

Abstract
Copper-zinc superoxide dismutase (Cu,ZnSOD) is the antioxidant enzyme that catalyzes the dismutation of superoxide (O2•−) to O2 and H2O2. In addition, Cu,ZnSOD also exhibits peroxidase activity in the presence of H2O2, leading to self-inactivation and formation of a potent enzyme-bound oxidant. We report in this study that lipid peroxidation of l-α-lecithin liposomes was enhanced greatly during the SOD/H2O2 reaction in the presence of nitrite anion (NO2−) with or without the metal ion chelator, diethylenetriaminepentacetic acid. The presence of NO2− also greatly enhanced α-tocopherol (α-TH) oxidation by SOD/H2O2 in saturated 1,2-dilauroyl-sn-glycero-3-phosphatidylcholine liposomes. The major product identified by HPLC and UV-studies was α-tocopheryl quinone. When 1,2-diauroyl-sn-glycero-3-phosphatidylcholine liposomes containing γ-tocopherol (γ-TH) were incubated with SOD/H2O2/NO2−, the major product identified was 5-NO2-γ-TH. Nitrone spin traps significantly inhibited the formation of α-tocopheryl quinone and 5-NO2-γ-TH. NO2− inhibited H2O2-dependent inactivation of SOD. A proposed mechanism of this protection involves the oxidation of NO2− by an SOD-bound oxidant to the nitrogen dioxide radical (•NO2). In this study, we have shown a new mechanism of nitration catalyzed by the peroxidase activity of SOD. We conclude that NO2− is a suitable probe for investigating the peroxidase activity of familial Amyotrophic Lateral Sclerosis-linked SOD mutants.
Copper-zinc superoxide dismutase (Cu,ZnSOD) is the antioxidant enzyme that catalyzes the dismutation of superoxide anion (O2•−) to O2 and H2O2 (1, 2). In the presence of high concentrations of H2O2, SOD slowly undergoes inactivation (k ≈ 3.1 M−1 s−1) (1–5) through formation of an enzyme-bound oxidant, SOD-Cu2+-•OH (also formulated as SOD-Cu2+OH) (Eqs. 1 and 2).
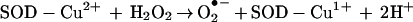 |
1 |
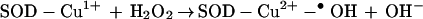 |
2 |
The reactivity of the SOD-Cu2+-•OH species was suggested to be similar to that of a “free” hydroxyl radical (2). The peroxidase activity of SOD, by using H2O2 as a substrate, generates a “bound” hydroxyl radical to the copper atom at the active site (Eq. 2) (1–5). This oxidant, SOD-Cu2+-•OH, formed at the active site of SOD presumably inactivates the enzyme by oxidative attack on a histidine residue (His-61) that binds the copper atom at the active site, forming an imidazole-derived radical (Eq. 3) that is subsequently oxidized to 2-oxo-histidine (1–8). This process is accompanied by the release of copper from the active site of SOD and inactivation of the enzyme (6).
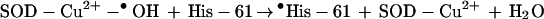 |
3 |
Inactivation of SOD was minimized in the presence of anionic radical scavengers or peroxidase substrates (such as formate, glutamate, urate, and azide) that have access to the active site of SOD because of the favorable electrostatic interaction (1–5). In contrast, neutral radical scavengers such as ethanol or mannitol, which are presumably not accessible to the active site of SOD, did not protect the enzyme against peroxidatic inactivation (1–5). For example, formate anion protected against H2O2-induced inactivation of SOD. This finding was suggested to be due to oxidation of formate to carbon dioxide radical anion (CO2•−) by the enzyme-bound oxidant (4, 5).
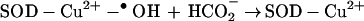 |
4 |
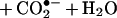 |
The objective of the present study was to investigate the peroxidase activity of SOD by using a small molecular weight anionic peroxidase substrate that forms diagnostic products. To this end, we chose nitrite anion (NO2−) because the resulting one-electron oxidation intermediate, nitrogen dioxide radical (•NO2), will react with phenolic compounds to yield diagnostic products (9–11). Because of the rapid hydrolysis of •NO2 to nitrite and nitrate in aqueous solution, we selected α-tocopherol (α-TH) and γ-tocopherol (γ-TH) incorporated into large unilamellar 1,2-dilauroyl-sn-glycero-3-phosphatidylcholine (DLPC) liposomes as •NO2 traps (12–16). The oxidation of NO2− to the nitrogen dioxide radical (•NO2) was followed by monitoring the formation of α-tocopheryl quinone (α-TQ) and 5-NO2-γ-TH (NGTH), the oxidation and nitration products of α-TH and γ-TH, respectively.
The mechanism of α-TQ and NGTH formation was investigated by the electron spin resonance (ESR) technique. Results show evidence for the formation of •NO2 radical from the peroxidase activity of SOD. The relevance of this reaction in the pathogenesis of familial Amyotrophic Lateral Sclerosis (FALS) disease is discussed.
