

Abstract
Matrix metalloproteinase-9 (MMP-9) activation plays an important role in blood-brain barrier dysfunction after central nervous system injury. Oxidative stress is also implicated in the pathogenesis after cerebral ischemia and spinal cord injury (SCI), but the relationship between MMP-9 activation and oxidative stress after SCI has not yet been clarified. We examined MMP-9 expression after SCI using copper/zinc-superoxide dismutase (SOD1) transgenic (Tg) rats. Our results show that MMP-9 activity significantly increased after SCI in both SOD1 Tg rats and their wild-type (Wt) littermates, although the increase was less in the SOD1 Tg rats. This pattern of MMP-9 expression was further confirmed by immunostaining and Western blot analysis. In situ zymography showed that gelatinolytic activity increased after SCI in the Wt rats, while the increase was less in the Tg rats. Evans blue extravasation increased in both the Wt and Tg rats, but was less in the SOD1 Tg rats. Inhibitor studies showed that with an intrathecal injection of SB-3CT, a selective MMP-2/MMP-9 inhibitor, MMP activity, Evans blue extravasation, and apoptotic cell death decreased after SCI. We conclude that increased oxidative stress after SCI leads to MMP-9 upregulation, blood-brain barrier disruption, and apoptosis, and that overexpression of SOD1 in Tg rats decreases oxidative stress and further attenuates MMP-9 mediated blood-brain barrier disruption.
Keywords: blood-brain barrier, copper/zinc-superoxide dismutase, endothelial, MMP-9, spinal cord injury
INTRODUCTION
Traumatic spinal cord injury (SCI) is a devastating condition that may result in permanent disability. Currently, steroids are the only effective drug, if administered within 8 h after injury. It is generally accepted that initial mechanical injury as well as various secondary injuries contribute to cell death and functional disability after SCI. Matrix metalloproteinases (MMPs) play important roles in limiting functional recovery after SCI (Noble et al., 2002).
MMPs are a family of proteolytic enzymes that degrade the extracellular matrix and junctional proteins and further increase endothelial permeability (Alexander and Elrod, 2002). MMP-9 (gelatinase B) is one of the major proteins, and active MMP-9 specifically degrades endothelial basal lamina and the tight junction proteins occludin and claudin (Rosenberg et al., 1998; Yang et al., 2007). It has been shown that active MMP-9 is increased and leads to the disruption of the blood-brain barrier (BBB) after focal cerebral ischemia (Gasche et al., 2001; Rosenberg et al., 1998). Studies using an in vitro BBB model with a human brain vascular endothelial cell line showed that BBB failure correlates with upregulation of MMP-9 (Cucullo et al., 2007; Haorah et al., 2007). It has also been demonstrated that treatment with an MMP inhibitor decreases endothelial gap formation and occludin loss (Reijerkerk et al., 2006).
MMPs can be regulated by reactive oxygen species (ROS) (Alexander and Elrod, 2002), which increase MMP-2 and MMP-9 activity (Rajagopalan et al., 1996). Moreover, oxidative stress also promotes an increase in MMP-2 and MMP-9 activity after focal cerebral ischemia (Gasche et al., 2001; Pustovrh et al., 2005). Activation of endothelial cell MMP by ROS through the vascular cell adhesion molecule 1 has been shown in a cell culture study (Deem and Cook-Mills, 2004). An in vitro study showed that ROS activated MMPs (-1, -2, and -9) and decreased tissue inhibitors of MMP in a BBB model (Haorah et al., 2007). Copper/zinc-superoxide dismutase (SOD1) is a crucial endogenous antioxidant enzyme responsible for eliminating superoxide. Overexpression of SOD1 in transgenic (Tg) rodents has been shown to decrease oxidative stress and attenuate induction and activation of MMPs after cold injury-induced brain trauma and focal cerebral ischemia (Kamada et al., 2007; Morita-Fujimura et al., 2000). We demonstrated that overexpression of SOD1 in Tg rats decreases oxidative stress and is neuroprotective after SCI (Sugawara et al., 2002). We hypothesize that MMP-9 activation by oxidative stress may cause BBB breakdown and neuronal damage in SCI. This study was designed to use SOD1 Tg rats to address this issue.
