

Abstract
A mechanism by which Ca2+/CaM-dependent protein kinase (CaMKII) is autophosphorylated by changes in extracellular calcium in the absence of detectable changes in cytoplasmic [Ca2+] has been identified. We find that when the external Ca2+ concentration ([Ca2+]O) is lowered, Ca2+ is released from intracellular stores to maintain a constant cytoplasmic Ca2+ level, gradually depleting the endoplasmic Ca2+ stores. Accompanying the store-depletion is a rapid decrease in CaMKII activity. Approximately 25% of the measured CaMKII autophosphorylation in DRG neurons in culture can be regulated by Ca2+ flux from intracellular stores caused by manipulating [Ca2+]O, as shown by blocking refilling of store-operated Ca2+-channels with SK&F 96365, Ruthenium Red, and a partial block with Ni2+. Blocking voltage-gated Ca2+-channels with either isradipine or SR 33805, had no effect on CaMKII autophosphorylation induced by restoring Ca2+O to normal after depleting the intracellular Ca2+ stores. These results show that removal of Ca2+O has profound effects on intracellular Ca2+ signaling and CaMKII autophosphorylation, in the absence of measurable changes in intracellular Ca2+. These findings have wide-ranging significance, because [Ca2+]O is manipulated in many experimental studies. Moreover, this explanation for the paradoxical changes in CaMKII phosphorylation in response to manipulating [Ca2+]O provides a possible mechanism linking activity-dependent depletion of Ca2+ from the synaptic cleft to a protein kinase regulating many neuronal properties.
Keywords: Calcium-induced calcium release, CaMKII, Capacitative calcium entry, Dorsal root ganglia, Extracellular calcium, LTP, Synaptic plasticity
1. Introduction
Ca2+/CaM-dependent protein kinase (CaMKII), is a highly abundant enzyme present in neurons, which can function as a molecular switch converting intracellular Ca2+ (Ca2+i) signals into short-, intermediate-, and long-term responses through phosphorylation of diverse substrates involved in exocytosis, transcriptional, and translational processes [1]. Curiously, several studies have reported that CaMKII autophosphorylation is also regulated by changes in the concentration of extracellular Ca2+ ([Ca2+]O) [2–6], but the mechanism is unknown. In several experimental systems, basal levels of the phosphorylated, autonomously active form of CaMKII are quite high, but lowering the [Ca2+]O reduces CaMKII autophosphorylation dramatically. The initial explanation was that this effect was secondary to a reduction in endogenous synaptic activity under low Ca2+O conditions. However, studies reporting the same phenomenon in cultured dorsal root ganglion neurons (DRG) lacking spontaneous activity and synapses in culture indicate that this cannot be the only mechanism [5].
Here we provide evidence that CaMKII autophosphorylation regulated by [Ca2+]O is not affected by blocking a wide range of voltage-gated Ca2+-channels (VGCC), but that Ca2+-flux from intracellular stores in response to changes in [Ca2+]O regulates CaMKII autophosphorylation. Manipulations that decrease stores, e.g. removing Ca2+O, do not affect bulk [Ca2+]i but act to rapidly decrease CaMKII phosphorylation. Additionally, re-addition of Ca2+O modestly increases resting [Ca2+]i and strongly activates CaMKII. Thus, this signaling pathway permits [Ca2+]O to directly regulate the activation state of CaMKII independent of neuronal depolarization and neurotransmitter-mediated activity. Rapid dephosphorylation of CaMKII over several minutes is within a physiological time frame. [Ca2+]O-mediated regulation of Ca2+i signaling involving CaMKII may underlie an overlooked but important mechanism contributing to synaptic plasticity. In order to address this issue we further studied [Ca2+]O-dependent regulation of CaMKII in DRG neurons.
2. Materials and methods
2.1. Cell culture
Campenot chambers were made of Teflon and attached to collagen-coated 35 mm culture dishes as described in Ref. [7]. DRG neurons were dissociated from 13.5-day old mouse embryos and plated at a density of 0.5 × 106 cells/ml into each side compartment in Eagle MEM with Earle's salts used for culturing neurons containing 5% horse serum and 100 ng/ml nerve growth factor as described previously [8]. For imaging studies, DRG neurons were plated at a density of 0.25 × 106 cells/ml onto glass coverslips. Non-neuronal cell division was inhibited by the addition of 13 μg/ml fluoro-2-deoxyuridine and uracyl 1 day following plating for 4–5 days. Cultures were subsequently used for experiments 3–4 weeks after plating at which time they display a mature axonal outgrowth.
2.2. Drugs
The following drugs were used: isradipine (Alomone Labs, Jerusalem, Israel), flunarizine, ionomycin, ω-agatoxin-IVA, and ω-conotoxin-GVIA (EMD Biosciences Inc., San Diego, CA), Fluo-4/AM and Indo-1/AM (Molecular Probes, Eugene OR), caffeine, EGTA, gadolinium chloride, lanthanum chloride, nickel chloride, and nifedipine (Sigma, St. Louis, MO), KB-R7943, MRS 1845, Ruthenium Red (RR), SK&F 96365, and SR 33805 (Tocris Cookson, Ellisville, MO). In cases where drugs were dissolved in DMSO, final concentrations did not exceed 0.1% except for MRS 1845 where the DMSO concentration was 0.16% which was included in controls.
2.3. Autophosphorylation of CaMKII at 286/287Thr
CaMKII autophosphorylation at 286/287Thr was analyzed by immunoblotting using a phosphorylation-site specific antibody that recognizes CaMKII only when it is autophosphorylated at Thr-286 (α) or Thr-287 (β, γ, δ) [9]. Neurons were incubated overnight in serum- and growth factor-free medium. The following day, the medium was exchanged four times with a physiological saline solution containing 50 nM free Ca2+O (referred to in the figures as 0 Ca2+O) [4]. This was obtained by substituting (in mM): 0.76 CaCl2, 1.13 MgCl2, and 2 EGTA in a HEPES-buffered saline (pH 7.38) containing 10 mM glucose. Free Ca2+ concentrations were calculated using Maxchelator [10]; EGTA was omitted in experiments with Gd3+, La3+, and Ni2+ as it binds these cations with much higher affinity than Ca2+. Following incubation in 50 nM free Ca2+O for 60 min, the [Ca2+]O was raised by rapidly exchanging the media within the compartment with media containing 1.8 mM free Ca2+O and incubating for 45 s (1.8 Ca2+). (Molloy and Kennedy [2] observed that CaMKII activity decreases to a stable baseline by 30 min in 0 Ca2+O.) Treated neurons were subsequently lysed in 100 μl 2× sample buffer for electrophoresis and immunoblot analysis.