MATERIALS AND METHODS
Cu,ZnSOD (bovine) was purchased from Boehringer Mannheim. DLPC was purchased from Avanti Polar Lipids. Soybean l-α-phospahtidyl-choline (l-α-lecithin), α-TH, γ-TH, and H2O2 were obtained from Sigma. MDL 101,002 was a gift from Craig Thomas of Lilly Research Laboratories (Indianapolis). 5-5′-dimethyl-1-pyrroline N-oxide, N-t-butyl-α-phenylnitrone (PBN), and α-(4-pyridyl-1-oxide)-N-t-butylnitrone (POBN) were purchased from Sigma.
Preparation of Unilamellar Liposomes and Incorporation of Tocopherols.
Liposomes were synthesized from DLPC and l-α-lecithin. α-TH, γ-TH, α-TQ, and NGTH (100 μM), each in methanol, were mixed with the phospholipids (10 mM) in chloroform, dried down under a stream of nitrogen, and further dried under vacuum for overnight. Multilammelar vesicles were prepared by hydration of lipid film in chelex-treated phosphate buffer with vigorous vortexing. The concentration of phosphate buffer was 20 mM for l-α-lecithin and 200 mM for DLPC liposomes. Five cycles of freezing and thawing preceded the extrusion through a 0.2-μm filter for preparing large unilammelar vesicles (17).
Conjugated Diene Measurements.
l-α-lecithin liposomes (250 μg/ml) were incubated with SOD (0.2 mg/ml) and H2O2 (1 mM) in chelex-treated phoshate buffer (20 mM, pH 7.4) with or without the transition metal ion chelator, diethylenetriaminepentacetic acid (DTPA) at 37°C. The changes in absorbance were continuously monitored at 234 nm to detect the formation of conjugated dienes (18).
Synthesis of α-TQ and NGTH.
α-TQ was synthesized as previously reported (13). NGTH was synthesized by nitration of γ-TH with nitrous acid (14). Briefly, γ-TH (10 mg) in ethanol (10 ml) was acidified with 4 ml of glacial acetic acid, followed by 6 ml of sodium nitrite (300 mM). After 4 min, the product was extracted into hexane (20 ml) and washed several times with deionized water. The hexane layer was dried over sodium bicarbonate, and the solvent was evaporated. NGTH was purified by HPLC on a semipreparative C18 column (5 μm, 1 × 25 cm, Beckman Instruments). The retention time of NGTH on this column was 54 min and was clearly separated from other oxidized products of γ-TH. NGTH was quantified by UV in CH3OH by using the following spectral characteristics: 264 nm (ɛmax = 2,483 M−1 cm−1), 312 nm (ɛmax = 2,464 M−1 cm−1), and 414 nm (ɛmax = 904 M−1 cm−1) (16).
HPLC.
Detection and separation of α-TH, γ-TH, and their oxidation products were performed on a HPLC system equipped with a UV detector. Samples (usually 300 μl) were taken up in water (200 μl) and ethanol (400 μl) and vortexed for 1 min. Heptane (400 μl) was added to the samples followed by vortexing for 1 additional minute. Samples were centrifuged for 10 min at 10,000 rpm to separate layers. The organic layer (top) was removed, dried under a stream of nitrogen, and stored overnight at −20°C. For analysis, the samples were dissolved in methanol and analyzed by HPLC. The mobile phase was methanol:water (95:5) for 10 min, after which it was graded to 100% methanol over a 5-min period. The stationary phase was a Partisil ODS-3 (0.46 × 25 cm, 4-μm particle size column from Whatman). α-TQ and NGTH were quantified with UV detection at λ = 266 nm and λ = 290 nm, respectively. UV detection (λ = 290 nm) also was used to monitor α-TH and γ-TH levels.
Measurement of SOD Activity.
SOD activity was measured by using the ferri cytochrome c (cyt c) reduction assay (19). Briefly, xanthine (0.5 mM) and xanthine oxidase (0.05 unit/ml) were incubated with cyt c (20 μM) in phosphate buffer (200 mM, pH 7.4). The rate of cyt c reduction by the superoxide anion was measured in the presence and absence of SOD. To investigate the effect of nitrite on SOD inactivation by H2O2, SOD (1 mg/ml) was incubated with H2O2 (1 mM) at 37°C in the presence and absence of nitrite. SOD activity was measured by diluting an aliquot (5 μl) of the reaction mixture with 1 ml of the assay mixture (phosphate buffer containing 1 mM DTPA and 1,000 units/ml of catalase) (20).
Measurement of Copper/Zinc Release During Inactivation of SOD by H2O2.