MATERIALS AND METHODS
SOD1 Tg Rats
Heterozygous SOD1 Tg rats with a Sprague-Dawley background, carrying human SOD1 genes, were derived from the founder stock and further bred with wild-type (Wt) Sprague-Dawley rats to generate heterozygous rats, as previously described by our group (Chan et al., 1998). The phenotype of these rats was identified by isoelectric focusing gel electrophoresis as described (Chan et al., 1998). There were no observable phenotypic differences in brain vasculature between the Tg and Wt rats.
Surgery
All animals were treated in accordance with Stanford University guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult female SOD1 Tg rats (250-300 gm) and their Wt littermates were used in this experiment. The animals were anesthetized with 2.0% isoflurane in 70% N2O and 30% O2 using a face mask. Lumbar enlargement was exposed by a partial laminectomy of the T13 vertebra, and a vascular clip (closing force 15 g; Ohwa Tsusho, Tokyo, Japan) was applied extradurally for 5 sec according to our previous study (Sugawara et al., 2002). The rectal temperature was controlled at 37.0 ± 0.5°C during surgery with a homeothermic blanket.
Drug Injection
SB-3CT is a selective and mechanism-based inhibitor of MMP-2 (Ki = 13.9 nM) and MMP-9 (Ki = 600 nM) and has been shown to be highly selective in inhibiting MMP-9 expression after transient focal cerebral ischemia (Gu et al., 2005). To investigate the role of MMP-9 after SCI, SB-3CT (EMD Biosciences, San Diego, CA) was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mg/ml and was further diluted to 1 mg/ml in filtered phosphate-buffered saline (PBS, pH 7.4) before use. A partial laminectomy was performed and lumbar enlargement was exposed. Six microliters of 1 mg/ml SB-3CT and the same amount of the vehicle (10% DMSO in PBS) were injected intrathecally 2 h before SCI. The animals were killed and the spinal cords were collected for gel zymography, a BBB permeability study, and an apoptotic cell death assay.
Gel Zymography
The spinal cords were removed 1, 3, and 7 days after SCI and frozen in powdered dry ice. They were then homogenized in a lysis buffer (1 M Tris-HCl, pH 7.6, 2.5 M NaCl, 1 M CaCl2, 30% BRIJ-35, 10% NaN3, and 1% Triton X-100). Sample supernatants (1.2 mg in 200 μl) were recovered and incubated with 50 μl of gelatin-Sepharose 4 B (Pharmacia Biotech, Uppsala, Sweden) for 60 min at 4°C with constant shaking. The samples were centrifuged at 500 g for 2 min at 4°C and then the pellets were incubated with 60 μl elution buffer (1 M Tris-HCl, pH 7.6, 2.5 M NaCl, 1 M CaCl2, 30% BRIJ-35, 10% NaN3, and 10% DMSO) at 4°C with constant shaking for 30 min. The supernatants were collected and saved for gel zymography. Ten microliters of a nonreducing sample buffer (0.4 M Tris, pH 6.8, 5% sodium dodecyl sulfate [SDS], 20% glycerol, 0.05% bromophenol blue) were added to the same volume of sample supernatants. The samples were loaded on 10% SDS-polyacrylamide electrophoresis gels, in which 0.1% porcine skin gelatin (Invitrogen, Carlsbad, CA) was co-polymerized, and run at 100 V for 2 h. The gels were incubated twice with 2.5% Triton-X 100 for 1 h at room temperature. They were then washed for 10 min in Tris-HCl/NaCl/CaCl2 buffer (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM CaCl2), and further incubated for 16 h at 37°C in the same buffer in a water bath. The gels were stained for 90 min in Coomassie blue (1% Coomassie brilliant blue, 30% methanol, 10% acetic acid) and destained in 30% methanol/10% acetic acid four times for 5, 15, 30, and 60 min. The gels were scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA) and a quantitative analysis was performed using Multi-Analyst software (Bio-Rad Laboratories). The optical density of each band was measured on the same gel and the results are presented as ± SEM.
