related protein (ALS2CL) has been characterized, which is highly homologous to the C-terminal half of ALS2, including the MORN motif and VSP9 domain (Hadano et al., 2004). ALS2CL has higher binding affinity to Rab5 than alsin, but it does not exhibit Rab5-GEF activity. Coexpression of ALS2CL and Rab5 in HeLa cells induces the reorganization of early endosomal antigen 1 (EEA1)-negative endosomes by forming some tabulated structures (Hadano et al., 2004). In contrast, coexpression of ALS2 and Rab5 induces the enlargement of EEA1-positive endosomes (Otomo et al., 2003), suggesting that ALS2 and ALS2CL play different roles in Rab5-mediated endosomal trafficking. Thus, whether ALS2CL can compensate for the loss of ALS2 remains to be established.
One of the observations in postmortem tissue from sporadic ALS patients is oxidative damage to proteins, lipids, and DNA induced by oxidative stress (Shaw et al., 1995). Although it remains unclear whether oxidative stress plays a primary role in the progression of ALS, aberrant production of ROS and nitrogen species may contribute to the demise of motor neurons. PQ is a common herbicide that has been implicated as a risk factor for Parkinson's disease in epidemiological studies (Suntres, 2002). The mechanism for PQ-induced toxicity is through generation of ROS, oxidation of NADPH, and peroxidation of polyunsaturated fatty acids (Suntres, 2002). Previous studies showed that mice deficient in SOD1 and its copper-loading protein CCS, which are critical for the removal of ROS from the cells, are more susceptible to the treatment of sublethal dosages of PQ (Wong et al., 2000). Recent studies showed that alsin plays a protective role against mutant SOD1-mediated cell death through a Rac1/phosphatidylinositol-3 kinase/Akt3 anti-apoptotic pathway, which is also involved in the anti-oxidative stress response in cells (Kanekura et al., 2005). In line with these studies, we found that loss of ALS2 rendered neurons more vulnerable to PQ-mediated cell death as evidenced by the decreased survival rate of ALS2-/- neurons compared with wild-type controls (Fig. 5E-G). Additional studies are required to clarify the role of alsin in oxidative stress, particular its DH/PH domain, which is believed to regulate Rac1 activity, and the MORN motif, which may function in the phospholipid signaling transduction pathway.
In summary, aged ALS2-/- mice exhibited elevated anxiety responses and were impaired in motor coordination and motor learning. However, we found no evidence in ALS2-/- mice that is consistent with classic motor neuron disease. Together, these observations indicate that loss of ALS2 function was not sufficient to cause motor neuron disease in a mouse model. However, the lack of ALS2 did predispose neurons to oxidative stress, implying that ALS2 might serve as a risk factor for motor neuron disease. In the future, it will be critical to further clarify the physiological function(s) of ALS2 in neurons and be interesting to assess any potential genetic interaction between alsin and SOD1.
Footnotes
This work was supported by a joint grant from the ALS Association and The Robert Packard Center for ALS Research at Johns Hopkins (H.C. and P.C.W.), by National Institute of Neurological Disorders and Stroke Grant R01 NS40014 (P.C.W.), and by the Intramural Research Program of the National Institute on Aging (H.C.). We thank all of the members of Drs. Cai and Wong's laboratories for their helpful discussions, Dr. Lee Martin for evaluation of histological sections, Dr. Lihong Cui for statistical analysis, Dr. M. Leid for kindly providing us the CTIP2 antibody, Dr. John Hardy and Jayanth Chandran for critical reading of this manuscript, and the assistance of the National Cancer Institute Fellows Editorial Board.