For quantification of 286/287Thr autophosphorylation, 15 μl of lysate from control and treated neurons were resolved in parallel by SDS-PAGE in duplicate 10% NOVEX Bis–Tris gels (Invitrogen, Carlsbad, CA) and electroblotted to PVDF membranes (Immobilon-P, from Millipore, Bedford, MA). Membranes were blocked in 5% non-fat dry milk in TBS-T for 2 h at room temperature, washed, and incubated in antibody that recognized total CaMKII at 1:2500 (mAb 38, from BD Biosciences Pharmingen, San Diego, CA) or phosphosite-specific antibody (1:10,000, from Dr. Y. Yamagata, Laboratory of Neurochemistry, Okazaki, Japan) overnight at 4 °C. Incubated membranes were washed and incubated with HRP-conjugated secondary antibodies (Amersham Pharmacia Biotech, Piscataway, NJ) for 2 h at room temperature. The immunocomplexes were visualized with ECL Plus substrate (Amersham Pharmacia Biotech, Piscataway, NJ) and quantified with ImageQuant and Storm image analysis system (Molecular Dynamics, Sunnyvale, CA). Relative autophosphorylation at 286/287Thr was compared by normalization of the RFU obtained with the phosphorylated enzyme to that of the total enzyme in the same sample from parallel experiments.
2.4. Immunocytochemistry
DRG neurons were fixed in 4% paraformalydehyde containing 10 mM EGTA and 4% sucrose. Following fixation, neurons were permeablized with 0.3% Triton X-100 for 5 min and free aldehydes were quenched with 50 mM ammonium chloride for 10 min. After blocking in 3% NGS, primary antibodies were incubated overnight at 4 °C. The following day, neurons were incubated for 2 h in secondary antibodies and mounted in Vectashield (Vector Labs, Burlingame, CA). The antibodies and dilutions used were: anti-phospho-CaMKII (1:500, Upstate USA Inc., Charlottesville, VA), anti-CaMKII (1:500, mAb 38, BD Biosciences Pharmingen), Alexa 488 goat anti-rabbit (1:500; Molecular Probes). All antibodies were diluted in 3% NGS.
2.5. Intracellular Ca2+ measurements
Caffeine-evoked Ca2+ transients in DRG neurons were measured using a Bio-Rad (Hercules, CA) 1024 visible/UV confocal microscope and a Nikon 40× 1.3 numerical aperture oil immersion objective on a Nikon inverted microscope. Quantitative Ca2+ measurements were made using ratiometric measurements of fluorescence intensity at 460 and 405 nm emission from DRG neurons loaded by incubation in 7.5 μM indo-1/AM and excited by an argon-ion laser at 350 nm [53]. Two-photon Ca2+ imaging with Fluo-4/AM were measured using a Bio-Rad Radiance 2100 MP confocal microscope. Relative changes in Ca2+ were made using measurements of fluorescence intensity from DRG neurons loaded by incubation in 9.1 μM Fluo-4/AM and excited by a Mira 900 tuned to 800 nm. Measurements were performed at room temperature in HEPES-buffered balanced salt solution (pH 7.38). In-cell calibration, as described in [54], was used to provide an estimate of the [Ca2+]i associated with the fluorescence ratios. Briefly, Rmin and Rmax were determined in neurons permeablized by 10 μM ionomycin, in solutions containing 1.8 mM Ca2+ and 0 mM Ca2+/10 mM EGTA, under the same intensifier gain and pinhole settings that were used during the experiments. Measurements of [Ca2+]i were made within an optical plane passing through the center of the nucleus in the area of the cytoplasm midway between cell membrane and the nucleus. The area of measurement comprised ∼1/8 of the area of cytoplasm in the plane of section. The measured responses were uniform within different regions of the cell on the time scale reported in these experiments. Normalized fluorescence values for Fluo-4 were calculated by first subtracting background and rationing to the fluorescence intensity immediately prior to re-addition of Ca2+O with the equation: F = (F − F0)/F0. To compensate for Fluo-4 photo-bleaching due to prolonged imaging, controls were performed where DRG neurons were imaged in 1.8 mM Ca2+O-containing saline.
3. Results
3.1. Ca2+O regulates CaMKII phosphorylation
We observed that the level of autophosphorylated CaMKII was dependent on the concentration of Ca2+O (Fig. 1). Removing Ca2+O for 60 min decreased CaMKII phosphorylation to very low levels (Fig. 1B and D). Re-addition of Ca2+O rapidly restored phosphorylation to previous levels (Fig. 1B and C); phosphorylation at 286/287Thr increased by 245 ± 27.6% relative to basal in 50 nM Ca2+O (n = 3). The increase in phosphorylation mediated by Ca2+O is much greater than that mediated by activity-evoked changes, e.g. action potentials, in [Ca2+]i, which result in ∼25% increase in CaMKII autophosphorylation and Ca2+-independent activity [5]. By immunocytochemistry, we found weak staining for 286/287Thr-CaMKII in DRG cell bodies incubated in low Ca2+O; immunoreactivity was entirely absent from axons (Fig. 1D1). Stimulation by 1.8 mM Ca2+O activated CaMKII as seen by strong immunoreactivity for 286/287Thr-CaMKII in both the cell bodies and axons (Fig. 1D2). Staining was predominately excluded from the nucleus in both unstimulated and stimulated neurons (Fig. 1D).
Fig. 1.
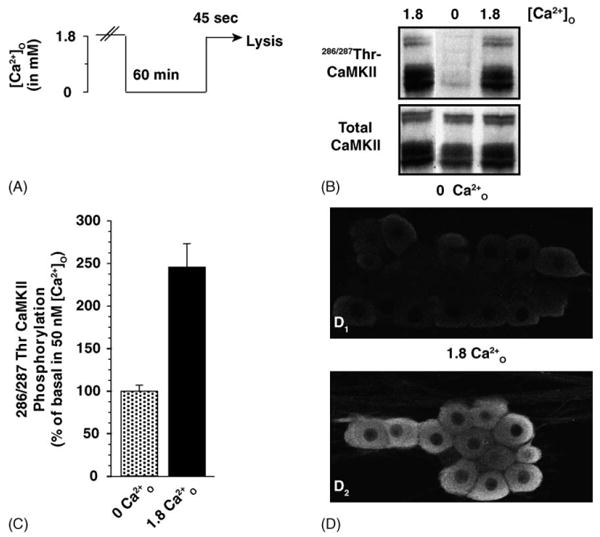
DRG neurons exhibit high basal CaMKII autophosphorylation at 286/287Thr that is regulated by Ca2+O. (A) In the Ca2+O-stimulation paradigm, DRG neurons were incubated in culture medium containing 50 nM free Ca2+ (0 Ca2+) for 60 min followed by a 45 s treatment with 1.8 mM Ca2+O (1.8 Ca2+). (B) Resting neurons in a medium containing 1.8 mM Ca2+O display strong CaMKII phosphorylation at 286/287Thr. Incubation for 60 min in 50 nM Ca2+O decreased the level of CaMKII phosphorylation; re-addition of extracellular calcium rapidly activated CaMKII. (C) Stimulation of DRG neurons with 1.8 mM Ca2+O increased phosphorylation at 286/287Thr-CaMKII by several-fold. Relative autophosphorylation of CaMKII at 286/287Thr was calculated by normalizing the relative immunoreactivity (RFU) of the phosphorylated sample to that of the total enzyme present in the same sample. Data are expressed as mean ± S.E.M. of percentage phosphorylation normalized to basal levels in 50 nM Ca2+O (n = 3). (D) DRG neurons were incubated in 0 Ca2+O. Ca2+O-stimulation was terminated by fixation in 4% paraformalydehyde containing 10 mM EGTA to block fixation-dependent Ca2+ influx [50,51]. Weak immunoreactivity for 286/287Thr-CaMKII is observed in unstimulated neurons and entirely absent from axons (D1 and D2). Re-addition of 1.8 mM Ca2+O activates CaMKII as seen by strong immunoreactivity for 286/287Thr-CaMKII in both the cell bodies and axons (D2). The boxed regions indicate the field of higher magnification. There appears to be an increase in staining in both the cytoplasmic and nuclear membrane compared to the intracellular compartment. Staining is predominately excluded from the nucleus.
