

Abstract
Vibrio cholerae is a Gram-negative bacterial pathogen that exports enterotoxins which alter host cells through a number of mechanisms resulting in diarrheal disease. Among the secreted toxins is the multifunctional, autoprocessing RTX toxin (MARTXVc), which disrupts actin cytoskeleton by covalently cross-linking actin monomers into oligomers. The region of the toxin responsible for cross-linking activity is the actin cross-linking domain (ACD). In this study, we demonstrate unambiguously that ACD utilizes G- and not F-actin as a substrate for the cross-linking reaction and hydrolyzes one molecule of ATP per cross-linking event. Furthermore, major actin binding proteins that regulate actin cytoskeleton in vivo do not block the cross-linking reaction in vitro. Cofilin inhibits the cross-linking of G- and F-actin at high mole ratio to actin, but accelerates F-actin cross-linking at low mole ratios. DNase I blocks completely the cross-linking of actin, likely due to steric hindrance with one of the cross-linking sites on actin. In the context of the holotoxin, the inhibition of Rho by the Rho-inactivating domain of MARTXVc (Sheahan, K.L., Satchell, K.J.F. 2007 Cellular Microbiology 9:1324-1335) would accelerate F-actin depolymerization and provide G-actin, alone or in complex with actin binding proteins, for cross-linking by ACD, ultimately leading to the observed rapid cell rounding.
The Gram-negative bacterial pathogen Vibrio cholerae is the causative agent of the diarrheal disease cholera. Following ingestion of V. cholerae from contaminated food or water, the bacterium colonizes the host intestine and secretes enterotoxins (1). In addition to Cholera Toxin, an ADP-ribosylating toxin that stimulates the adenylate cyclase complex, V. cholerae secretes a number of accessory toxins that contribute to pathogenesis, including the multifunctional, autoprocessing RTX toxin of V. cholerae (MARTXVc) (2)
Active MARTXVc is a potent toxin produced by the pathogenic O1 El Tor and O139 strains responsible for the current cholera pandemic and a broad range of non-O1, non-O139 clinical and environmental isolates (3). The full-length toxin is >450,000 Da and is comprised of a series of glycine-rich repeat regions at the N- and C-termini and activity domains located within the central portion of the toxin (4) These activity domains carry two distinct cell rounding activities, one of which leads to cell rounding through inactivation of Rho GTPases (5) and the other by the novel mechanism of covalent actin cross-linking (6).
The actin cross-linking domain (ACD) located between a.a. residues 1963-2375 has been identified as the region of MARTXVc responsible for actin cross-linking activity (7). Recently, a fusion protein of the ACD with the N-terminal portion of Bacillus anthracis lethal factor (LFN) was tested for actin cross-linking activity both in vivo and in vitro. Purified LFNACD was translocated to the host cell cytoplasm via the entry mechanism for anthrax toxin, and cross-linked actin proteins were detected in cell lysates. An in vitro actin cross-linking assay was developed to further investigate the role of the ACD in the cross-linking reaction, and it has been demonstrated that LFNACD directly catalyzes the covalent cross-linking of purified actin in the absence of host protein intermediates. The cross-linking reaction requires both Mg2+ and ATP as cofactors, but it remains unclear whether these cofactors are essential for the enzymatic function of the ACD or for the initiation of actin polymerization (8).
Actin exists in dynamic equilibrium between globular monomers (G-actin) and filamentous polymers (F-actin) and this equilibrium is regulated by multiple actin binding proteins (ABPs) (9). Actin has a high affinity binding site for a divalent cation, specifically Mg2+ or Ca2+, in complex with a nucleotide, ATP or ADP (10;11). As the salt concentration increases to physiological levels, G-actin spontaneously polymerizes into F-actin, and the ATP molecule bound to G-actin is hydrolyzed to ADP and Pi. The latter dissociates from F-actin with a delay time of few minutes (12-14). The nucleation of polymerization in the absence of specific nucleators (gelsolin, formins, ARP2/3 complex, spire) is a thermodynamically unfavorable, rate limiting process of self-association of actin monomers into a stable trimer complex, to which further monomers are added during filament elongation (15).
Actin is a dynamic protein involved in various cellular processes, and the versatility of actin makes it an attractive and vulnerable target for bacterial toxins (16). For example, Clostridium botulinum C2 toxin and the C. perfringens iota toxin modify G-actin by adding ADP-ribose moiety to the Arg-177 residue. This modification disrupts the ability of monomers to assemble into filaments. As a result, the dynamic equilibrium shifts toward G-actin, ultimately causing actin depolymerization and host cell rounding (17). The distinct correlation between MARTXVc-mediated actin cross-linking into oligomers and host cells rounding suggests that MARTXVc may manifest its action through a similar mechanism of shifting the equilibrium between soluble and polymeric actin.
In this report, we further characterize the actin cross-linking reaction catalyzed by the ACD of MARTXVc. We find that G-actin is the substrate for ACD and that F-actin is not cross-linked in vitro. Moreover, we demonstrate that major ABPs, which interact with G-actin under in vivo conditions, allow for actin cross-linking by ACD in vitro. In addition, we show that ATP is hydrolyzed directly by ACD. These results contribute to our overall understanding of the actin cross-linking activity of MARTXVc, and suggest a mechanism by which the covalent cross-linking of actin results in host cell rounding through the disruption of actin polymerization.
Experimental Procedures
Chemical reagents
Analytical grade chemicals were purchased from Sigma Chemical (St. Louis, MO), unless specified differently. ATP and HEPES were from Merck (Darmstadt, Germany). Tetramethylrhodamine-5-maleimide (TMR) was purchased from Invitrogen (Carlsbad, CA). Kabiramide C (KabC) was a generous gift from Dr. Gerard Marriott (University of Wisconsin).
Proteins
Rabbit skeletal actin was purified from rabbit back muscle as described in Spudich and Watt (18). G-actin was maintained in 5.0 mM TRIS (pH8.0), 0.2 mM ATP, 0.2 mM CaCl2 buffer with 5.0 mM β-mercaptoethanol, and was used within two weeks of purification. β-Thymosin was kindly donated by Dr. Safer, University of Pennsylvania. Wild-type human profilin-1 cDNA in an expression vector pMW172 was provided by Dr. Henry N. Higgs of Dartmouth Medical School. The pET28(+) plasmids encoding C-terminally truncated mouse twinfilin (a.a. residues 1-328) and full length twinfilin, both with the N-terminal His6-tag, were donated by Dr. Roberto Dominguez (University of Pennsylvania). Recombinant twinfilins were expressed in E.coli BL21(DE3) cells and were purified on His-bind affinity column (Novagen, Madison, WI) according to the manufacturer's protocol. Profilin, gelsolin segment 1, and yeast and human cofilins were expressed in E.coli and purified as described earlier (19-21). Full length gelsolin was purified from bovine blood plasma as described before (22). Recombinant fusion protein of ACD with the Bacillus anthracis lethal factor (LFNACD) was purified as previously described by Cordero et al. (8). LFN does not interfere with actin-cross-linking activity of ACD (8). Ultra pure DNaseI was purchased from Bio-World (Dublin, OH).