SOD (1 mg/ml) was incubated with H2O2 (1 mM) in chelex-treated phosphate buffer (20 mM, pH 7.4) at 37°C in the presence of 4-pyridylazaresorcinol (100 μM). The change in absorbance because of Cu2+- and Zn2+-4-pyridylazaresorcinol complex formation was followed for 3 hr at 500 nm. The amount of zinc released during this process was calculated by adding 1.6 mM of nitrilotriacetic acid at the end of the reaction. The decrease in absorbance corresponds to Zn2+-nitrilotriacetic acid complex formation (21). The amount of copper released was calculated by the subsequent addition of 0.8 mM EDTA to the incubation mixture as previously reported (21).
ESR Measurements.
The formation of α-tocopheroxyl (α-T•), γ-T•, and 5-nitro-γ-tocopheroxyl (NGT•) radicals were monitored by ESR (22). ESR measurements were performed at ambient temperature by using a Varian E-109 spectrometer operating at 9.5 GHz (X-band) by using 100 kHz field modulation. A typical incubation for ESR experiments consisted of 10 mg/ml SOD, nitrite (10 mM), H2O2 (10 mM), and α-TH, γ-TH, or NGTH (2 mM) incorporated into DLPC liposomes (100 mM) in 0.25 ml of phosphate buffer (200 mM, pH 7.4) containing DTPA (1 mM). Samples were analyzed in a quartz flat cell.
RESULTS
Lipid Peroxidation by SOD/H2O2/NO2−.
SOD/H2O2-mediated lipid peroxidation was monitored by measuring conjugated diene formation at 234 nm in l-α-lecithin liposomes. Incubation of liposomes in the presence of SOD (200 μg/ml) and H2O2 (1 mM) resulted in conjugated diene formation following a sigmoidal curve (Fig. 1A), typical of transition metal ion-mediated lipid peroxidation. Addition of NO2− (10 μM) but not NO3− accelerated lipid peroxidation in the presence of SOD/H2O2.
Figure 1.

Effect of nitrite on SOD/H2O2-mediated lipid peroxidation. (A) l-α-lecithin liposomes (250 μg/ml) were incubated in the absence of DTPA with SOD (200 μg/ml) at 37°C in chelex-treated phosphate buffer (20 mM, pH 7.4) and monitored at 234 nm for conjugated diene formation in the presence or absence of H2O2 (1 mM) and NO2− (10 μM). (B) Liposomes were incubated with SOD (200 μg/ml), H2O2 (1 mM), and DTPA (100 μM), and conjugated diene formation was measured as a function of NO2− concentration; a, 100 μM; b, 40 μM; c, 20 μM; d, 10 μM; and e, 0 μM. (Inset) The effect of α-TH and γ-TH (10 μM each) on SOD/H2O2/NO2− initiated conjugated diene formation.
Fig. 1B shows SOD/H2O2-mediated lipid peroxidation in the presence of DTPA (100 μM) as a function of NO2− concentration. In contrast to Fig. 1A, lipid oxidation was slower and occurred without any lag-time (23). In the absence of NO2−, conjugated diene formation was negligible in the presence of DTPA (Fig. 1B, trace e). However, addition of NO2− enhanced conjugated diene formation in a concentration dependent manner. Lipid peroxidation resulting from the formation of the oxidant generated by SOD/H2O2/NO2− was inhibited in the presence of the phenolic antioxidants, α-TH and γ-TH (10 μM, Fig. 1B Inset).
Oxidation of α-TH and Nitration of γ-TH.
To investigate the nature of the oxidant formed in the SOD/H2O2/NO2− system, we monitored both α-TH and γ-TH depletion and product formation during incubation with SOD/H2O2/NO2−. α-TH was incorporated into large unilamellar vesicles of saturated lipid (DLPC) that cannot undergo peroxidation. Fig. 2 shows HPLC analysis of the reaction of SOD/H2O2 with α-TH in the presence of NO2−. Analysis of samples showed the presence of two peaks, characteristic of α-TH and α-TQ (Fig. 2A). Formation of α-TQ was not observed in the absence of either SOD, H2O2, or NO2−. α-TQ formed from the reaction between α-TH and SOD/H2O2/NO2−, increased linearly from 0 to 15 μM in 4 hr, and then began to plateau (Fig. 2B). Total yield of α-TQ after 1 hr ranged from 0 to 10 μM with increasing concentration of H2O2 (0–1 mM, Fig. 2C).
Figure 2.