3.2. Basal CaMKII phosphorylation decays rapidly in 0 Ca+O
We more thoroughly characterized the response of DRGs to changes in Ca2+O by examining both the time course of CaMKII phosphorylation due to increasing time in 0 Ca2+O as well as sensitivity to increasing [Ca2+]O. CaMKII autophosphorylation at 286/287Thr rapidly decreased with increasing time in low Ca2+O (Fig. 2A) [2–6]. We observed that phosphorylation increased in a dose-dependent manner; half-maximal activation of CaMKII occurred at 2.6 mM Ca2+O (Fig. 2B). Despite intense research in this ubiquitous protein kinase, the mechanism regulating CaMKII autophosphorylation in response to changes in Ca2+O have remained obscure for many years.
Fig. 2.
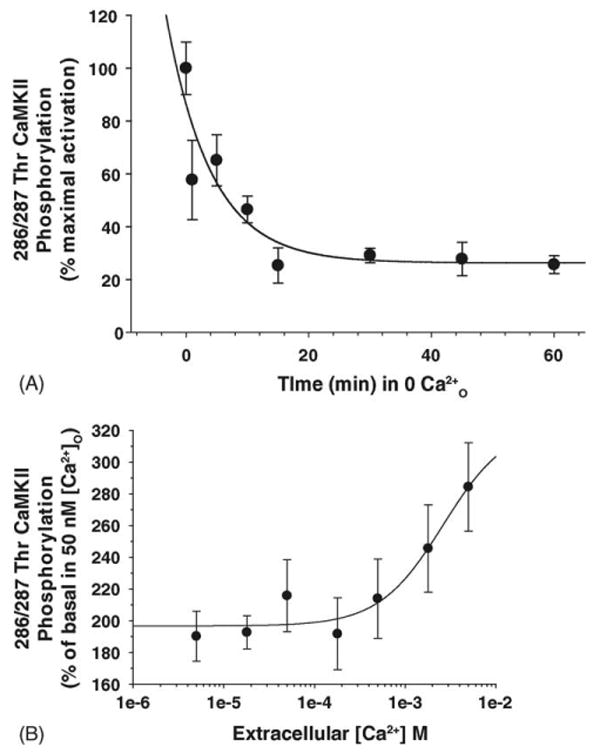
Ca2+O-regulation of CaMKII phosphorylation decreases rapidly and is dose-dependently activated by Ca2+O. (A) Phosphorylation of CaMKII at 286/287Thr rapidly decreased with increasing incubation in 50 nM free Ca2+. DRG neurons were incubated in low Ca2+ for different time periods. The minimum time to transfer cells from normal Ca2+ to low Ca2+ is 1 min. Data were fit to three-parameter exponential decay to obtain the decay rate constant, 0.137 s−1. CaMKII phosphorylation relative to control was decreased to less than 50% by 7 min in 50 nM free Ca2+ (n = 4). (B) DRG neurons were incubated in a medium containing 50 nM free Ca2+ for 60 min and then stimulated for 45 s with increasing [Ca2+]O. As in (Fig. 1(C)), relative autophosphorylation of CaMKII at 286/287Thr was compared for each condition by normalizing the RFU of the phosphorylated sample to that of the total enzyme present in the same sample. The ratio of phospho:total CaMKII for each [Ca2+] was then normalized to stimulation by 50 nM free Ca2+; data were fit to a four-parameter logistic equation to obtain an EC50 of 2.6 mM as mean ± S.E.M. (n = 3).
3.3. Intracellular Ca2+i does not change during incubation in 0 Ca2+O
Confirming previous results [5], two-photon Ca2+-imaging with Fluo-4 showed that decreasing Ca2+O produced no measurable changes in bulk [Ca2+]i over 60 min (Fig. 3). Previously, Eshete and Fields [5] did not observe an increase in [Ca2+]i with re-addition of Ca2+O. We utilized two-photon Ca2+–Ca2+-imaging with Fluo-4 (Fluo-4 is more sensitive to Δ[Ca2+]i as compared with Indo-1) to minimize both photo-bleaching and cellular damage due to prolonged imaging. We were able to detect a small increase in [Ca2+]i following re-addition of Ca2+O (Fig. 3, inset). For comparison, application of 10 mM caffeine elicited a large, rapid increase in [Ca2+]i (11.5-fold higher compared with Ca2+O-stimulation) that decayed to basal levels over time.
Fig. 3.
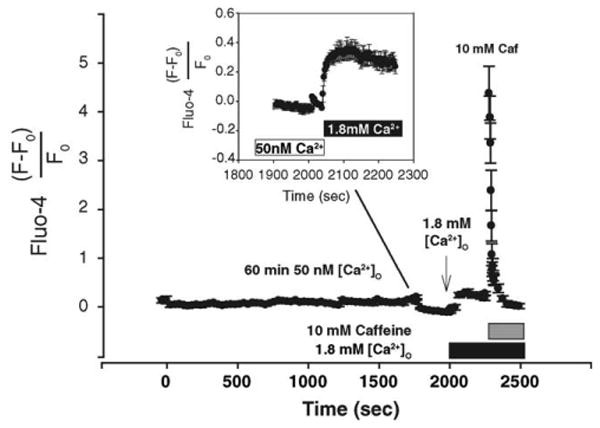
Incubation in 0 Ca2+O does not alter intracellular Ca2+. Incubation of DRG neurons in 0 Ca2+ did not decrease intracellular calcium measured by Fluo-4. DRG neurons were imaged at 30 s intervals in 50 nM free Ca2+ for 65 min followed by re-addition of 1.8 mM Ca2+O. Control neurons were incubated in 1.8 mM Ca2+O and imaged to adjust for photo-bleaching under constant Ca2+ levels. Bleaching was not adjusted for later time points, e.g. prior to re-addition of Ca2+O as the acquisition rate was changed to 3 s intervals. Fluorescence values were calculated by subtracting background and normalizing to the fluorescence (F0) immediately prior to 1.8 mM Ca2+. Re-addition of Ca2+O rapidly increased Ca2+i to a new baseline. 10 mM caffeine rapidly increased Ca2+i that decayed to baseline. Fluorescence values are the mean ± S.E.M. of 14 cells. Inset: larger time resolution demonstrates that re-addition of 1.8 mM Ca2+O rapidly increases Ca2+i. The initial decrease in fluorescence is due to photo-bleaching of the dye. Compared to the effects of 10 mM caffeine, the relative changes in Ca2+i corresponding to Ca2+O are small but not negligible.