Actin cross-linking assay
All reactions, unless specified differently, were performed at 22°C in G-actin buffer (5 mM HEPES, pH7.5, 0.2 mM ATP, 0.2 mM CaCl2, 5mM β-mercaptoethanol) supplemented with 0.1 mM MgCl2 and 0.4 mM EGTA. 10 minutes before the initiation of cross-linking, Ca-G-actin was converted to Mg-G-actin by adding 0.1 mM MgCl2 and 0.4 mM EGTA. Latrunculin A (LatA) and kabiramide C (KabC) were added to Mg-G-actin at a molar ratio of 1.5:1, 20 minutes prior to the addition of LFNACD. ABPs were also added to Mg-G-actin 20 minutes before ACD. The mole ratios of ABPs to actin were as following (unless specified otherwise): profilin – 1.5:1; cofilins – 1.5:1; thymosin-β4 – 2:1; DNaseI – 1.2:1; gelsolin segment 1 (GS1): 1.2:1; full gelsolin 1.2:1; twinfilin – 1.5:1. Actin was modified at Cys-374 with TMR as described by Otterbein et al. (23), with minor modifications (24). Pre-polymerized F-actin was prepared by adding 1.0 mM MgCl2 and 50 mM KCl to Mg-G-actin, 2 to 3 hours prior to actin cross-linking by ACD. LFNACD was added to non-polymerizable and fully polymerized actin substrates at mole ratios specified in the text and in the figure legends.
To replace ATP in the nucleotide pocket with AMP-PNP, ATP-actin was passed through PD-10 column equilibrated with nucleotide-free buffer and supplemented with a 25 fold molar excess of AMP-PNP. After 45 minutes incubation on ice, GS1 was added to actin at 1.25:1 mole ratio to lock AMP-PNP in the nucleotide pocket. Nucleotide excess was then removed on a PD-10 column and the desired nucleotide (ATP or AMP-PNP) was added to the reaction mixture. All cross-linking reactions were conducted at 22°C. Reactions were stopped at the indicated time points with the SDS-PAGE sample buffer. Samples were boiled for 3 minutes, analyzed by SDS-PAGE, and the extent of actin cross-linking was measured by gel densitometry of actin bands stained with Coomassie Brilliant Blue R-250.
Measurements of inorganic phosphate (Pi) release
The release of inorganic phosphate was monitored via color reaction with the EnzChek Phosphate assay kit (Invitrogen, Carlsbad, CA). To initiate the cross-linking, KabC-actin complex was added to the EnzChek reaction mixture containing 1.0 mM MgCl2, 0.2 mM ATP, and 0.01 μM LFNACD, so that the final concentration of actin was 10 μM. The accumulation of the chromophoric reporter of Pi was monitored at 360 nm on a Hewlett Packard 8453 spectrophotometer. 20 μl time point aliquots were taken for SDS-gel analysis directly from the experimental mixture during the course of Pi measurements. Densitometry analysis of gels was done using the Scion image software (Scion corporation, Frederick, MD). It was assumed that n-1 covalent cross-link bonds were formed per each n-mer of cross-linked actin.
Purification of the cross-linked actin dimer
For the preparation of purified cross-linked actin dimer, the extent of cross-linking reaction was limited by adding 10 mM ethylenediamide to the mixture of actin and LFNACD (1000 to 1 mole ratio). The reaction was initiated by the addition of 1.0 mM MgCl2 and stopped after 1.0 hour incubation at 22°C by removing Mg2+ on a PD10 column (Amersham Biosciences, Uppsala, Sweden) equilibrated with Ca-G-buffer. This preparation procedure had no effect on the polymerization properties of monomeric actin, as tested in a parallel experiment without LFNACD. The cross-linked actin dimer was separated from monomers and a small population of higher oligomers by gel filtration chromatography on a High Load 16/60 Superdex 200 column (Amersham biosciences, Uppsala, Sweden).
Results
G-actin is the substrate for the ACD-catalyzed actin cross-linking reaction
We have shown previously that Mg2+ is an essential cofactor for cross-linking of actin by the ACD of MARTXVc (8). As Mg2+ is also the key factor modulating conformational transitions on G-actin to a “pre-polymerization” state (25;26), it has been difficult to assess whether it is required to support the enzymatic function of ACD itself, or to induce actin nucleation and polymerization. Drugs stabilizing actin monomers and filaments have been used in vivo to separate actin polymerization from cross-linking and the results suggested that G-actin was the substrate of the cross-linking reaction (6;8). However, the possibility that actin oligomers, or even short actin filaments, may serve as ACD substrates could not be excluded.
To determine unambiguously the substrate for actin cross-linking with ACD, individual contributions of G- and F-actin to this reaction were characterized under in vitro conditions. Non-polymerizable G-actin substrates were obtained by binding either LatA or KabC to actin, or by covalently modifying it with TMR at Cys374. LatA binds to the nucleotide cleft on actin between subdomains (SDs) 2 and 4, while KabC binds to the cleft between SDs 1 and 3 (27;28); both drugs, as well as TMR-labeling, efficiently prevented actin polymerization under our experimental conditions (data not shown). F-actin was prepared by adding 1.0 mM MgCl2 and 50 mM KCl to Mg-ATP G-actin 2 hours prior to the cross-linking reaction.
To initiate the cross-linking, LFNACD was added either alone (to F-actin) or together with 1.0 mM MgCl2 and 50 mM KCl (to G-actin). As shown in Figure 1A, the formation of covalently cross-linked actin oligomers was detected for all forms of G-, but not for F-actin – even after two-fold longer incubation time. The minimal cross-linking observed in F-actin samples can be explained by the presence of a small fraction of G-actin during treadmilling – the process of preferential dissociation of actin monomers from the pointed end of the filament and their re-assembly at the filament's barbed end. Indeed, further inhibition of the cross-linking of F-actin was noted in the presence of phalloidin, consistent with a role of this drug as an inhibitor of actin treadmilling. LatA and KabC had no effect on the rates of G-actin cross-linking even at shorter incubation times, during initial phase of the cross-linking reaction (data not shown).