Immunohistochemistry
Anesthetized animals were perfused with 10 U/ml heparin saline and subsequently with 3.7% formaldehyde in PBS 1, 3, and 7 days after SCI. Spinal cords at the lumbar enlargement were collected and postfixed with the same fixative for 24 h. The tissue was embedded in paraffin and sectioned 6 μm thick with a microtome. The sections were deparaffinized and incubated with 3% H2O2 in PBS. We used a 5% blocking serum and then incubated the sections with mouse polyclonal anti-MMP-9 antibody (1:50; EMD Biosciences) at 4°C overnight. The sections were then reacted with biotin-conjugated goat anti-mouse immunoglobulin G (1:100; Vector Laboratories, Burlingame, CA) at room temperature for 1 h, then incubated with avidinbiotin horseradish peroxidase solution (ABC kit; Vector Laboratories) for 30 min at room temperature and visualized using 0.025% 3,3′-diaminobenzidine hydrochloride (DAB kit; Vector Laboratories). Finally, the sections were counterstained with methyl green and mounted with Permount (Fisher Scientific, Pittsburgh, PA).
Western Blot Analysis
To obtain the whole cell fraction, the spinal cords were collected 1, 3, and 7 days after SCI and gently homogenized by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ) in 5 volumes of cold suspension buffer (20 mM HEPES-KOH, pH 7.5, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, l tablet of cocktail inhibitor [Roche Diagnostics, Mannheim, Germany] in 25 ml buffer). Approximately 30 μg of total protein per lane were subjected to SDS-polyacrylamide gel electrophoresis on a Tris-glycine gel (Invitrogen, Carlsbad, CA) and transferred to a polyvinylidene difluoride membrane (Invitrogen). The membrane was incubated in polyclonal rabbit anti-MMP-9 antibody (1:1000; Chemicon International, Temecula, CA) and then incubated in a peroxidase-conjugated secondary antibody. The signals were detected with a chemiluminescence kit (Amersham Biosciences, Uppsala, Sweden) and exposed on x-ray film. To confirm equal loading, the membranes were then stained with β-actin. After the film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories), a quantitative analysis was performed using Multi-Analyst software (Bio-Rad Laboratories). The optical density of each band was measured on the same membrane and the results are presented as ± SEM.
Measurement of BBB Disruption
BBB disruption was evaluated in the Wt and SOD1 Tg rats, as well as in the SB-3CT– and DMSO-treated rats. Twenty hours after SCI, 2.5 ml/kg of 4% Evans blue (Sigma-Aldrich, St. Louis, MO) in 0.9% saline were injected through the jugular vein. The animals were anesthetized and perfused with 200 ml of 10 U/ml heparin saline. To quantify Evans blue extravasation, the spinal cords were removed and homogenized in 400 μl of N, N-dimethylformamide (Sigma-Aldrich) and then incubated for 72 h at 55°C and centrifuged at 20,000 × g for 20 min. The Evans blue level was determined in the supernatants at 620 nm using a spectrophotometer (Molecular Devices, Sunnyvale, CA). The results are presented as micrograms of Evans blue per spinal cord. For qualitative examination of Evans blue extravasation, the animals were perfused with 10 U/ml heparin saline and subsequently with 3.7% formaldehyde in PBS as described above. The spinal cords were sectioned 20 μm thick with a cryostat. The sections were mounted and nuclear counterstaining was done with 4′,6 diamidino-2-phenylindole (DAPI) (Vector Laboratories). The sections were observed with a fluorescence microscope.
In Situ Zymography
To examine in situ gelatinolytic activity, in situ zymography was performed. Animals were sacrificed 24 h after SCI and the spinal cords were collected and frozen on dry ice. Frozen sections were cut at 20 μm using a cryostat. Sections were incubated with 200 μg/mL DQ gelatin fluorescein conjugate (Enzecheck Gelatinase Assay kit; Invitrogen) for 2 h at 37°C. The sections were mounted and observed with a fluorescence microscope.
In Situ Detection of Superoxide Anions and Double Labeling with In Situ Zymography
To examine the colocalization of superoxide anion and in situ gelatinolytic activity, we used the hydroethidine (HEt) method. HEt is specifically oxidized by superoxide anions into ethidium inside the cell, which then has a red fluorescence (Bindokas et al., 1996). One milliliter of 1 mg/ml HEt in PBS was injected through the jugular vein 4 h after SCI. The animals were sacrificed 6 h after SCI and the spinal cords were collected. In situ zymography was performed as described above. The nuclei were counterstained with DAPI and observed with a fluorescence microscope.