3.4. Caffeine-sensitive stores are depleted during incubation in 0 [Ca2+]O
We suspected that microdomains of intracellular Ca2+ might be changing in response to Ca2+O manipulation, but that they were undetectable with standard confocal Ca2+ imaging. One possibility was that if Ca2+ were leaking across the membrane under low Ca2+O conditions, it might be replenishable from Ca2+ supplied from intracellular stores to maintain the [Ca2+]i at a physiologically stable level. We tested the hypothesis that Ca2+ may be released from intracellular stores in DRG neurons incubated in reduced [Ca2+]O, by measuring the release of Ca2+ from caffeine-sensitive stores using ratiometric Indo-1 confocal microscopy, in DRG neurons incubated in reduced external [Ca2+] for 5–60 min.
We found that although there were no measurable changes in resting [Ca2+]i with prolonged incubation in 0 Ca2+i for up to 60 min, the peak amplitude of the Ca2+i transient induced by 10 mM caffeine decreased the longer DRG neurons were incubated in low external Ca2+ concentrations (Fig. 4A and B). Accompanying this decline in peak Ca2+ response, the caffeine-induced Ca2+i-transient decayed more rapidly, consistent with the hypothesis that the stores were becoming depleted of Ca2+ and less able to sustain release (Fig. 4A). These two effects are consistent with a partial depletion of Ca2+i stores in reduced extracellular [Ca2+]O. Comparison of the CaMKII phosphorylation level over time in reduced Ca2+O (Fig. 2A) and the decline in the peak Ca2+i response to caffeine stimulation (Fig. 4B) showed that the decrease in Ca2+i response and CaMKII phosphorylation followed a similar time course and were well correlated (Fig. 5 and inset). The extent of store-depletion depletion may act to regulate the basal autophosphorylation state of CaMKII. We therefore tested the hypothesis that Ca2+ entry through SOC contributes to CaMKII activation.
Fig. 4.
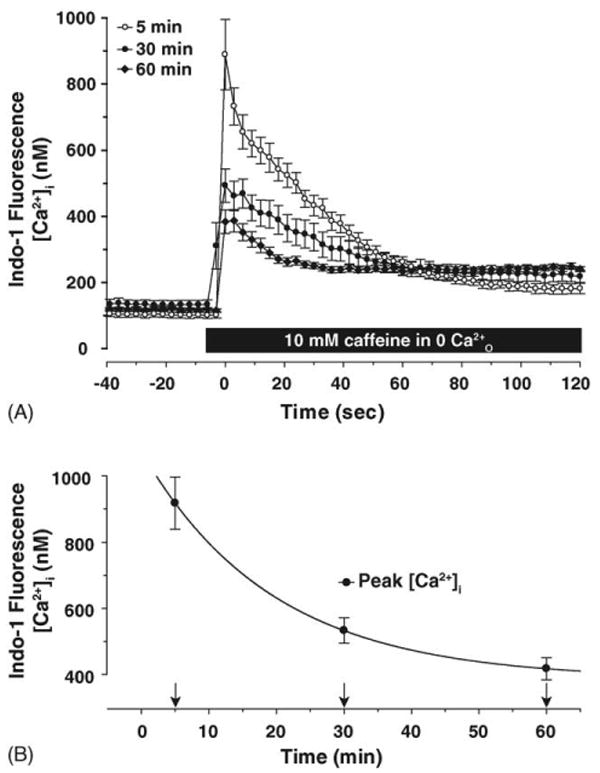
Increasing incubation time in 0 Ca2+O depletes caffeine-sensitive intracellular Ca2+ stores. (A) Brief exposure of DRG neurons to 10 mM caffeine rapidly elicits Ca2+i transients that slowly decayed over tens of seconds. Increasing the incubation time in 0 Ca2+O increases the decay rate of the caffeine-induced Ca2+i-transient. Baseline [Ca2+]i was measured until stable and then 10 mM caffeine was applied. Raw fluorescence data obtained from selected ROI was converted to [Ca2+] using the following equation: [Ca2+] = Kd × β(R − Rmin)/(Rmax − R) [52]. Data for Ca2+i transients were fit to a three-parameter exponential decay function to obtain decay rates for caffeine-induced transients; the decay rates were: 0.033 s−1 for 5 min (n = 7 cells), 0.050 s−1 for 30 min (n = 7 cells), and 0.095 s−1 for 60 min (n = 8 cells) (one-way ANOVA testing the decay rate for 5, 30, and 60 min incubation in 0 Ca2+O: F2,21 = 16.31, p < 0.001). Although incubation in 0 Ca2+ for 30 min did not significantly increase the decay rate over 5 min, by 60 min, the decay rate was significantly increased. (B) The peak Ca2+i response decreased with longer incubation in 0 Ca2+O with a decay rate of 0.058 s−1 (one-way ANOVA testing peak [Ca2+]i at 5, 30, and 60 min: F2,21 = 24.65, p < 0.001). Baseline [Ca2+]i prior to caffeine treatment was not significantly affected with prolonged incubation in 0 Ca2+O (p ≫ 0.05).
Fig. 5.
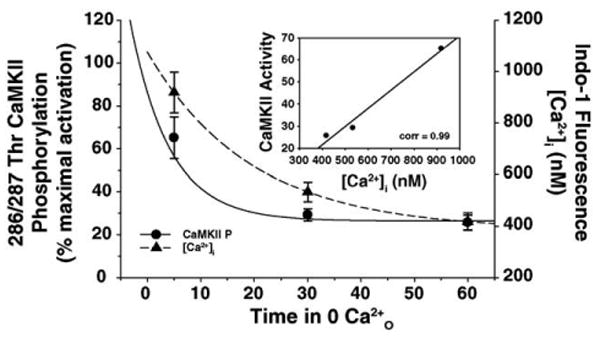
The decay in CaMKII phosphorylation correlates with the decrease in the peak Ca2+i response. Data presented in Fig. 2A and Fig. 4B were compared to demonstrate that CaMKII phosphorylation (●) decays proportionally to the decrease in the peak Ca2+i transient (▲) produced by 10 mM caffeine. Inset: scatter plot of CaMKII activity and peak Ca2+i demonstrate that both functions are well correlated (correlation value = 0.99).