Figure 1. G- but not F-actin is a substrate for the ACD-induced cross-linking.
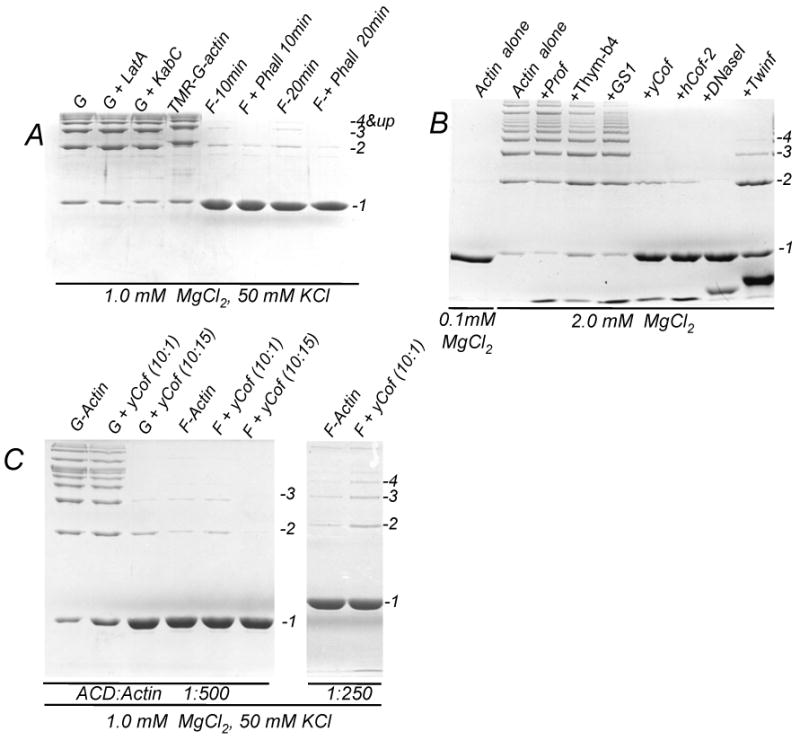
(A) Cross-linking of 10 μM actin in G- (G) and F-(F) states was initiated by adding LFNACD (1 to 500 mole ratio of ACD to actin) in the presence of 1.0 mM MgCl2 and 50 mM KCl. TMR-labeled actin (TMR-G-actin), as well as actin in the presence of Lat A (G+LatA) and KabC (G+KabC) were used to block the polymerization of Mg-G-actin into filaments. Phalloidin (15 μM) was added to F-actin (F+Phall) to inhibit filaments treadmilling. The extent of the cross-linking was assessed after 10 min incubation with LFNACD. For F-actin, even after 20 min incubation only a small amount of actin was cross-linked (F-20min and F+Phall 20 min), likely as a result of treadmilling. The cross-linking reaction was stopped with a sample buffer and analyzed by 10% SDS-PAGE.
(B) Actin alone and in the presence of ABPs was cross-linked for 15 minutes in the presence 2.0 mM MgCl2 at 1 to 500 mole ratio of LFNACD to actin. No cross-linking was observed in the presence of 0.1 mM MgCl2 (left lane). The ABPs are annotated as follows: profilin (+Prof), thymosin-β4 (+Thym-β4), gelsolin segment 1 (+GS1), yeast cofilin (+YCof), human cofilin-2 (HCof-2), DNaseI (+DNaseI), twinfilin with truncated C-terminus (+Twinf). Full length twinfilin showed the same inhibitory effect as its shortened counterpart (data not shown).
(C) 10 μM G-actin and 10 μM F-actin, polymerized for 2 hours before the cross-linking, was pre-incubated for 20 minutes with 1.0 μM or 15 μM yeast cofilin. The cross-linking reaction was initiated by adding LFNACD at 1 to 500 (left panel) or 1 to 250 (right panel) mole ratio to actin and stopped after 30 minutes (left panel) or 60 minutes (right panel) after the initiation.
Therefore, we exclude F-actin as potential substrate for the in vitro cross-linking by ACD, as the reaction proceeded only in the absence of polymerized actin. These data agree with the identification of G-actin as the substrate of the actin cross-linking reaction in vivo (6;8) and clearly indicate that F-actin does not participate in the cross-linking by MARTXVc.
Cross-linking of actin bound to ABPs
It is well recognized that in the living cell G-actin does not exist in a free form and is typically bound to one of the several ABPs (9). Therefore, we tested how the most abundant ABPs (profilin, thymosin-β4, cofilin, twinfilin, gelsolin, and DNase I) affect the cross-linking of G-actin by LFNACD. Remarkably, profilin, thymosin-β4, the G-actin binding segment of gelsolin (GS1) (Fig. 1B), as well as intact gelsolin (data not shown), did not have any noticeable effect on the cross-linking reaction. Among the proteins tested, only DNase I blocked completely the formation of actin oligomers (Fig. 1B). Both S. cerevisiae cofilin and human cofilin-2 (at 1.5 to 1 mole ratio to actin) strongly inhibited the cross-linking, but did not block it completely (Fig. 1B). To explore whether this inhibition is due to acceleration of bulk actin polymerization by cofilins (29-32), or to their effect on G-actin, we employed a modified form of twinfilin, an ABP homologous to cofilin that has two tandem ADF-domains (33). It has been shown recently that a truncated twinfilin, with the C-terminus residues 317-353 deleted, retains the capacity of the full molecule to bind and sequester G-actin, but its ability to interact with barbed ends of F-actin is strongly reduced (34). Therefore, we tested the C-terminally truncated form of twinfilin (1-328 a.a.) - as well as full length twinfilin - and found that both proteins inhibit strongly the cross-linking of actin with LFNACD. This suggests that cofilin effects are also caused, at least partially, through its binding to G-actin.
Because cofilin is a key regulator of the equilibrium between G- and F-actin in living cells, we compared its effects on the cross-linking of monomeric and filamentous actin. At 1.5 to 1 mole ratio of cofilin to actin, the cross-linking of both G- and F-actin was inhibited strongly (Fig. 1C, left panel). At a low mole ratio (1 to 10), the effect of cofilin depends on the polymerization state of actin: cofilin inhibits slightly the cross-linking of G-actin but accelerates slightly, but reproducibly the cross-linking of F-actin (Fig. 1C). This slight acceleration of F-actin cross-linking was more obvious when the experiment was repeated at a higher mole ratio of the LFNACD to F-actin (1 to 250) (Fig.1C, right panel). The increased F-actin cross-linking at low mole ratios of cofilin to actin is most likely due to the higher critical concentration for actin polymerization and the equilibrium shift towards G-actin, which is the substrate for cross-linking.