In Situ Zymography and Double Labeling with Fluorescent Probes
To study the cellular localization of gelatinolytic activity, in situ zymography with double fluorescent staining, and double fluorescent staining of the cellular markers neuron-specific nuclear protein (NeuN, for neurons), CD31 (for endothelial cells), myeloperoxidase (MPO, for neutrophils), and glial fibrillary acidic protein (GFAP, for astrocytes) were performed. In situ zymography was done as described above. The sections were then fixed in 3.7% formaldehyde in PBS for 10 min. The sections were incubated with mouse polyclonal anti-NeuN (1:500; Chemicon International), mouse monoclonal anti-CD31 (1:20; BD Biosciences, San Jose, CA), mouse monoclonal anti-MPO (1:5000; EMD Biosciences), or rabbit anti-GFAP (1:5000; Sigma-Aldrich) at 4°C overnight. The sections were then reacted with Texas Red-conjugated anti-mouse immunoglobulin G (1:100; Jackson Immunoresearch Laboratories, West Grove, PA) or Cy3-conjugated anti-rabbit immunoglobulin (1:100; Jackson Immunoresearch Laboratories) at room temperature for 1 h. Nuclei were counterstained with DAPI (Vector Laboratories) and observed with a fluorescence microscope.
Apoptotic Cell Death Assay
To quantify apoptosis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Diagnostics). This assay detects apoptotic but not necrotic cell death (Bonfoco et al., 1995). Samples were obtained from the spinal cord of the SB-3CT–treated rats and control rats 3 days after SCI. Fresh tissue was taken and homogenized with a Teflon homogenizer in 5 volumes of ice-cold buffer (50 mM KH2PO4 and 0.1 mM EDTA, pH 7.8) and centrifuged for 10 min at 750 × g at 4°C. The supernatant was collected and centrifuged for 20 min at 10,000 × g at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, using the manufacturer's protocol. A statistical study was carried out in the same manner as mentioned under Western blot analysis.
Quantification and Statistical Study
Significance was determined by analysis of variance followed by Fisher's protected least significant difference test and accepted when p < 0.05.
RESULTS
MMP-9 Expression after SCI
To investigate MMP-9 activity, we first performed gel zymography after SCI. MMP-9 activity increased 1 day after SCI in both the Wt and SOD1 Tg rats. It decreased by 3 days and continued to decrease at 7 days. There was a significant difference in the level of MMP-9 between the Wt and SOD1 Tg rats 1 day after SCI (Fig. 1; n = 5, p < 0.05). MMP-2 activity slightly increased 3 and 7 days after SCI in both the Wt and SOD1 Tg rats, but there was no difference in the level of MMP-2 between the Wt and SOD1 Tg rats. Active MMP-2 increased in a time-dependent pattern 1, 3, and 7 days after SCI in both the Wt and SOD1 Tg rats, while there was a greater increase 3 and 7 days after SCI in the SOD1 Tg rats (Fig. 1; n = 5, p < 0.05). Further study with the use of immunostaining showed that MMP-9 was not visible in either the Wt or the SOD1 Tg control rats. There was increased expression 1 day after SCI in the vascular structure of both the Wt and SOD1 Tg rats, but the staining was stronger in the Wt rats. Three days after SCI, expression of MMP-9 was decreased in both the Wt and Tg rats. Seven days after SCI, MMP-9 expression was the same in both the Wt and Tg rats (Fig. 2). Western blot analysis showed that MMP-9 was transiently increased 1 day after SCI in the Wt rats, whereas such an increase in the SOD1 Tg rats was not obvious. There was a significant difference in the level of MMP-9 between the Wt and SOD1 Tg animals 1 day after SCI (Fig. 3; n = 6, p < 0.05). Active MMP-9 increased 1, 3, and 7 days after SCI in the Wt rats, while there was no increase in the SOD1 Tg rats. There were significant differences in the level of active MMP-9 between the Wt and SOD1 Tg rats 1, 3, and 7 days after SCI (Fig. 3; n = 6, p < 0.05). In situ zymography was performed to further examine gelatinolytic activity. There was no fluorescent staining in the control rats. In the Wt rats, some vessels and cells were positive for in situ gelatinolytic activity, whereas there was less in the SOD1 Tg rats (Fig. 4).