3.5. Store-operated Ca2+-permeable channels contribute to CaMKII activation by Ca2+O
DRG neurons express all of the components that underlie capacitative Ca2+-entry (CCE) and Ca2+-induced Ca2+ release (CICR) commonly observed in non-excitable cells [11–14]. Both RyR- and IP3R-sensitive Ca2+ stores have been demonstrated to be functionally coupled in DRG neurons and contribute to intracellular Ca2+ signaling [15]. Additionally, DRG neurons express a full complement of non-selective cation channels that form the transient receptor potential channel family (TRP), which responds to stimuli such as cold, heat, and stretch. Depletion of intracellular stores and subsequent activation of these channels would provide a route of Ca2+ entry in resting neurons, independent of VGCC-mediated Ca2+ influx.
DRG neurons were incubated in 50 nM free Ca2+ in the presence of inhibitors of store-operated influx. After 60 min, the concentration of Ca2+O was increased to 1.8 mM. Ruthenium Red and SK&F 96365, which are blockers of non-voltage gated Ca2+-permeable channels, including store-operated channels (SOC), inhibited the activation of CaMKII significantly. Although not statistically significant, Ni2+ modestly decreased CaMKII phosphorylation by 8 ± 6% (n = 10; Fig. 6). (Note that Ni2+ occludes Ca2+ efflux from DRG neurons [12], therefore DRG neurons were first incubated in 0 Ca2+O and then switched to a medium containing 1.8 mM Ca2+O in the presence of 500 μM Ni2+.)
Fig. 6.
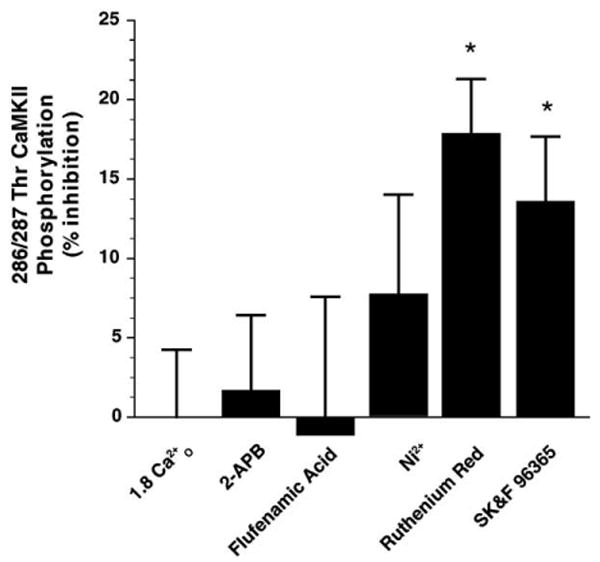
CaMKII activation by Ca2+O is sensitive to inhibitors of SOCs. DRG neurons were incubated in SOC blockers prior to stimulation by 1.8 Ca2+O. Phosphorylation of CaMKII in 0 Ca2+O was 11 ± 2% of maximal activation. Ruthenium Red (RR) at 100 μM and SK&F 96365 at 10 μM modestly inhibited the activation of CaMKII by 19 ± 4% (n = 16) and 14 ± 4%, respectively (n = 16). 2-APB (n = 12) and flufenamic acid (n = 9) at 100 μM did not block CaMKII activation. Because EGTA effectively chelates Ni2+, EGTA was excluded from the buffer. Following incubation in 0 Ca2+ for 1 h in the absence of blockers, DRG neurons were then stimulated with 1.8 mM Ca2+O in the presence of Ni2+ (500 μM). Basal CaMKII phosphorylation in this nominally Ca2+-free saline was 31 ± 3% of maximal phosphorylation by 1.8 Ca2+O (n = 6) which is higher than in experiments performed in the presence of EGTA to buffer [Ca2+]O to low nM levels. Both RR and SK&F 96365 significantly decreased CaMKII activation (one-way ANOVA testing the effect of SOC blockers on Ca2+O stimulation: F5,86 = 2.64, p = 0.029).
Other drugs that can block SOC were tested, but the results were inconclusive, possibly because of their known actions on other Ca2+-permeable channels. 2-APB blocks several SOC and IP3-mediated Ca2+ release; however, 2-APB at increasing concentrations, activates TRPV-channels [16] so the effects of this drug are more difficult to interpret. We found that 100 μM 2-APB did not block CaMKII activation (Fig. 6). Flufenamic acid, both a broad inhibitor of non-selective cation channels and several TRP-channels, failed to block CaMKII activation (Fig. 6). However, there have been reports of the related compound niflumic acid releasing Ca2+ from ryanodine-sensitive stores that might act to reverse the affects on Ca2+-influx [17]. We also tested several di- and trivalent cations; neither Gd3+ nor La3+ at 300 μM blocked activation of CaMKII. Mg2+ at 5 mM was ineffective (not shown).
We used Ca2+imaging to test the alternative hypothesis that the inhibitory effect of SK&F 96365 on CaMKII was caused by blocking Ca2+-influx that occurs following re-addition of 1.8 mM Ca2+O. DRG neurons were incubated in low Ca2+O for 65 min and then switched to saline containing 10 μM SK&F 96365. Normalized fluorescence increased following superfusion of the drug and reached a new plateau after several minutes (Fig. 7). SK&F 96365 did not block the increase in [Ca2+]i due to re-addition of Ca2+O. The decrease in Ca2+-stimulated CaMKII, specifically by RR and SK&F 96365 suggest that components of the store-operated Ca2+-influx pathway contribute to Ca2+O-mediated modulation of CaMKII.
Fig. 7.

SK&F 96365 does not affect Ca2+O-mediated influx. As in Fig. 1, incubation of DRG neurons in 50 nM free Ca2+O did not decrease [Ca2+]i. After incubating DRG neurons in 50 nM free Ca2+O for 60 min, neurons were incubated in 10 μM SK&F 96365 for 5 min prior to re-addition of 1.8 mM Ca2+O. Incubation in drug modestly increased Ca2+i above resting levels. Fluorescence values are the mean ± S.E.M. of 13 cells. Inset: larger time resolution demonstrates that SK&F 96365 increased Ca2+i. Note that the increase in Ca2+i in the presence of drug (○) was indistinguishable from control (●).