Nitrite-dependent α-TQ formation during the oxidation of α-TH by SOD/H2O2. (A) DLPC liposomes containing α-TH (50 μM) were incubated with SOD (1 mg/ml), H2O2 (1 mM), and NO2− (1 mM) in phosphate buffer (200 mM, pH 7.4) at 37°C for 1 hr. An HPLC trace showing the formation of α-TQ during the incubation of α-TH (50 μM) with SOD, H2O2, and NO2− (Aa). A typical HPLC trace showing the result when the above incubation was performed in the absence of SOD, or H2O2, or NO2− (Ab). HPLC trace showing the retention time for an authentic α-TH standard (Ac) and an α-TQ standard (Ad). (B) Kinetics of α-TQ formation as a function of time when liposomes were incubated with SOD (1 mg/ml), H2O2 (1 mM), and NO2− (1 mM) in phosphate buffer (200 mM, pH 7.4) at 37°C. (C) the same as B but as a function of H2O2 concentration.
Nitration of γ-TH incorporated into DLPC liposomes was observed during the reaction of γ-TH (50 μM) with SOD/H2O2 in the presence of NO2− (Fig. 3). HPLC analysis of samples resulted in the detection of two major peaks, γ-TH and its nitrated product, NGTH (Fig. 3A). NGTH concentration steadily increased as a function of time from 0 to 12 μM in 3 hr, followed by a plateau (Fig. 3B). A decrease in NGTH concentration was observed after 8 hr, suggesting NGTH was further oxidized by the oxidant formed during the reaction (Fig. 3B). The yield of NGTH increased from 0 to 6 μM during the incubation of γ-TH with SOD/NO2− for 1 hr in the presence of increasing concentrations of H2O2 (0 μM to 1 mM, Fig. 3C). Under identical conditions, nitration of γ-TH was at least 10-fold greater than that of tyrosine, indicating a greater propensity for SOD-catalyzed nitration of lipophilic phenols.
Figure 3.

Nitrite-dependent nitration of γ-TH during the oxidation of γ-TH by SOD/H2O2. (A) DLPC liposomes containing γ-TH (50 μM) were incubated with SOD (1 mg/ml), H2O2 (1 mM) and NO2− (1 mM) in phosphate buffer (200 mM, pH 7.4) at 37°C for 1 hr. HPLC trace showing the formation of NGTH during the incubation of γ-TH (50 μM) with SOD, H2O2 and NO2− (Aa), HPLC trace showing the result when the above incubation was performed in the absence of SOD, or H2O2, or NO2− (Ab), HPLC trace showing the retention time for an authentic γ-TH standard (Ac), and a NGTH standard (Ad). (B) Kinetics of NGTH formation as a function of time when liposomes were incubated with SOD (1 mg/ml), H2O2 (1 mM) and NO2− (1 mM) in phosphate buffer (200 mM, pH 7.4) at 37°C and C same as B but as a function of H2O2 concentration.
ESR Detection of Tocopheroxyl Radicals.
The mechanism of α-TQ and NGTH formation was investigated by ESR (22, 24, 25). Addition of H2O2 (10 mM) to an incubation mixture containing α-TH (2 mM), SOD (10 mg/ml), and nitrite (10 mM) produced a seven-line ESR spectrum (Fig. 4Aa) characteristic of the α-T• radical (aCH35 (3H) = 5.93 G; aCH37 (3H) = 4.55 G, simulation shown as b of Fig. 4A) (24–25). Because of poor signal-to-noise, a relatively high modulation amplitude was used, reducing our ability to resolve couplings from the methyl protons at carbon-8 and methylene protons at carbon-4. In the absence of nitrite, there was a marked reduction in the signal intensity (Fig. 4Ac). If either SOD or H2O2 were excluded, there was no detectable ESR signal (Fig. 4Ad).
Figure 4.

Nitrite-enhanced formation of α-T•, γ-T•, and NGT• radicals during the oxidation of α-TH, γ-TH, and NGTH by SOD/H2O2. DLPC liposomes containing α-TH, γ-TH, or NGTH (2 mM each) were incubated in the presence or absence of SOD (10 mg/ml), H2O2 (10 mM), and NO2− (10 mM) in phosphate buffer (200 mM, pH 7.4) at ambient temperature. (A) ESR spectrum of α-T• generated in the presence of SOD, H2O2, and NO2− (Aa), simulation of the spectrum by using the spectral parameters for α-T• as given in the text (Ab), spectrum generated in the presence of SOD and H2O2 without NO2− (Ac), and the ESR spectrum generated in the presence of SOD and NO2− without H2O2 (Ad). (Ba–d) Spectra correspond to the conditions used in Aa–d by using γ-TH and (C) ESR spectrum of standard NGT radical obtained on incubation of NGTH (1 mM) in methanol with 5 mg of silver oxide in 200 μl of sample volume under anaerobic conditions (Ca), computer simulation by using spectral parameters given in the text (Cb), spectrum obtained on incubating SOD (10 mg/ml) with H2O2 (10 mM) in the presence of nitrite (10 mM) (Cc), at higher modulation amplitude (Cd), and in the absence of nitrite (Ce). Spectrometer conditions: time constant, 0.128 s; microwave power, 20 mW; scan range, 40 G; and scan time, 1 min, modulation amplitude 0.5 G. Modulation amplitude for Ca was 0.125 G and 0.2 G for Cc.