FIG. 1.
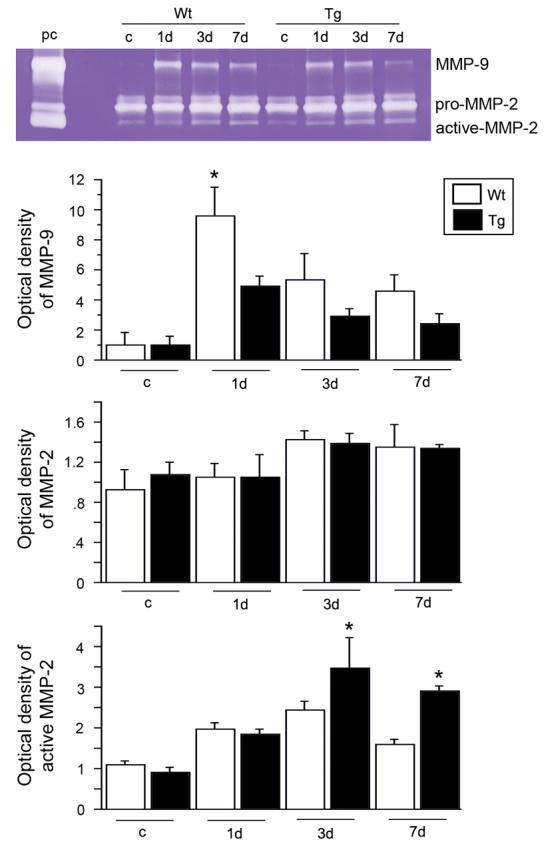
Gel zymography after SCI. MMP-9 activity acutely increased 1 day after SCI in both the Wt and SOD1 Tg rats. It decreased by 3 days and continued to decrease at 7 days. There was a significant difference in the level of MMP-9 between the Wt and SOD1 Tg rats 1 day after SCI (n = 5, p < 0.05). MMP-2 activity slightly increased 3 and 7 days after SCI in both the Wt and SOD1 Tg rats. Active MMP-2 increased in a time-dependent pattern 1, 3, and 7 days after SCI in both the Wt and SOD1 Tg rats, while there was a greater increase 3 and 7 days after SCI in the SOD1 Tg rats (n = 5, p < 0.05). pc, MMP-2 and MMP-9 standards; c, control. *p < 0.05.
FIG. 2.
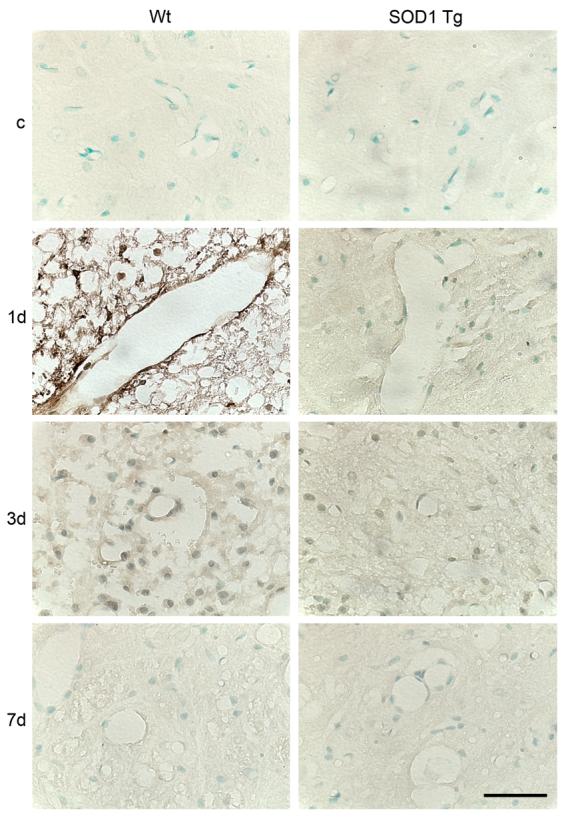
Representative photomicrographs of MMP-9 immunostaining after SCI. MMP-9 immunostaining was not visible in either the Wt or SOD1 Tg control (c) rats. There was increased expression 1 day after SCI in the vascular structure in both the Wt and SOD1 Tg rats, while there was stronger staining in the Wt rats than in the Tg rats. Three days after SCI, expression of MMP-9 was decreased in both the Wt and Tg rats. Seven days after SCI, MMP-9 expression was the same in both the Wt and Tg rats. Scale bar = 50 μm.