3.6. Ca2+O-dependent activation of CaMKII in resting DRG neurons does not require influx through voltage-gated Ca2+-channels
Ca2+ influx through voltage-gated Ca2+-channels (VGCC) is the primary means of Ca2+ entry in excitable cells. However, these channels may also mediate Ca2+-influx at rest, and the changes in Ca2+ in micro-domains near the membrane channels could couple to CaMKII activation and provide another mechanism for regulating CaMKII by [Ca2+]O. We further tested whether Ca2+O activated CaMKII through either direct modulation of VGCC or influx through these channels but found no evidence to support this hypothesis. DRG neurons were incubated in inhibitors of VGCC, isradipine and SR 33805. Neither compound at 10 μM had an effect on CaMKII activation (Fig. 8). We also tested a cocktail of Ca2+-channel inhibitors at concentrations known to block activation of L-, N-, P/Q-, and T-type channels [18]. In the presence of the VGCC cocktail, we observed a modest 14 ± 7% (n = 3) reduction in CaMKII activation which was not statistically significant. As a control, we tested whether reverse mode operation of the Na+/Ca2+-exchanger contributes to CaMKII activation [19,20]. However, KB-R7943 at 10 μM was inactive in modulating the CaMKII response to Ca2+O (Fig. 8). The dihydropyridine MRS 1845, which displays low μM affinity for both L-type channels and some SOC [21], did not significantly decrease Ca2+-activation of CaMKII (6 ± 8% decrease from control (n = 15)).
Fig. 8.
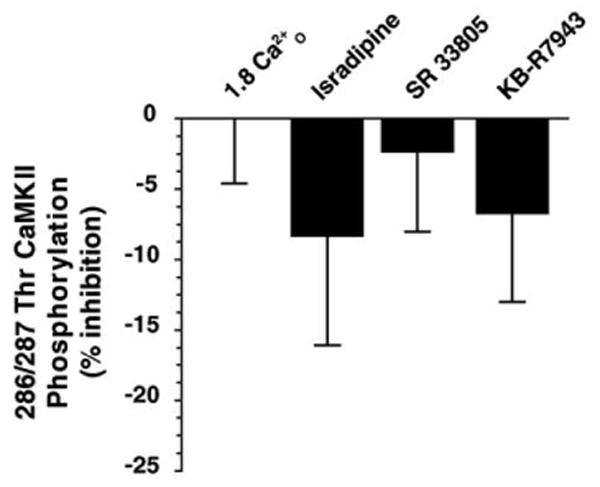
Activation of CaMKII is independent of VGCC-mediated Ca2+ influx. Pre-incubation with either the L-type channel blockers isradipine or SR 33805 (n = 6) at 10 μM failed to block CaMKII activation. Additionally, the Na+/Ca2+-exchanger inhibitor KB-R7943 at 10 μM (n = 6) also did not affect activation of CaMKII by Ca2+O-stimulation. Basal phosphorylation at 286/287Thr in 0 Ca2+ was 12 ± 1% of stimulation by 1.8 Ca2+O. Neither compound significantly affected Ca2+-stimulation (n = 6) suggesting that changes in [Ca2+]O act at a site independent of VGCC.
4. Discussion
Reduction in [Ca2+]O is a widely used experimental manipulation in studies of synaptic physiology and gap junction coupling, and the [Ca2+]O can be depleted naturally under several physiological and pathophysiological conditions. We have found that changes in [Ca2+]O act to regulate the autophosphorylation state of CaMKII through intracellular store-depletion and subsequent influx through non-voltage-gated channels. This provides a mechanism to link depletion of Ca2+O to intracellular signaling pathways to regulate cellular processes mediated by CaMKII. Activity-dependent changes in [Ca2+]O thus may contribute to Ca2+ signaling through this novel mechanism to modulate intracellular stores and continuously tune CaMKII activity based on the extracellular environment. In this context, Ca2+O can function as an activity-dependent signaling molecule.
Many laboratories working in varied systems have demonstrated that free Ca2+ in the extrasynaptic space is regulated during trains of synaptic activity [22–26]. Additionally, mathematical modeling suggests that the degree of [Ca2+]O depletion is highly frequency dependent [27]. In this context, action potential-induced fluctuations in [Ca2+]O may act as an extracellular activity-dependent signal that modulates the balance between inactive and active CaMKII. However, it is less clear how depletion of [Ca2+]O is linked to intracellular signaling. More recently, synaptic activity in addition to driving influx of Ca2+ through both VGCC and NMDA receptors also activates capacitative Ca2+-entry (CCE) invoking both inositol 1,4,5-trisphosphate receptor (IP3R)- and ryanodine receptor (RyR)-sensitive Ca2+ stores [28,29] to regulate presynaptic release properties [30–32].
Non-voltage gated Ca2+-influx pathways mediated by SOC (including TRP channels), originally identified in non-excitable cells [33] are widely expressed in neurons and contribute to signaling via detection of various extracellular stimuli [34,35]. The experimental manipulation of removing Ca2+O should be used cautiously as it impacts on CaMKII signaling in neurons.
4.1. CaMKII de-phosphorylation is regulated by intracellular store-depletion
We found by ratiometric Indo-1 Ca2+ imaging that prolonged incubation in 0 Ca2+O reduces caffeine-sensitive stores without net change in bulk cytoplasmic [Ca2+] (Fig. 4A and B). The decrease in CaMKII phosphorylation is regulated by the filled state of the intracellular stores. We also observed that re-addition of Ca2+O produced a modest increase in Ca2+i that was insensitive to SK&F 96365, a compound that partially decreases CaMKII activation by Ca2+O and occludes Ca2+i transients induced by intracellular store release. As intracellular stores are progressively depleted, a signal is generated that activates SOC and the Ca2+-release activated current (ICRAC). The pharmacology for these channels has not been clearly worked out as they comprise a large family that may form heterodimeric channels with greatly different pharmacologies and properties dependent upon the cell type [36]. They are also regulated to some extent by [Ca2+]O in the physiological range of synaptic cleft Ca2+ [33,37,38]. As observed in non-neuronal cells, re-addition of Ca2+ activates CCE through these SOC and then regulates downstream Ca2+-signaling pathways. We have found that RR and SK&F 96365, compounds that block store-mediated influx, partially inhibit the activation of CaMKII (Figs. 6 and 7).
4.2. Store-operated channels may underlie an important influx mechanism regulating CaMKII
Our observations on the role of Ca2+O in regulating Ca2+i-dependent processes strongly argue that activity-dependent alterations in Ca2+O directly regulate intracellular Ca2+-dependent processes. We have observed that removal of Ca2+O depletes ryanodine-sensitive stores (both IP3R and RyR stores are coupled in DRG neurons [15]). The store-depletion paradigm that we used in these experiments required a prolonged incubation (30–60 min) in 0 Ca2+O; however, both the decrease in peak Ca2+i and CaMKII phosphorylation occur within 10 min and plateau by 30 min (Figs. 2A, 4B and 5). The time course of CaMKII activation induced by re-addition of Ca2+O is very rapid, occurring in less than 45 s [5].