Fig. 4B shows a similar series of experiment with γ-TH. Spectrum in Fig. 4Ba is characteristic of the γ-T• radical (aH5 (1H) = 4.55 G; aCH37 (3H) = 5.93 G, simulation shown in Fig. 4Bb). Again, additional hyperfine couplings could not be resolved. Exclusion of nitrite reduced signal intensity (Fig. 4Bc) and there was no ESR detectable signal if either SOD or H2O2 were excluded (Fig. 4Bd).
Incubation of DLPC liposomes containing NGTH with SOD/H2O2/NO2− produced a four-line spectrum (Fig. 4Cd ) with a 1:3:3:1 intensity ratio, typical for an electron interacting with three equivalent protons. Exclusion of nitrite reduced the intensity of this signal (Fig. 4Ce). At lower modulation amplitudes, the hyperfine couplings due to methyl protons at carbon-8 and nitrogen at carbon-5 were resolved (Fig. 4Cc). An authentic spectrum of NGT radical was obtained by oxidizing NGTH in methanol with silver oxide (Fig. 4Ca). The computer simulation of this spectrum is shown in Fig. 4Cb (aCH37 (3H) = 4.4 G; aCH38 (3H) = 1.2 G; aN5 (1) = 1.0 G). ESR results clearly demonstrate that an oxidant derived from the nitrite anion, most likely •NO2, reacts with tocopherols to produce the corresponding tocopheroxyl radicals. The rapid reaction between •NO2 and α-T• or γ-T• may explain the decrease in their steady–state concentrations. ESR detectable radicals were not observed with nitrite alone, or in the absence of SOD or H2O2.
Effect of Spin Traps on SOD/H2O2/NO2−-Mediated Oxidation and Nitration of Tocopherols.
Fig. 5 shows the effect of various spin traps on α-TQ and NGTH formation from α-TH and γ-TH by the SOD/H2O2/NO2− system. MDL 101,002 and POBN were most effective at inhibiting the formation of α-TQ and NGTH. POBN (50 mM) inhibited α-TQ and NGTH formation by ≈60%. Nitrone spin traps are effective radical scavengers, and it is very likely that nitrones inhibit SOD/H2O2/NO2−-catalyzed oxidation and nitration of α-TH and γ-TH by scavenging the •NO2 radical.
Figure 5.

Nitrone spin traps inhibit oxidation of α-TH and nitration of γ-TH by SOD/H2O2/NO2−. DLPC liposomes containing α-TH or γ-TH (50 μM) were incubated with SOD (1 mg/ml), H2O2 (1 mM), and NO2− (1 mM) in phosphate buffer (200 mM, pH 7.4) containing DTPA (100 μM) at 37°C for 1 hr in the presence and absence of spin traps (n = 3 ± SD).
Protection of H2O2-Mediated SOD Inactivation by Nitrite.
The rate of superoxide-dependent cyt c reduction was monitored at 550 nm. As shown in Fig. 6, addition of SOD inhibited the increase in absorbance. Cyt c reduction was not inhibited by SOD that was incubated with H2O2 for 2 hr. The presence of nitrite (1 mM) protected SOD from H2O2-dependent inactivation. SOD activity was calculated from the initial rate of cyt c reduction. Incubation of SOD with H2O2 (1 mM) for 5 hr resulted in an ≈80% reduction in SOD activity (Fig. 6 Inset). However, the addition of nitrite (1 mM) inhibited inactivation over the entire 5-hr period. Under similar conditions, NO3− did not protect the enzyme (data not shown). Inactivation of SOD by H2O2 was accompanied by the release of copper and zinc from the active site as determined by 4-pyridylazaresorcinol assay (21). NO2− (1 mM) inhibited the release of copper and zinc during H2O2-mediated inactivation of SOD (data not shown).
Figure 6.

Nitrite inhibits H2O2-mediated SOD inactivation. Superoxide-dependent cyt c (20 μM) reduction during the reaction of xanthine (500 μM) and xanthine oxidase (0.05 unit/ml) at 37°C in 200 mM phosphate buffer containing 1,000 units/ml of catalase (——). Inhibition of cyt c reduction by SOD (5 μg/ml, – – –); effect of H2O2-inactivated SOD (——), H2O2-treated SOD in the presence of nitrite (1 mM, – – –). (Inset) Effect of nitrite (1 mM) on H2O2-dependent SOD inactivation.
DISCUSSION
Oxidation of Nitrite to the Nitrogen Dioxide (•NO2) Radical.