FIG. 3.

Western blot analysis of MMP-9 after SCI. MMP-9 was visualized as a 108-kDa band, while active MMP-9 was seen as a 88-kDa band. MMP-9 was transiently increased 1 day after SCI in the Wt rats, while there was no obvious increase in the SOD1 Tg rats. There was a significant difference in the level of MMP-9 between the Wt and SOD1 Tg rats 1 day after SCI (n = 6, p < 0.05). Active MMP-9 increased 1, 3, and 7 days after SCI in the Wt rats, while there was no obvious increase in the SOD1 Tg rats. There were significant differences in the level of active MMP-9 between the Wt and SOD1 Tg rats 1, 3, and 7 days after SCI (n = 6, p < 0.05). β-actin was used as an internal control and no difference in loading was observed between the samples. pc, positive control; c, control. *p < 0.05.
FIG. 4.

Representative photomicrographs of in situ zymography after SCI. There was no fluorescence in the control rats. In the Wt rats, some vessels and cells showed green fluorescence, which indicates gelatinolytic activity, while in the SOD1 Tg rats, there was less fluorescence. Scale bar = 50 μm.
BBB Disruption after SCI
There was no Evans blue extravasation in the control rats 1 day after SCI. There was intense red fluorescence, which indicates that Evans blue leaked into the tissue, while there was less red fluorescence in the SOD1 Tg rats (Fig. 5, Upper panel). A quantitative study showed that there was very little Evans blue in the tissue of the Wt and SOD1 Tg control animals. It was significantly increased 1 day after SCI in both groups, however, the amount of Evans blue was significantly less in the SOD1 Tg rats compared with the Wt rats (Fig. 5, Lower panel; n = 4, p < 0.05).
FIG. 5.

Representative photomicrographs of an Evans blue extravasation study. There was no Evans blue extravasation in the control rats. One day after SCI, there was intense red fluorescence, which indicates that Evans blue leaked into the tissue, while there was less red fluorescence in the SOD1 Tg rats (Upper panel). A quantitative study showed that there was very little Evans blue in the tissue of the Wt and SOD1 Tg control animals. It was significantly increased 1 day after SCI in both the Wt and SOD1 Tg rats, although there was significantly less in the SOD1 Tg rats compared with the Wt rats (Lower panel) (n = 4, p < 0.05). *p < 0.05. Scale bar = 50 μm.
In Situ Gelatinolytic Activity Colocalized with Oxidized HEt
In situ zymography showed that gelatinolytic activity presented on the vessel as green fluorescence. Punctate red fluorescence indicated that HEt was oxidized by superoxide anions. Merged photos showed that in situ gelatinolytic activity colocalized with oxidized HEt signals on the vessel wall (Fig. 6).
FIG. 6.

Representative photomicrograph of in situ zymography and oxidized HEt staining. In situ gelatinolytic activity was seen as a green fluorescence. Oxidized HEt was seen as a punctate red fluorescence. Merged photos showed in situ gelatinolytic activity colocalized with the oxidized HEt signals on the vessel wall. Scale bar = 50 μm.
In Situ Gelatinolytic Activity Localized in Neutrophils, Endothelial Cells, and Neurons
Double staining of the different cell markers (GFAP, NeuN, MPO, or CD31) and in situ zymography showed that GFAP staining did not colocalize with in situ gelatinolytic activity. Some NeuN staining and MPO staining colocalized with in situ gelatinolytic activity. CD31 staining colocalized with in situ gelatinolytic activity on the vessel wall (Fig. 7).
FIG. 7.