Therefore, our data indicate that the most abundant G-actin binding proteins - thymosin-β4, profilin, cofilin, and gelsolin - do not block the ACD-induced cross-linking. In fact, most of these proteins do not even inhibit the cross-linking reaction. This further confirms the conclusion that monomeric actin is the substrate for the MARTXVc -induced cross-linking of actin under in vitro and, most likely, in vivo conditions.
ACD hydrolyzes ATP during the cross-linking reaction
Recently, we have shown that ATP and Mg2+ are required for actin cross-linking with LFNACD. However, it remained uncertain whether ATP is required for the catalytic activity of LFNACD or is needed to support the specific, cross-linking competent, Mg-ATP conformation of G-actin. To clarify whether ACD hydrolyzes ATP during the cross-linking reaction, we monitored the production of inorganic phosphate (Pi) under in vitro conditions. In these experiments, we selected to use the complex of G-actin with KabC as a substrate for LFNACD for two reasons: i), to inhibit nucleotide release from G-actin by a factor of ∼5 ((35); DK unpublished results); and ii), to block actin polymerization and the concomitant Pi release (Fig. 2; red trace), thereby ensuring that the ATP hydrolysis was due solely to the ACD-catalyzed reaction. As shown in Fig. 2, the extent and rate of Pi accumulation increased with the amount of LFNACD added to the actin samples.
Figure 2. Actin cross-linking by ACD is accompanied by the release of inorganic phosphate.

Inorganic phosphate (Pi) release during the course of cross-linking of 10 μM actin was measured with the EnzChek Phosphate kit (Invitrogen). When added to 10 μM Mg-G-actin, KabC (closed black circles) blocks actin polymerization as well as the subsequent ATP hydrolysis and Pi release - which normally accompany actin polymerization (open black circles). Addition of LFNACD to non-polymerizable actin-KabC complex at 1 to 200 (cyan), 1 to 500 (magenta), and 1 to 1000 (green) mole ratios causes dose- and time-dependent release of Pi. LFNACD alone does not produce any noticeable amount of Pi under conditions used here (blue).
The intrinsic ATPase activity of LFNACD was below the detection limit under these conditions (Fig. 2). However, at higher concentration of LFNACD (1.0 μM) and at a higher temperature (30°C) we measured ∼ 0.2 μmole of Pi produced per min per each μmole of LFNACD (data not shown). This is ∼ 570 times slower than actin-activated reaction (∼115-135 μmole of Pi/min/μmole) estimated from the initial, linear stage of cross-linking at 1 to 500 and 1 to 1000 mole ratio of LFNACD to actin.
Next, we compared the rates of Pi release and the cross-linking reaction by examining on SDS-gels aliquots of the reaction mixture that was monitored for Pi release (Fig. 3). We estimated the extent of cross-linking from SDS-gels (Fig. 3A), assuming that n-1 covalent bonds were formed per each oligomer consisting of n actin protomers. We found good correlation between rates of ATP hydrolysis and the cross-linking, with 1 mole of covalent bonds produced per mole of Pi released (Fig. 3A, B).
Figure 3. Pi release correlates with the level of actin cross-linking.
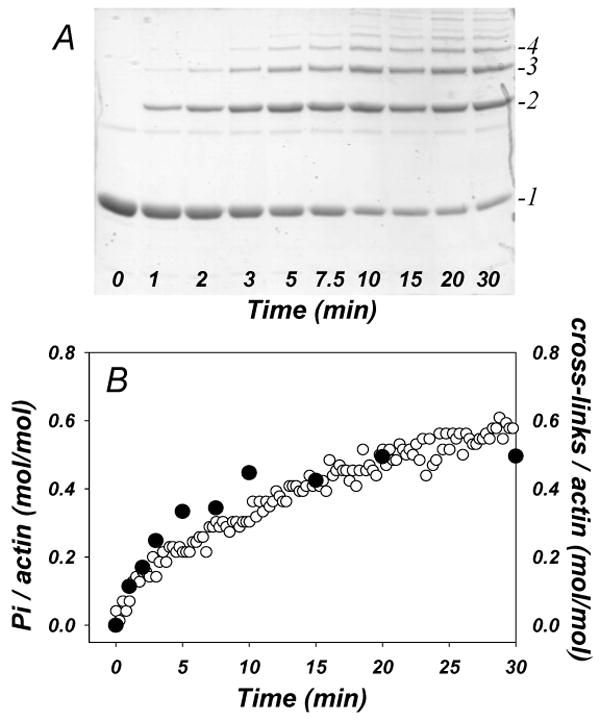
The LFNACD catalyzed cross-linking of 10 μM actin (at 1 to 1000 mole ratio of enzyme to actin) and the Pi release during this reaction were measured simultaneously by SDS-PAGE (A) and color reaction at 560 nm (B – open circles), respectively. The number of the cross-linking events was determined by gel densitometry assuming that oligomers of n actin subunits contain n-1 covalent cross-links. The ratio of cross-linking events to total actin (closed circles on B) shows good correlation with the fraction of Pi release during the cross-linking reaction.
To confirm that during cross-linking ATP is hydrolyzed by LFNACD and not by actin, we compared actin cross-linking under conditions of ATP excess and in the absence of free nucleotide i.e., when all ATP present is bound to the nucleotide cleft on actin. If ATP were hydrolyzed by actin and not LFNACD during the cross-linking, we would expect to see little difference between these two reaction conditions. In preliminary experiments we found that the cross-linking was strongly inhibited in the absence of free ATP, although not abolished completely (data not shown), apparently because of ATP leaking from the nucleotide binding site of actin. It has been shown previously (24;36), and we confirmed it again for the conditions of these experiments, that GS1 blocks efficiently the nucleotide release from actin. Therefore, we supplemented ATP-actin with a 1.25 fold molar excess of GS1, passed the actin-GS1 complex through a PD10 column, and then incubated it with Dowex 1X8-100 ion exchange resin for 20 minutes to ensure complete removal of any free ATP from the solution (37). We observed almost no cross-linking and no Pi release in the sample without free ATP, while efficient cross-linking - accompanied by ATP hydrolysis and Pi release - occurred in the sample supplemented with 0.25 mM free ATP (Fig 4 A, B). This suggests that solution ATP is a critical component of the cross-linking reaction.
Figure 4. Solution ATP, and not ATP in the nucleotide cleft of actin, is required for actin cross-linking with LFNACD.