Under physiological conditions, action-potential firing alone is sufficient to partially deplete intracellular stores [39]; however, activity itself activates voltage-gated Ca2+-influx pathways. As DRG neurons are not spontaneously active, we utilized the 0 Ca2+O paradigm to reproduce store-depletion independent of voltage-gated channel activation. We also observed that depletion of caffeine-sensitive stores decreased CaMKII phosphorylation. Influx of Ca2+O may occur through a store-operated influx mechanism to act on CaMKII that is situated in subplasmalemmal domains or ER cistern that oppose the cell membrane [40,41]. As well, re-addition of Ca2+O increases Ca2+i to a much higher level directly beneath the membrane than within the cytoplasm [42]. Other experimental systems have demonstrated that intracellular stores act to regulate CaMKII activation [43] and CaMKII acts to potentiate SOC currents [37].
4.3. Additional mechanisms regulating CaMKII by Ca2+O
These studies do not rule out the possibility that additional mechanisms may contribute to the sensitivity of CaMKII to Ca2+O. In these experiments, however, blocking VGCC with either isradipine or SR 33805 did not block the increase in CaMKII phosphorylation upon restoring Ca2+O to normal. Additional routes of Ca2+ influx that may contribute to the remaining CaMKII phosphorylation may be due to other Ca2+-permeable channels that are insensitive to RR and SK&F 96365. Smith et al. [26] recently reported an extracellular Ca2+ sensor that functions to regulate the excitability of presynaptic terminals via regulation of a non-selective cation channel. Although DRG neurons express a CaSR ([44] and unpublished observations), we did not observe activation of CaMKII with increasing [Gd3+], which activates the CaSR, arguing against its involvement in CaMKII regulation.
4.4. Implications for Ca2+O-regulated CaMKII phosphorylation in synaptic plasticity
We found that activation of CaMKII was saturable with increasing [Ca2+]O (Fig. 2B) and the EC50 for stimulation was within the range of Ca2+ that is observed within the synaptic cleft, making it highly sensitive to respond to changes in Ca2+O that is modulated by synaptic activity [22,25,27]. Synaptic activity regulates intracellular Ca2+ stores through both IP3- and RyR-sensitive stores that exert effects on both short- and long-term plasticity [28,30,32,45–48] (see Berridge [49] for review). However, what is not clear is the degree to which Ca2+O-modulation of intracellular signaling, e.g. CaMKII, contributes to the overall processes during periods of synaptic activity. However, as we and others have observed, a large amount of this basal autonomous activity is present in resting DRG neurons and highly dependent on Ca2+O. Manipulations that deplete intracellular stores act to decrease CaMKII activity. We have also observed that compounds that interfere with these intracellular stores, decrease CaMKII activation. Our observations on the role of Ca2+O in regulating Ca2+i-dependent processes strongly argue that activity-dependent alterations in Ca2+O directly regulate intracellular Ca2+-dependent processes that would have lasting effects on neuronal signaling.
Acknowledgments
This work was supported by the National Institute of Child Health and Human Development. We thank Dr. Yoko Yamagata for the phosphosite-specific antibodies used in the study.
References
- 1.Fink CC, Meyer T. Molecular mechanisms of CaMKII activation in neuronal plasticity. Curr Opin Neurobiol. 2002;12:293–299. doi: 10.1016/s0959-4388(02)00327-6. [DOI] [PubMed] [Google Scholar]
- 2.Molloy SS, Kennedy MB. Autophosphorylation of type II Ca2+/calmodulin-dependent protein kinase in cultures of postnatal rat hippocampal slices. Proc Natl Acad Sci U S A. 1991;88:4756–4760. doi: 10.1073/pnas.88.11.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukunaga K, Rich DP, Soderling TR. Generation of the Ca2(+)-independent form of Ca2+/calmodulin-dependent protein kinase II in cerebellar granule cells. J Biol Chem. 1989;264:21830–21836. [PubMed] [Google Scholar]
- 4.Scholz WK, Palfrey HC. Activation of Ca2+/calmodulin-dependent protein kinase II by extracellular calcium in cultured hippocampal pyramidal neurons. J Neurochem. 1998;71:580–591. doi: 10.1046/j.1471-4159.1998.71020580.x. [DOI] [PubMed] [Google Scholar]
- 5.Eshete F, Fields RD. Spike frequency decoding and autonomous activation of Ca2+-calmodulin-dependent protein kinase II in dorsal root ganglion neurons. J Neurosci. 2001;21:6694–6705. doi: 10.1523/JNEUROSCI.21-17-06694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ocorr KA, Schulman H. Activation of multifunctional Ca2+/calmodulin-dependent kinase in intact hippocampal slices. Neuron. 1991;6:907–914. doi: 10.1016/0896-6273(91)90231-n. [DOI] [PubMed] [Google Scholar]
- 7.Fields RD, Yu C, Neale EA, Nelson PG. Recording chambers in cell culture. In: Kettenmann H, Grantyn R, editors. Electrophysiological Methods for In Vitro Studies in Vertebrate Neurobiology. Liss; New York: 1992. pp. 67–76. [Google Scholar]
- 8.Sheng HZ, Fields RD, Nelson PG. Specific regulation of immediate early genes by patterned neuronal activity. J Neurosci Res. 1993;35:459–467. doi: 10.1002/jnr.490350502. [DOI] [PubMed] [Google Scholar]
- 9.Yamagata Y, Obata K. Dynamic regulation of the activated, autophosphorylated state of Ca2+/calmodulin-dependent protein kinase II by acute neuronal excitation in vivo. J Neurochem. 1998;71:427–439. doi: 10.1046/j.1471-4159.1998.71010427.x. [DOI] [PubMed] [Google Scholar]
- 10.Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Ayar A, Scott RH. The actions of ryanodine on Ca2+-activated conductances in rat cultured DRG neurones; evidence for Ca2+-induced Ca2+ release. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:81–91. doi: 10.1007/pl00005335. [DOI] [PubMed] [Google Scholar]
- 12.Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;519(Pt 1):115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razani-Boroujerdi S, Partridge LD, Sopori ML. Intracellular calcium signaling induced by thapsigargin in excitable and inexcitable cells. Cell Calcium. 1994;16:467–474. doi: 10.1016/0143-4160(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 14.Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. J Physiol. 1995;489(Pt 3):627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solovyova N, Verkhratsky A. Neuronal endoplasmic reticulum acts as a single functional Ca2+ store shared by ryanodine and inositol-1,4,5-trisphosphate receptors as revealed by intra-ER [Ca2+] recordings in single rat sensory neurones. Pflugers Arch. 2003;446:447–454. doi: 10.1007/s00424-003-1094-z. [DOI] [PubMed] [Google Scholar]