The present data suggest that NO2− is oxidized by the oxidant, SOD-Cu2+-•OH, to a free radical, most likely •NO2 (Eq. 5).
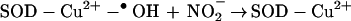 |
5 |
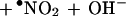 |
The peroxidase activity of SOD has been shown to be nonspecific and is capable of oxidizing several substrates with a range of redox potentials, as follows:E° (•N3/N3−) = 1.3 V; E° (•NO2/NO2−) = 1.03 V; E° (urate radical/urate) = 0.26 V (26–28). Thus, the oxidation potential of the bound oxidant generated from SOD and H2O2 should be considerably higher than those reported for conventional peroxidases such as horseradish peroxidase, E° (HRP-II/HRP) = 0.97 V (27). Previous investigations have concluded that the oxidant generated is a site-specific copper-bound hydroxyl radical, not a freely diffusible hydroxyl radical (1–2, 29, 30). Further support for this mechanism came from studies showing that H2O2-dependent inactivation of SOD was prevented by urate, histidine, azide, and formate but not by alcohols or spin traps (1, 2, 29, 30). The present results show that nitrite anion also inhibits the inactivation of SOD by H2O2.
Mechanism of Conjugated Diene Formation.
Nitrogen dioxide-initiated oxidation of polyunsaturated fatty acids is well established (31–33). Scheme S1 illustrates how •NO2 reacts with a linoleate-type fatty acid (k ≈ 105 − 106 M−1 s−1) (9, 34) to produce the corresponding conjugated diene.
Scheme 1.

•NO2-mediated oxidation of unsaturated fatty acids.
According to this mechanism, •NO2 abstracts an allylic hydrogen atom to form a resonance-stabilized, carbon-centered radical. This radical can combine with oxygen to form a peroxyl radical, which can propagate lipid oxidation. In the absence of oxygen, the allylic radical can combine with a molecule of •NO2 to form an allylic nitro or nitrite compound. Alternatively, •NO2 also can add to the double bond and form a carbon-centered radical, which can react with oxygen to form a nitro-peroxyl radical or, in the absence of oxygen, react with another molecule of •NO2 to form a dinitro compound (35).
NO2•-mediated autoxidation of unsaturated lipid is different from copper- and hydroperoxide-dependent oxidation as follows: (i) DTPA completely inhibits copper-mediated oxidation of unsaturated fatty acids, (ii) the copper-dependent process is auto-catalytic, and (iii) •NO2-mediated lipid oxidation is not auto-catalytic and is not dependent on “seeded” hydroperoxides.
Mechanism of α-TQ and NGTH Formation.
•NO2, a potent lipid-soluble oxidant, can abstract a hydrogen atom from the phenolic hydroxyl group of α-TH and γ-TH to form the corresponding α-T• and γ-T• radicals (36). The steady–state concentration of •NO2 in hydrophobic membranes should be higher compared with the aqueous phase due to decreased hydrolysis (37). Based on the published data for trolox, a water soluble analog of α-TH, the rate constant for these reactions (Eqs. 6 and 8) are expected to vary with pH, from ≈105 M−1 s−1 at pH 7 to 109 M−1 s−1 at pH 11 (38) (Scheme 2). The reaction between the α-T• radical and •NO2 is very rapid (Eq. 7) (3 × 109 M−1 s−1), forming an intermediate (due to radical-radical recombination at C-8a) which rearranges to α-TQ (9, 11). The reaction between the γ-T• radical and •NO2 (Eq. 9) results in nitration at C-5 due to radical-radical recombination. HPLC data show that the presence of nitrite anion is required for product formation (Figs. 2 and 3). The rapid reaction between •NO2 and the α-T• and γ-T• radicals may account for their low steady–state concentrations as measured by ESR (Fig. 4). ESR data show that •NO2 can abstract a hydrogen atom from NGTH (Eq. 10), to form the corresponding tocopheroxyl radical, •NGT. Consistent with this, NGTH concentration decreased with time during the reaction (Fig. 3B).
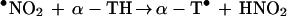 |
6 |
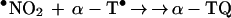 |
7 |
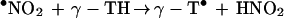 |
8 |
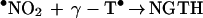 |
9 |
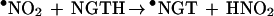 |
10 |
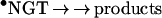 |
11 |
Scheme 2. Reactions between •NO2 and tocopherols and tocopheroxyl radicals.
Implications in ALS.