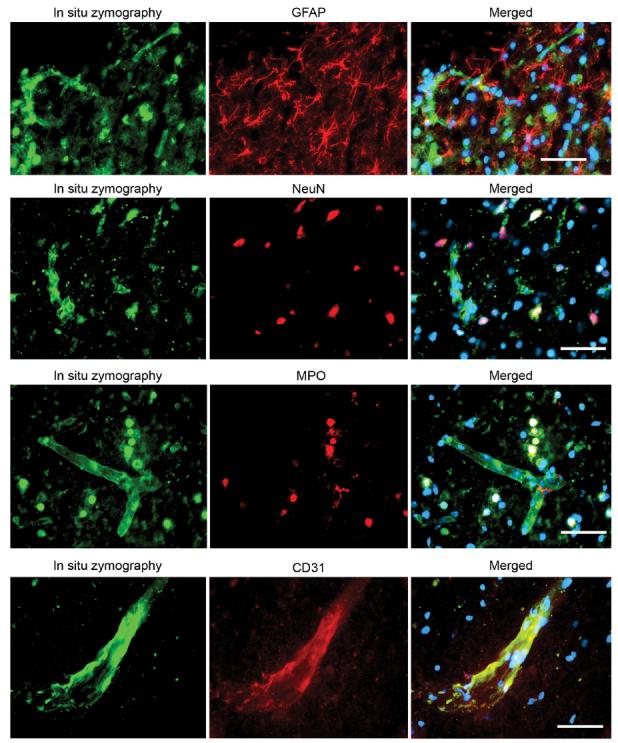
Representative photomicrographs of double fluorescent staining of in situ zymography and different cell markers. In situ gelatinolytic activity was seen as green fluorescence. GFAP, NeuN, MPO, and CD31 were seen as red fluorescence. Merged photos showed that GFAP staining did not colocalize with in situ gelatinolytic activity. Some NeuN staining colocalized with in situ gelatinolytic activity. Most of the MPO staining colocalized with in situ gelatinolytic activity. CD31 staining colocalized with in situ gelatinolytic activity on the vessel wall. Scale bar = 50 μm.
MMP-9 Activation Led to BBB Disruption and Apoptosis
To study the mechanism of MMP-9 activation on BBB disruption and apoptosis, we performed gel zymography, an Evans blue extravasation study, and an apoptotic cell death assay after intrathecal injection of SB-3CT after SCI. Gel zymography showed that MMP-9 activity decreased significantly in the group of rats injected with SB-3CT 1 day after SCI, compared with the group injected with the vehicle (DMSO) (Fig. 8A; n = 4, p < 0.05). The Evans blue study showed that there was significantly less Evans blue extravasation in the group of rats injected with SB-3CT, compared with the group injected with the vehicle after SCI (Fig. 8B; n = 4, p < 0.05). The cell death assay showed that the apoptotic cell death signal was significantly decreased in the group of rats injected with SB-3CT, compared with the group injected with the vehicle after SCI (Fig. 8C; n = 4, p < 0.05).
FIG. 8.