- 16.Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 17.Cruickshank SF, Baxter LM, Drummond RM. The Cl(−) channel blocker niflumic acid releases Ca(2+) from an intracellular store in rat pulmonary artery smooth muscle cells. Br J Pharmacol. 2003;140:1442–1450. doi: 10.1038/sj.bjp.0705571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilaire C, Diochot S, Desmadryl G, Richard S, Valmier J. Toxin-resistant calcium currents in embryonic mouse sensory neurons. Neuroscience. 1997;80:267–276. doi: 10.1016/s0306-4522(97)00101-2. [DOI] [PubMed] [Google Scholar]
- 19.Brown AM, Wender R, Ransom BR. Ionic mechanisms of aglycemic axon injury in mammalian central white matter. J Cereb Blood Flow Metab. 2001;21:385–395. doi: 10.1097/00004647-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Lehning EJ, Doshi R, Isaksson N, Stys PK, LoPachin RM., Jr Mechanisms of injury-induced calcium entry into peripheral nerve myelinated axons: role of reverse sodium–calcium exchange. J Neurochem. 1996;66:493–500. doi: 10.1046/j.1471-4159.1996.66020493.x. [DOI] [PubMed] [Google Scholar]
- 21.Harper JL, Camerini-Otero CS, Li AH, Kim SA, Jacobson KA, Daly JW. Dihydropyridines as inhibitors of capacitative calcium entry in leukemic HL-60 cells. Biochem Pharmacol. 2003;65:329–338. doi: 10.1016/s0006-2952(02)01488-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicholson C, Bruggencate GT, Steinberg R, Stockle H. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci U S A. 1977;74:1287–1290. doi: 10.1073/pnas.74.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinemann U, Stabel J, Rausche G. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res. 1990;83:197–214. doi: 10.1016/s0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- 25.Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol. 1999;521(Pt 1):123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron. 2004;41:243–256. doi: 10.1016/s0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- 27.Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J. 1997;72:2103–2116. doi: 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose CR, Konnerth A. Stores not just for storage. Intracellular calcium release and synaptic plasticity. Neuron. 2001;31:519–522. doi: 10.1016/s0896-6273(01)00402-0. [DOI] [PubMed] [Google Scholar]
- 30.Unni VK, Zakharenko SS, Zablow L, DeCostanzo AJ, Siegelbaum SA. Calcium release from presynaptic ryanodine-sensitive stores is required for long-term depression at hippocampal CA3–CA3 pyramidal neuron synapses. J Neurosci. 2004;24:9612–9622. doi: 10.1523/JNEUROSCI.5583-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 32.Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22:21–28. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 35.Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–344. doi: 10.1016/s0143-4160(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 36.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium. 2003;33:311–321. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 37.Machaca K. Ca2+-calmodulin-dependent protein kinase II potentiates store-operated Ca2+ current. J Biol Chem. 2003;278:33730–33737. doi: 10.1074/jbc.M305023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bird GJ, Oliver KG, Horstman DA, Obie J, Putney JW., Jr Relationship between the calcium-mobilizing action of inositol 1,4,5-trisphosphate in permeable AR4-2J cells and the estimated levels of inositol 1,4,5-trisphosphate in intact AR4-2J cells. Biochem J. 1991;273(Pt 3):541–546. doi: 10.1042/bj2730541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Usachev YM, Thayer SA. All-or-none Ca2+ release from intracellular stores triggered by Ca2+ influx through voltage-gated Ca2+ channels in rat sensory neurons. J Neurosci. 1997;17:7404–7414. doi: 10.1523/JNEUROSCI.17-19-07404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle Sarcoplasmic reticulum. Trends Pharmacol Sci. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- 41.Blaustein MP, Golovina VA. Structural complexity and functional diversity of endoplasmic reticulum Ca(2+) stores. Trends Neurosci. 2001;24:602–608. doi: 10.1016/s0166-2236(00)01891-9. [DOI] [PubMed] [Google Scholar]
- 42.Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997;16:1575–1581. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matifat F, Fournier F, Lorca T, Capony JP, Brule G, Collin T. Involvement of the Ca2+/calmodulin-dependent protein kinase II pathway in the Ca2+-mediated regulation of the capacitative Ca2+ entry in Xenopus oocytes. Biochem J. 1997;322(Pt 1):267–272. doi: 10.1042/bj3220267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Awumey EK, Chatterjee PK, Somasundaram C, Bian K, Rogers KV, Dunn C, Bukoski RD. Molecular cloning and characterization of a rat sensory nerve Ca2+-sensing receptor. Am J Physiol Cell Physiol. 2003;285:C64–C75. doi: 10.1152/ajpcell.00543.2002. [DOI] [PubMed] [Google Scholar]
- 45.Cong YL, Takeuchi S, Tokuno H, Kuba K. Long-term potentiation of transmitter exocytosis expressed by Ca2+-induced Ca2+ release from thapsigargin-sensitive Ca2+ stores in preganglionic nerve terminals. Eur J Neurosci. 2004;20:419–426. doi: 10.1111/j.1460-9568.2004.03492.x. [DOI] [PubMed] [Google Scholar]
- 46.Nakano M, Yamada S, Udagawa R, Kato N. Frequency-dependent requirement for calcium store-operated mechanisms in induction of homosynaptic long-term depression at hippocampus CA1 synapses. Eur J Neurosci. 2004;19:2881–2887. doi: 10.1111/j.0953-816X.2004.03390.x. [DOI] [PubMed] [Google Scholar]
- 47.Bengtson CP, Tozzi A, Bernardi G, Mercuri NB. Transient receptor potential-like channels mediate metabotropic glutamate receptor EPSCs in rat dopamine neurones. J Physiol. 2004;555:323–330. doi: 10.1113/jphysiol.2003.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S, Mikoshiba K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron. 1999;24:701–713. doi: 10.1016/s0896-6273(00)81123-x. [DOI] [PubMed] [Google Scholar]
- 49.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 50.Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW. Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci U S A. 2001;98:15342–15347. doi: 10.1073/pnas.211563998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim KM, Herrera GA, Battarbee HD. Role of glutaraldehyde in calcification of porcine aortic valve fibroblasts. Am J Pathol. 1999;154:843–852. doi: 10.1016/S0002-9440(10)65331-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 53.Fields RD, O'Donovan MJ. Current Protocols Neuroscience. John Wiley; New York: 1997. Imaging activity of the nervous system; pp. 2.3.1–2.3.12. [Google Scholar]
- 54.Fields RD, Guthrie PG, Russell JT, Kater SB, Malhotra BS, Nelson PG. Accommodation of mouse DRG growth cones to electrically induced collapse: Kinetic analysis of calcium transients and set-point theory. J Neurobiol. 1993;24:1080–1098. doi: 10.1002/neu.480240807. [DOI] [PubMed] [Google Scholar]