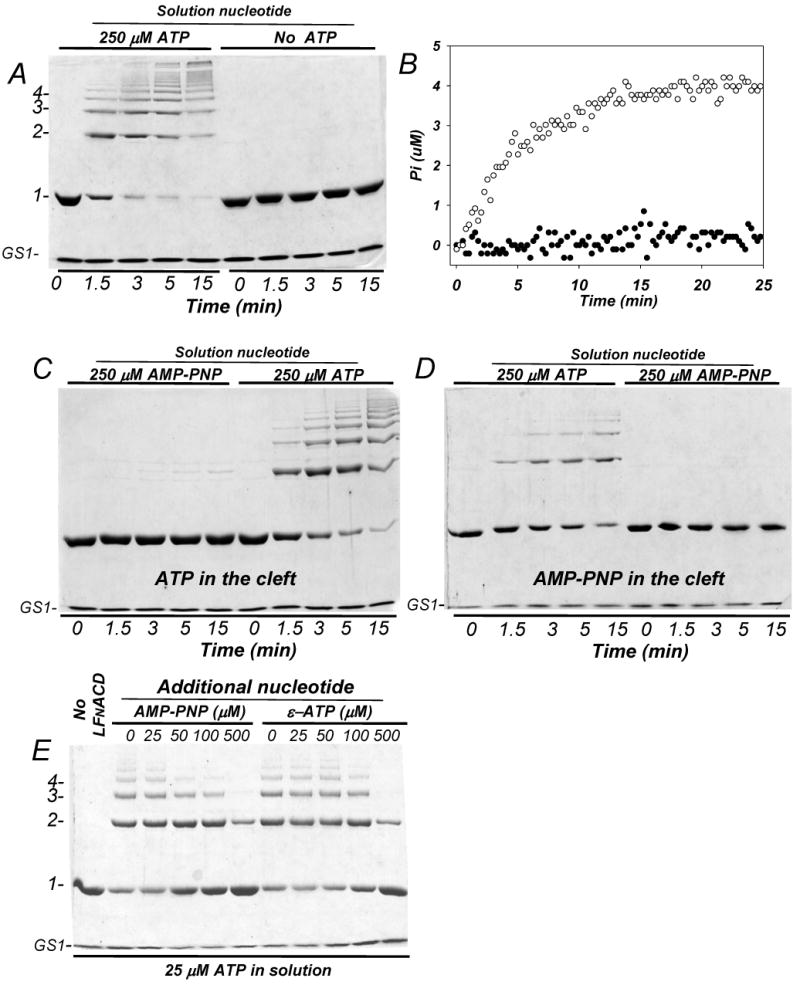
(A) The cross-linking of 9 μM Mg-G-actin-GS1 complex with LFNACD (1 to 250 mole ratio) was conducted in the absence and the presence of 250 μM ATP in the reaction solution. The cross-linking reaction was initiated by adding 1.0 mM MgCl2. (B) The cross-linking of 5 μM ATP-G-actin-GS1 complex with 0.02 μM LFNACD (1 to 250 mole ratio to actin) in the presence (open circles) and absence (closed circles) of 100 μM free ATP in solution was measured with the EnzChek Phosphate kit. Neither cross-linking (A) nor Pi release (B) occurred in the absence of free ATP in the solution. (C) A complex of ATP-actin and GS1 (10 and 12.5 μM final concentrations, respectively) was transferred into the buffer free of ATP via a PD10 size exclusion column and then supplemented with 250 μM of either AMP-PNP or ATP (as indicated). The cross-linking was initiated by adding 0.04 μM of LFNACD and 1.0 mM MgCl2. (D) A complex of GS1 (7.5 μM) with G-actin (6.0 μM) with AMP-PNP bound at the nucleotide binding cleft was supplemented with 250 μM of either ATP or AMP-PNP and 0.024 μM LFNACD. (C) and (D) show that free ATP in solution is required to support the cross-linking, while either ATP or non-hydrolysable AMP-PNP bound to the actin nucleotide-binding cleft allow the reaction. (E) ATP-Actin (10 μM) in complex with GS1 (15 μM) was cross-linked for 10 minutes with 0.04 μM of LFNACD (1 to 250 mole ratio to actin) in the presence of 25 μM free ATP and increasing concentrations of either AMP-PNP or εATP. Although εATP can be hydrolyzed to Pi and εADP by some enzymes, ACD dose not appear to use it as an energy source.
We have shown recently that actin cannot be cross-linked by ACD if ATP in the solution is replaced by excess of non-hydrolysable nucleotide analog AMP-PNP. Although, this result suggested that ATP was required for the cross-linking, it did not discriminate between ATP as an energy source for ACD or a factor defining actin conformation. To eliminate this ambiguity, we used now GS1 to lock AMP-PNP (or ATP) in the nucleotide binding cleft, while supplementing the experimental mixture with free ATP (or AMP-PNP) (Fig. 4 C,D). The results of this experiment demonstrate unambiguously that free ATP in solution is a mandatory factor for the cross-linking reaction, while the presence of ATP in the nucleotide cleft is not required for the cross-linking, although it appears to improve it.
If ATP is required for ACD activity, we would expect that structurally related, but functionally inactive nucleotides should compete with ATP for ACD. To test this prediction, we observed the cross-linking of ATP-actin in the presence of 25 μM ATP and increasing amounts of AMP-PNP and εATP (Fig 4E). As above, to ensure that ATP is locked inside the actin cleft, we added excess of GS1. As expected, both ATP analogs inhibited the cross-linking, with AMP-PNP being slightly more effective than εATP in that task.
Altogether, our results strongly suggest that LFNACD is an ATPase and consumes ATP as the energy source for the cross-linking reaction.
To clarify the requirement of the reaction for MgCl2, we carried out cross-linking of 10 μM actin-KabC complex in the presence of 200 μM ATP and different concentrations of MgCl2. When plotted as percentage of covalent cross-linked bonds formed in 20 minutes versus free Mg2+ concentration in the solution, the cross-linking shows a typical hyperbolic dependence on MgCl2, with an estimated apparent Kd = 0.54 mM (Fig. 5), which is in the range of Mg2+ affinity for actin. In addition to the high affinity cation binding site in the nucleotide cleft, actin has several (5 to 9) low affinity binding sites for divalent cations. The dissociation constants for those sites were estimated by different groups to be between 0.018 to 0.5 mM (38-40). It is possible that, similarly to the requirements for actin polymerization (26), certain number of divalent cations must be bound to actin to compensate for the repulsive interactions between negatively charged G-actin molecules, and thus allow actin monomers to approach each other and be cross-linked by ACD. However, at concentrations high enough to cause actin polymerization, CaCl2 and KCl do not support the cross-linking by ACD, even in the presence of 0.1 mM MgCl2 (data not shown). Thus, it is difficult to distinguish between the possibilities that Mg2+ is required for actin to adopt a cross-linking competent conformation, or it is needed to activate the ACD or, perhaps, serves both purposes.