ALS, also known as Lou Gehrig’s disease, is a condition in which there is degeneration of the motor neurons of the spinal cord, brain stem, and cerebral cortex (39, 40). The genetic defect in 20% of FALS cases has now been linked to SOD1, the gene encoding the cytosolic Cu,ZnSOD enzyme. It has been hypothesized that an aberrant in vivo property of a FALS SOD mutant was responsible for increased formation of 3-nitrotyrosine and FALS pathogenesis (41, 42). Nitration of tyrosine residues in neurofilament proteins was suggested to be catalyzed by SOD and peroxynitrite (21). Recently, it was shown that •NO2 generated by the myeloperoxidase/H2O2/nitrite system was able to nitrate tyrosine (10, 11). These findings suggest a functional role for SOD and metabolic nitrite in ALS pathogenesis. The same mechanism, i.e., SOD/H2O2/nitrite-dependent formation of •NO2, also may explain the formation of increased lipid oxidation products in ALS tissues (43). Recently, using DMPO as a peroxidase substrate, it was shown that FALS-associated SOD mutants exhibit greater peroxidase activity (44, 45). However, the mechanism of oxidation of DMPO in this system was more complex (29) than previously reported (44, 45). Clearly, there is a definite need to directly probe the peroxidase activity of SOD1 mutants. Although the peroxidase activity can be indirectly monitored by following the rate of inactivation of SOD1 mutants (20), the present results indicate that the relative peroxidase activities of FALS-linked SOD1 mutants can be measured by using NO2− as a peroxidase substrate.
CONCLUSIONS
We conclude that the peroxidase activity of SOD in the presence of nitrite will initiate both peroxidation and nitration reactions. We postulate that •NO2 generated from NO2− during its reaction with SOD/H2O2 is capable of oxidizing α-TH to α-TQ and nitrating γ-TH to NGTH. ESR studies suggest that both α-TQ and NGTH are formed through the intermediate production of α-T• and γ-T• radicals. Results from this study may be relevant to the understanding of oxidation and nitration reactions catalyzed by extracellular SOD and SOD1 mutants in neuronal disease processes, in particular, ALS.
Acknowledgments
This work was supported by grants from the National Institutes of Health RR01008 and GM22923 and from the Amyotrophic Lateral Sclerosis Association. We express thanks to Dr. Neil Hogg and Dr. Jeannette Vásquez-Vivar for their help in data interpretation and Dr. Joseph Beckman for stimulating discussions.
ABBREVIATIONS
- α-TH
α-tocopherol
- α-T•
α-tocopheroxyl radical
- FALS
famalial amyotrophic lateral sclerosis
- DLPC
1,2-dilauroyl-sn-glycero-3-phosphatidylcholine
- DMPO
5–5′-dimethyl-1-pyrroline N-oxide
- ESR
electron spin resonance
- γ-TH
γ-tocopherol
- γ-T•
γ-tocopheroxyl radical
- •NO
nitric oxide
- NGTH
5-nitro-γ-TH
- •NGT
5-nitro-γ-T radical
- •NO2
nitrogen dioxide
- POBN
α-(4-pyridyl-1-oxide)-N-t-butylnitrone
- SOD
superoxide dismutase
- NO2−
nitrite anion
- DTPA
diethylenetriaminepentacetic acid
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Fridovich I. Annu Rev Biochem. 1975;44:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 2.Fridovich I. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 3.Hodgson E K, Fridovich I. Biochemistry. 1975;14:5299–5303. doi: 10.1021/bi00695a011. [DOI] [PubMed] [Google Scholar]
- 4.Hodgson E K, Fridovich I. Biochemistry. 1975;14:5294–5298. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 5.Cabelli D E, Allen D, Bielski B H, Holcman J. J Biol Chem. 1989;264:9967–9971. [PubMed] [Google Scholar]
- 6.Sato K, Akaike T, Kohno M, Ando M, Maeda H. J Biol Chem. 1992;267:25371–25377. [PubMed] [Google Scholar]
- 7.Uchida K, Kawakishi S. J Biol Chem. 1994;269:2405–2410. [PubMed] [Google Scholar]
- 8.Uchida K, Kawakishi S. Arch Biochem Biophys. 1990;283:20–26. doi: 10.1016/0003-9861(90)90606-y. [DOI] [PubMed] [Google Scholar]
- 9.Prutz W A, Mönig H, Butler J, Land E J. Arch Biochem Biophys. 1985;243:125–134. doi: 10.1016/0003-9861(85)90780-5. [DOI] [PubMed] [Google Scholar]