Correspondence should be addressed to Dr. Huaibin Cai, Section of Transgenesis, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Building 35, Room 1A116, MSC 3707, 35 Convent Drive, Bethesda, MD 20892-3707. E-mail: caih@mail.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257567-08$15.00/0
References
- Andreasson KI, Savonenko A, Vidensky S, Goellner JJ, Zhang Y, Shaffer A, Kaufmann WE, Worley PF, Isakson P, Markowska AL (2001) Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J Neurosci 21: 8198-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45: 207-221. [DOI] [PubMed] [Google Scholar]
- Ben Hamida M, Hentati F, Ben Hamida C (1990) Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 113: 347-363. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2: 806-819. [DOI] [PubMed] [Google Scholar]
- Crawley JN (1999) Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res 835: 18-26. [DOI] [PubMed] [Google Scholar]
- Devon RS, Helm JR, Rouleau GA, Leitner Y, Lerman-Sagie T, Lev D, Hayden MR (2003) The first nonsense mutation in alsin results in a homogeneous phenotype of infantile-onset ascending spastic paralysis with bulbar involvement in two siblings. Clin Genet 64: 210-215. [DOI] [PubMed] [Google Scholar]
- Eymard-Pierre E, Lesca G, Dollet S, Santorelli FM, Di Capua M, Bertini E, Boespflug-Tanguy O (2002) Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am J Hum Genet 71: 518-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown Jr RH, Scherer SW, Rouleau GA, Hayden MR, et al. (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29: 166-173. [DOI] [PubMed] [Google Scholar]
- Hadano S, Otomo A, Suzuki-Utsunomiya K, Kunita R, Yanagisawa Y, Showguchi-Miyata J, Mizumura H, Ikeda JE (2004) ALS2CL, the novel protein highly homologous to the carboxy-terminal half of ALS2, binds to Rab5 and modulates endosome dynamics. FEBS Lett 575: 64-70. [DOI] [PubMed] [Google Scholar]
- Handley SL, Mithani S (1984) Effects of alpha-adrenoceptor agonists and antagonists in a maze-exploration model of `fear'-motivated behaviour. Naunyn Schmiedebergs Arch Pharmacol 327: 1-5. [DOI] [PubMed] [Google Scholar]
- Heine W, Conant K, Griffin JW, Hoke A (2004) Transplanted neural stem cells promote axonal regeneration through chronically denervated peripheral nerves. Exp Neurol 189: 231-240. [DOI] [PubMed] [Google Scholar]
- Hentati A, Bejaoui K, Pericak-Vance MA, Hentati F, Speer MC, Hung WY, Figlewicz DA, Haines J, Rimmler J, Ben Hamida C (1994) Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33-q35. Nat Genet 7: 425-428. [DOI] [PubMed] [Google Scholar]
- Kanekura K, Hashimoto Y, Kita Y, Sasabe J, Aiso S, Nishimoto I, Matsuoka M (2005) A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J Biol Chem 280: 4532-4543. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20: 83-90. [DOI] [PubMed] [Google Scholar]
- Lalonde R (2002) The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev 26: 91-104. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Bensoula AN, Filali M (1995) Rotarod sensorimotor learning in cerebellar mutant mice. Neurosci Res 22: 423-426. [DOI] [PubMed] [Google Scholar]
- Otomo A, Hadano S, Okada T, Mizumura H, Kunita R, Nishijima H, Showguchi-Miyata J, Yanagisawa Y, Kohiki E, Suga E, Yasuda M, Osuga H, Nishimoto T, Narumiya S, Ikeda JE (2003) ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum Mol Genet 12: 1671-1687. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463: 3-33. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE (1997) Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8: 711-713. [DOI] [PubMed] [Google Scholar]
- Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364: 362. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Ince PG, Falkous G, Mantle D (1995) Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol 38: 691-695. [DOI] [PubMed] [Google Scholar]
- Suntres ZE (2002) Role of antioxidants in paraquat toxicity. Toxicology 180: 65-77. [DOI] [PubMed] [Google Scholar]
- Topp JD, Gray NW, Gerard RD, Horazdovsky BF (2004) Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J Biol Chem 279: 24612-24623. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14: 1105-1116. [DOI] [PubMed] [Google Scholar]
- Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD (2000) Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA 97: 2886-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Vande VC, Eymard-Pierre E, Bertini E, Boespflug-Tanguy O, Cleveland DW (2003) Unstable mutants in the peripheral endosomal membrane component ALS2 cause early onset motor neuron disease. Proc Natl Acad Sci USA 100: 16041-16046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Lemon RN (2003) An electron microscopic examination of the corticospinal projection to the cervical spinal cord in the rat: lack of evidence for cortico-motoneuronal synapses. Exp Brain Res 149: 458-469. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben Hamida M, Pericak-Vance M, Hentati F, Siddique T (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29: 160-165. [DOI] [PubMed] [Google Scholar]