Figure 5. The efficiency of the cross-linking depends on Mg2+ concentration.

Actin-KabC (10 μM) complex was cross-linked by LFNACD (1 to 500 mole ratio) for 20 minutes in the presence of 200 μM ATP and increasing concentrations of MgCl2. The amount of actin cross-linked was analyzed as in Fig. 3B, normalized to 100%, and plotted as a function of free MgCl2 concentration in the solution (calculated by WinMAXC software, Version 2.40, Stanford University). Solid line represents single ligand binding fitting, with an apparent Kd = 0.54 mM.
The above results demonstrate that ATP hydrolysis occurs simultaneously with the formation of cross-linked actin species, and that ATP bound to the nucleotide cleft of actin is not sufficient to support the cross-linking. Therefore, we conclude that ATP is hydrolyzed by LFNACD in the course of energy-consuming formation of covalent bonds between actin protomers.
Actin monomers are a better substrate than dimers for cross-linking
As it can be noticed from Figure 3, the cross-linking reaction appears to reach a plateau before all actin is cross-linked. One possible explanation for this could be that actin oligomers are not cross-linked as efficiently as monomers, if at all, due to their size, shape, diffusion, and orientation limitations. To examine this supposition, we purified ACD-cross-linked actin dimer and tested its susceptibility to the cross-inking (Fig. 6). We found that actin dimers disappear during the cross-linking at ∼4 folds slower rate than the monomers (kdimer=0.021 min-1 and kmon=0.083 min-1, the exponential decay rates for dimers and monomers, respectively). This result indicates that although ACD-cross-linked oligomers can participate in further cross-linking to higher species, the efficiency of such a reaction diminishes with the increase in size of oligomers. These data further indicate that the increasing size of oligomers occurs predominantly by addition of monomers to oligomers rather than the joining of oligomers.
Figure 6. The purified ACD-cross-linked actin dimers are less good substrate for cross-linking than actin monomers.
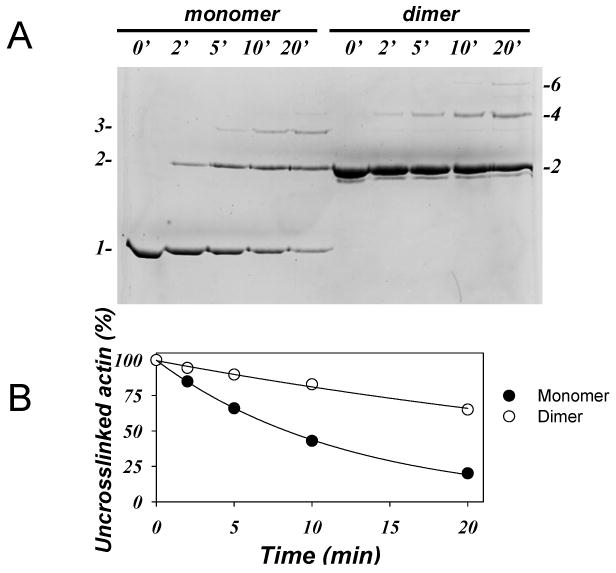
5.0 μM actin monomers and 5.0 μM actin dimers, purified as described in the Experimental procedures, were cross-linked in the presence of 10 μM KabC, 0.01 μM LFNACD (1 to 1000 mole ratio to actin), and 2.0 mM MgCl2. The decay of the initial uncross-linked actin on 7.5% SDS-gel (A) was used as a measure of the cross-linking efficiency and plotted in (B). Closed circles represent the decay of uncross-linked monomeric actin, open circles represent decay of purified actin dimers. Experimental data were fitted to a single exponential expression (solid lines).
Discussion
There is growing evidence that in order to compromise cell integrity, a diverse group of pathogens and toxins targets and impairs the actin cytoskeleton. One of such pathogenic factors is the ACD of MARTXVc (6). The cross-linking of actin with ACD, both under in vivo and in vitro conditions, results in the formation of a set of oligomers, appearing as a ladder of actin bands on SDS-gels ((8), and Fig. 1). Recently, we showed that the ACD is an enzyme that directly catalyzes the cross-linking of actin dependent only upon addition of Mg2+ and ATP to the in vitro reaction. In the present work, we investigate the catalytic requirements for the cross-linking reaction and demonstrate that the ACD cross-links monomeric G-actin and consumes ATP during the in vitro reaction.
A ladder with a very similar pattern of oligomers can be produced by a covalent cross-linking of actin with N'-azidonitrophenyl maleimide (ANP; (41)). In the latter case, ANP-labeled actin must be pre-polymerized and the cross-linking occurs only in the polymeric state. Therefore, it was important to test whether the ACD-induced cross-linking also requires actin polymerization. Our experiments with actins, for which the polymerization was blocked with actin-specific drugs (LatA and KabC) or by a covalent modification with TMR, clearly demonstrated that G-actin can be cross-linked efficiently by LFNACD (Fig. 1A). In contrast, the cross-linking of pre-polymerized F-actin is inhibited (Fig 1C), indicating that polymerization competes with the cross-linking reaction. The cross-linking of F-actin was further inhibited by phalloidin likely due to the inhibition of treadmilling and a decrease in the critical concentration of G-actin for polymerization, which would diminish the amount of monomeric actin available to participate in the cross-linking reaction.
Since G-actin in the living cell is normally bound to one of the ABPs, it was important to test how those proteins affect the ACD-induced cross-linking. Intriguingly, we found the most abundant ABPs that interact with G-actin in vivo had no effect on cross-linking in vitro indicating that association of G-actin with other proteins does not provide a cell protection against MARTXVc-mediated cross-linking. Indeed, only DNaseI completely abolished the formation of actin oligomers (Fig. 1B). This result suggests that binding of DNase I to actin subdomain 2 either blocks the site for binding of ACD to actin binding or it blocks the ACD-mediated actin-actin interface that would form during the introduction of cross-link.