- 10.Eiserich J P, Hristova M, Cross C E, Jones A D, Freeman B A, Halliwell B, Van der Vliet A. Nature (London) 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 11.Eiserich J P, Butler J, Van der Vliet A, Cross C E, Halliwell B. Biochem J. 1995;310:745–749. doi: 10.1042/bj3100745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooney R V, Franke A A, Harwood P J, Hatch-Pigott V, Custer L J, Mordan L J. Proc Natl Acad Sci USA. 1993;90:1771–1775. doi: 10.1073/pnas.90.5.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogg N, Joseph J, Kalyanaraman B. Arch Biochem Biophys. 1994;314:153–158. doi: 10.1006/abbi.1994.1423. [DOI] [PubMed] [Google Scholar]
- 14.Christen S, Woodall A A, Shigenaga M K, Southwell-Keely P T, Duncan M W, Ames B N. Proc Natl Acad Sci USA. 1997;94:3217–3222. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoglen N C, Waller S C, Sipes I G, Liebler D C. Chem Res Toxicol. 1997;10:401–407. doi: 10.1021/tx960200h. [DOI] [PubMed] [Google Scholar]
- 16.Liebler D C, Kaysen K L, Burr J A. Chem Res Toxicol. 1991;4:89–93. doi: 10.1021/tx00019a012. [DOI] [PubMed] [Google Scholar]
- 17.Singh R J, Feix J B, Pintar T J, Girroti A W, Kalyanaraman B. Photochem Photobiol. 1991;53:493–500. doi: 10.1111/j.1751-1097.1991.tb03661.x. [DOI] [PubMed] [Google Scholar]
- 18.Goss S P A, Hogg N, Kalyanaraman B. Chem Res Toxicol. 1995;8:800–806. doi: 10.1021/tx00047a021. [DOI] [PubMed] [Google Scholar]
- 19.Hodgson E K, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 20.Liochev S I, Chen L L, Hallewell R A, Fridovich I. Arch Biochem Biophys. 1998;352:237–239. doi: 10.1006/abbi.1998.0616. [DOI] [PubMed] [Google Scholar]
- 21.Crow J P, Sampson J B, Zhuang Y, Thompson J A, Beckman J S. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 22.Iwatsuki M, Tsuchiya J, Komuro E, Yamamoto Y, Niki E. Biochem Biophys Acta. 1994;1200:19–26. doi: 10.1016/0304-4165(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 23.Esterbauer H, Striegl G, Puhl H, Rotheneder M. Free Radical Res Commun. 1989;6:67–75. doi: 10.3109/10715768909073429. [DOI] [PubMed] [Google Scholar]
- 24.Kalyanaraman B, Darley-Usmar V M, Wood J, Joseph J, Parthasarathy S. J Biol Chem. 1992;267:6789–6795. [PubMed] [Google Scholar]
- 25.Lambelet P, Loliger J. Chem Phys Lipids. 1984;35:185–198. doi: 10.1016/0009-3084(84)90045-8. [DOI] [PubMed] [Google Scholar]
- 26.Stanbury D M. Adv Inorg Chem. 1989;33:69–138. [Google Scholar]
- 27.Buettner G. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 28.Jovanovic S V, Simic M G. J Phys Chem. 1986;90:974–978. [Google Scholar]
- 29.Singh R J, Karoui H, Gunther M R, Beckman J S, Mason R P, Kalyanaraman B. Proc Natl Acad Sci USA. 1998;95:6675–6680. doi: 10.1073/pnas.95.12.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jewett S L, Abel J M. Abstracts of the Annual Meeting of the Oxygen Society, November 20–24, 1997. San Francisco, CA: Oxygen Society; 1997. [Google Scholar]
- 31.Pryor W A, Lightsey J W. Science. 1981;214:435–437. doi: 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- 32.Gallon A A, Pryor W A. Lipids. 1994;29:171–176. doi: 10.1007/BF02536725. [DOI] [PubMed] [Google Scholar]
- 33.Gallon A A, Pryor W A. Lipids. 1993;28:125–133. doi: 10.1007/BF02535776. [DOI] [PubMed] [Google Scholar]
- 34.Forni L J, Mora-Arellano V O, Packer J E, Willson R L. J Chem Soc Perkin Trans. 1986;2:1–6. [Google Scholar]
- 35.Pryor W A, Castle L, Church F D. J Am Chem Soc. 1985;107:211–217. [Google Scholar]
- 36.Brunton G, Cruse H W, Riches K M, Whittle A. Tetrahedron Lett. 1979;12:1093–1094. [Google Scholar]
- 37.Liu X, Miller M J S, Joshi M S, Thomas D D, Lancaster J R., Jr Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies M J, Forni L G, Willson R L. Biochem J. 1988;255:513–522. [PMC free article] [PubMed] [Google Scholar]
- 39.Price D L, Cleveland D W, Koliatsos V E. Neurobiol Dis. 1994;1:3–11. doi: 10.1006/nbdi.1994.0002. [DOI] [PubMed] [Google Scholar]
- 40.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 41.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584–584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 42.Bruijn L I, Beal M F, Becher M W, Schulz J B, Wong P C, Price D L, Cleveland D W. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrante R J, Shinobu L A, Schulz J B, Matthews R T, Thomas C E, Kowall NW, Gurney M E, Beal M F. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 44.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 45.Yim M B, Kang J H, Yim H S, Kwak H S, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]