Among other proteins tested, cofilin and twinfilin also showed significant inhibition of actin cross-linking by LFNACD (Fig. 1B). These proteins belong to the actin destabilizing factor (ADF) family of proteins, which are major regulators of actin-dependent processes in living cells. Twinfilin contains two homologous ADF-like domains while cofilin has only one. In spite of the strong inhibitory effect of both proteins on G-actin cross-linking in vitro, their in vivo effect is difficult to predict because at least one of them, cofilin, causes slight enhancement of F-actin cross-linking (Fig. 1C), probably via an increased critical concentration for polymerization and/or increased turnover of F-actin (42).
The mechanism of ACD cross-linking inhibition by cofilin is yet unclear and requires further investigation. This inhibition of cross-linking can be partially explained by the ability of cofilin to accelerate in vitro polymerization of actin (29-32) – the process which competes strongly with the cross-linking reaction (Fig. 1A). However, twinfilin, which does not accelerate actin polymerization, also inhibits significantly the cross-linking (Fig. 1B). Alternatively, the effects of cofilin and twinfilin could be explained by steric hindrance of the cross-linking sites by these proteins and/or by allosteric alterations in one or both actin sites involved in the cross-linking. Interestingly, ADF-homology domains are structurally similar to the repeating domains of gelsolin (43). Although the detailed binding interface of cofilin and twinfilin with actin is still uncertain, it was predicted that ADF-homology domain and GS1 interact with actin through a similar interface (44-46). Yet, cofilin and twinfilin, in contrast to GS1, show strong protection against ACD-induced cross-linking (Fig. 1B). This result suggests some differences between the binding interfaces of ADF-homology proteins and GS1 on G-actin.
Although it is difficult to predict the effects of ADF-family proteins on ACD-induced cross-linking of actin in vivo, these can be tested experimentally. The mechanism of twinfilin regulation in living cells is yet unknown, but cofilin activities are appear to be mainly regulated by Slingshot phosphatases and LIM-kinases (47). Therefore, it would be interesting to probe the effects of cofilin activation/inhibition on actin cross-linking in the living cells by tuning the activity of these regulatory enzymes.
To date, little is known about the MARTXVc-induced cross-linking in actin and the particular residues involved in the reaction. Recently, we showed that ATP is required for the cross-linking of actin with LFNACD, and that neither ADP nor the non-hydrolyzable ATP analog AMP-PNP could substitute for ATP in the cross-linking reaction (8). However, actin itself is an ATPase whose conformational state depends on the nucleotide bound to the nucleotide-binding cleft. Thus, our previous study left unresolved the possibility that ATP is needed to support the appropriate conformational state of actin and not for the catalytic activity of ACD. The results of this study demonstrate unequivocally that free Mg-ATP in solution is required to support efficient actin cross-linking by LFNACD (Fig. 4) and suggest that MARTXVc hydrolyzes one molecule of ATP per each cross-linking event (Fig. 3). Apparently, ATP is required to produce an activated intermediate product in a course of the cross-linking reaction; however the details of this reaction remain to be clarified. Actin monomers are apparently a better substrate for the cross-linking reaction than the cross-linked oligomers (Fig. 6). Therefore, after monomer depletion, the reaction slows down, appearing to reach a plateau prior to forming large cross-linked polymers. To cross-link, ACD is most likely binding to two actins. At this stage, it is premature to speculate whether the two actin sites on ACD are equivalent, and if the faster cross-linking of monomers than oligomers is not just a result of steric interference between the oligomers.
Overall, we can propose a more detailed model of how MARTXVc accomplishes the rounding of cells through the cross-linking of actin. Recently we have found evidence that MARTXVc undergoes autocatalytic cleavage and we proposed that this autoprocessing releases independent activity domains from the large toxin freely to the cytosol (48). In this study, we have shown that the ACD then functions to bind monomeric G-actin and catalyzes the formation of actin oligomers while consuming ATP to energize the reaction. Apparently the reaction can proceed despite the binding of G-actin in vivo to ABPs. Ultimately, the sequestering of G-actin through cross-linking would accelerate F-actin depolymerization and lead to cell rounding. In the context of the holotoxin, the inhibition of Rho by the MARTXVc Rho-inactivating domain (5) would likewise accelerate F-actin depolymerization and increase the concentration of G-actin available for cross-linking leading to the observed rapid cell rounding.
Acknowledgments
This work was supported by an Investigator in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund and United States Public Health Service Grant AI051490 (to K.J.F.S.) and by United States Public Health Service Grant GM-077190 and National Science Foundation Grant MCB 0316269 (to E.R.). D.K. was supported by American Heart Association Western States Affiliates fellowship 0425119Y. C.L.C. was supported by National Research Service Award Predoctoral Fellowship F31-AI52490. We would like to thank Sari Juwono for excellent technical assistance. We are thankful to Dr. Safer for kind donation of thymosin-β4 peptide, Dr. Roberto Dominguez for twinfilin plasmid, Dr. Henry N. Higgs for profilin plasmid, and Dr. Gerard Marriott for the gift of KabC.
The abbreviations used are
- ABP
actin binding protein
- ACD
actin cross-linking domain
- GS1
gelsolin segment 1
- KabC
kabiramide C
- LatA
latrunculin A
- LFN
Bacillus anthracis lethal factor
- MARTXVc
Multifunctional, autoprocessing RTX (repeats-in-toxin) toxin of V. cholerae
- SD
subdomains of actin
- TMR
tetramethylrhodamine-5-maleimide
Reference List
- 1.Kaper JB, Morris JG, Levine MM. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olivier V, Haines GK, III, Tan Y, Satchell KJF. Infect Immun. 2007;75:5043–5055. doi: 10.1128/IAI.00506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cordero CL, Sozhamannan S, Satchell KJF. J Clin Microbiol. 2007;45:2289–2292. doi: 10.1128/JCM.00349-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satchell KJF. Infect Immun. 2007 published online 7/23/07. [Google Scholar]
- 5.Sheahan KL, Satchell KJF. Cell Microbiol. 2007;9:1324–1335. doi: 10.1111/j.1462-5822.2006.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fullner KJ, Mekalanos JJ. Embo J. 2000;19:5315–5323. doi: 10.1093/emboj/19.20.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheahan KL, Cordero CL, Satchell KJF. P Natl Acad Sci USA. 2004;101:9798–9803. doi: 10.1073/pnas.0401104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordero CL, Kudryashov DS, Reisler E, Satchell KJF. J Biol Chem. 2006;281:32366–32374. doi: 10.1074/jbc.M605275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard TD, Borisy GG. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 10.Gershman LC, Selden LA, Estes JE. Biochem Bioph Res Co. 1986;135:607–614. doi: 10.1016/0006-291x(86)90036-7. [DOI] [PubMed] [Google Scholar]
- 11.Kinosian HJ, Selden LA, Estes JE, Gershman LC. J Biol Chem. 1993;268:8683–91. [PubMed] [Google Scholar]
- 12.Carlier MF. Biochem Bioph Res Co. 1987;143:1069–1075. doi: 10.1016/0006-291x(87)90361-5. [DOI] [PubMed] [Google Scholar]
- 13.Carlier MF, Pantaloni D, Korn ED. J Biol Chem. 1987;262:3052–3059. [PubMed] [Google Scholar]
- 14.Korn ED, Carlier MF, Pantaloni D. Science. 1987;238:638–644. doi: 10.1126/science.3672117. [DOI] [PubMed] [Google Scholar]
- 15.Sept D, McCammon JA. Biophys J. 2001;81:667–674. doi: 10.1016/S0006-3495(01)75731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steele-Mortimer O, Knodler LA, Finlay BB. Traffic. 2000;1:107–118. doi: 10.1034/j.1600-0854.2000.010203.x. [DOI] [PubMed] [Google Scholar]
- 17.Barth H, Aktories K, Popoff MR, Stiles BG. Microbiol Mol Biol R. 2004;68:373–+. doi: 10.1128/MMBR.68.3.373-402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spudich JA, Watt S. J Biol Chem. 1971;246:4866–71. [PubMed] [Google Scholar]
- 19.Bobkov AA, Muhlrad A, Kokabi K, Vorobiev S, Almo SC, Reisler E. J Mol Biol. 2002;323:739–750. doi: 10.1016/s0022-2836(02)01008-2. [DOI] [PubMed] [Google Scholar]
- 20.Goldsmith SC, Guan JQ, Almo S, Chance M. J Biomol Struct Dyn. 2001;19:405–18. doi: 10.1080/07391102.2001.10506750. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser DA, Goldschmidtclermont PJ, Levine BA, Pollard TD. Cell Motil Cytoskel. 1989;14:251–262. doi: 10.1002/cm.970140211. [DOI] [PubMed] [Google Scholar]
- 22.Kurokawa H, Fujii W, Ohmi K, Sakurai T, Nonomura Y. Biochem Bioph Res Co. 1990;168:451–457. doi: 10.1016/0006-291x(90)92342-w. [DOI] [PubMed] [Google Scholar]
- 23.Otterbein LR, Graceffa P, Dominguez R. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 24.Kudryashov DS, Reisler E. Biophys J. 2003;85:2466–2475. doi: 10.1016/s0006-3495(03)74669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuler H. BBA-Protein Struct M. 2001;1549:137–47. doi: 10.1016/s0167-4838(01)00255-2. [DOI] [PubMed] [Google Scholar]
- 26.Rouayrenc JF, Travers F. Eur J Biochem. 1981;116:73–77. doi: 10.1111/j.1432-1033.1981.tb05302.x. [DOI] [PubMed] [Google Scholar]
- 27.Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I. Nat Struct Biol. 2003;10:1058–1063. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- 28.Morton WM, Ayscough KR, McLaughlin PJ. Nat Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- 29.Blanchoin L, Pollard TD. J Biol Chem. 1999;274:15538–15546. doi: 10.1074/jbc.274.22.15538. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Bernstein BW, Sneider JM, Boyle JA, Minamide LS, Bamburg JR. Biochemistry. 2004;43:7127–7142. doi: 10.1021/bi049797n. [DOI] [PubMed] [Google Scholar]
- 31.Du JY, Frieden C. Biochemistry. 1998;37:13276–13284. doi: 10.1021/bi981117r. [DOI] [PubMed] [Google Scholar]
- 32.Kudryashov DS, Galkin VE, Orlova A, Phan M, Egelman EH, Reisler E. J Mol Biol. 2006;358:785–797. doi: 10.1016/j.jmb.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Goode BL, Drubin DG, Lappalainen P. J Cel Biol. 1998;142:723–733. doi: 10.1083/jcb.142.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paavilainen VO, Hellman M, Helfer E, Bovellan M, Annila A, Carlier MF, Permi P, Lappalainen P. P Natl Acad Sci USA. 2007;104:3113–3118. doi: 10.1073/pnas.0608725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito S, Karaki H. Clin Exp Pharmacol P. 1996;23:743–746. doi: 10.1111/j.1440-1681.1996.tb01770.x. [DOI] [PubMed] [Google Scholar]
- 36.Bryan J. J Cel Biol. 1988;106:1553–62. doi: 10.1083/jcb.106.5.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollard TD, Goldberg I, Schwarz WH. J Biol Chem. 1992;267:20339–20345. [PubMed] [Google Scholar]
- 38.Carlier MF, Pantaloni D, Korn ED. J Biol Chem. 1986;261:778–784. [PubMed] [Google Scholar]
- 39.Tellam R. Biochemistry. 1985;24:4455–4460. doi: 10.1021/bi00337a029. [DOI] [PubMed] [Google Scholar]
- 40.Zimmerle CT, Patane K, Frieden C. Biochemistry. 1987;26:6545–6552. doi: 10.1021/bi00394a039. [DOI] [PubMed] [Google Scholar]
- 41.Kim E, Phillips M, Hegyi G, Muhlrad A, Reisler E. Biochemistry. 1998;37:17793–17800. doi: 10.1021/bi9812874. [DOI] [PubMed] [Google Scholar]
- 42.Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. J Cel Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatanaka H, Ogura K, Moriyama K, Ichikawa S, Yahara I, Inagaki F. Cell. 1996;85:1047–1055. doi: 10.1016/s0092-8674(00)81305-7. [DOI] [PubMed] [Google Scholar]
- 44.Lappalainen P, Fedorov EV, Fedorov AA, Almo SC, Drubin DG. Embo J. 1997;16:5520–5530. doi: 10.1093/emboj/16.18.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Troys M, Dewitte D, Verschelde JL, Goethals M, Vandekerckhove J, Ampe C. J Biol Chem. 1997;272:32750–32758. doi: 10.1074/jbc.272.52.32750. [DOI] [PubMed] [Google Scholar]
- 46.Wriggers W, Tang JX, Azuma T, Marks PW, Janmey PA. J Mol Biol. 1998;282:921–932. doi: 10.1006/jmbi.1998.2048. [DOI] [PubMed] [Google Scholar]
- 47.Huang TY, DerMardirossian C, Bokoch GM. Curr Opin Cel Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Sheahan KL, Cordero CL, Satchell KJF. Embo J. 2007;26:2552–2561. doi: 10.1038/sj.emboj.7601700. [DOI] [PMC free article] [PubMed] [Google Scholar]