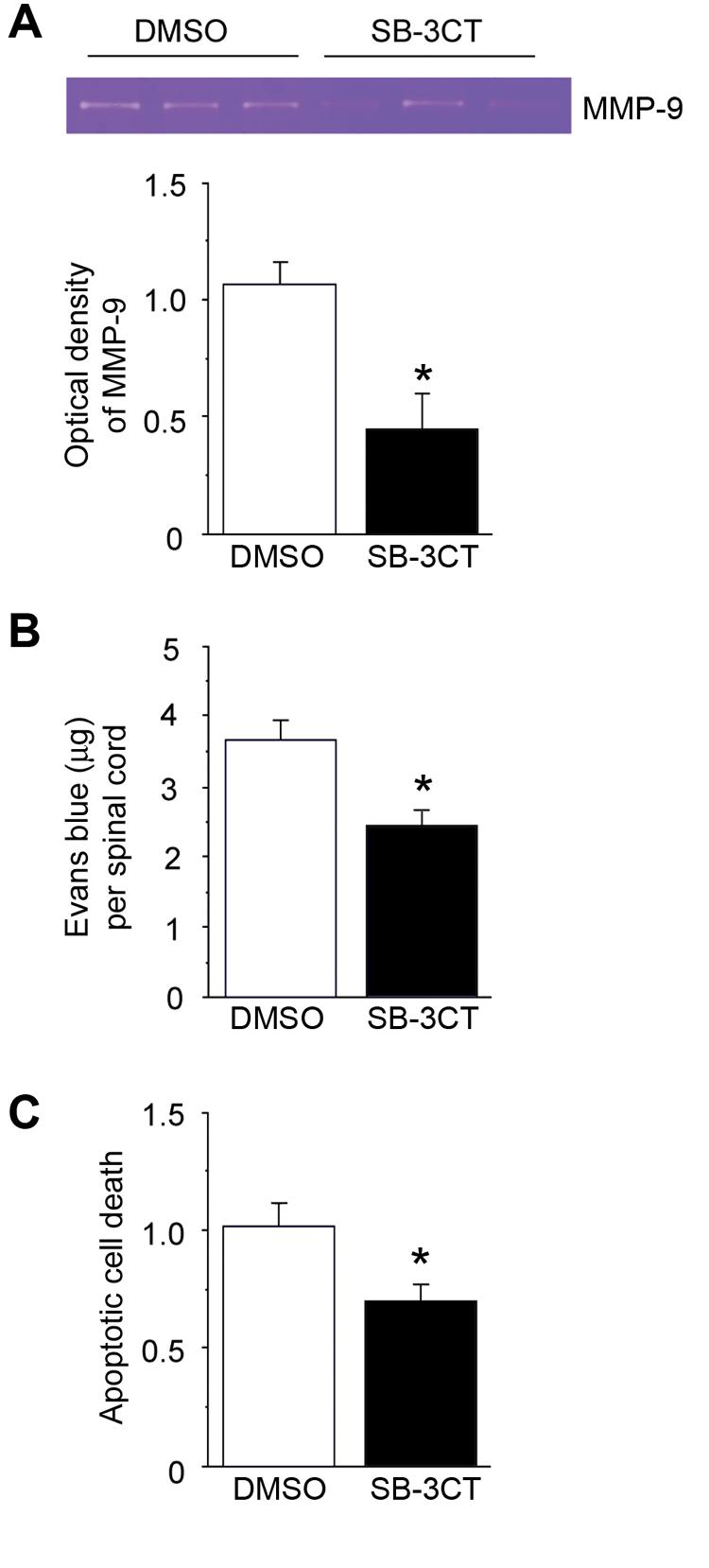
Gel zymography, BBB permeability study, and apoptotic cell death assay after intrathecal injection of SB-3CT 1 day after SCI. (A) Gel zymography showed that MMP-9 activity decreased in the group of rats with intrathecal injection of SB-3CT 1 day after SCI. A statistical study showed that there was a significant difference compared with the group subjected to an intrathecal injection of DMSO (n = 4, p < 0.05). (B) BBB permeability study showed that there was significantly less Evans blue extravasation in the group of rats injected with SB-3CT compared with the group injected with DMSO after SCI (n = 4, P < 0.05). (C) Cell death assay showed that the apoptotic cell death signal was significantly decreased in the rats subjected to intrathecal injection of SB-3CT, compared with the group injected with DMSO after SCI (n = 4, p < 0.05). *p < 0.05.
DISCUSSION
In the present study, we demonstrated the following: 1) MMP-9 activity increased 1 day after SCI and gradually decreased from 3 to 7 days after SCI in the Wt rats. MMP-9 expression was further confirmed by immunostaining and Western blot analysis. In situ gelatinolytic activity also increased in vessels and some cells after SCI. BBB disruption was evident from a study of Evans blue extravasation. 2) Double staining showed that in situ gelatinolytic activity colocalized with superoxide anion production. On a cellular level, it localized with endothelial cells, neutrophils, and neurons, but not with astrocytes. 3) Inhibitor studies showed that MMP activity, Evans blue extravasation, and apoptotic cell death decreased after intrathecal injection of SB-3CT. 4) MMP-9 expression and gelatinolytic activity, as well as Evans blue extravasation, were decreased in the SOD1 Tg rats compared with the Wt rats.
Microvascular endothelial permeability is related to occludin, focal adhesive bond, and activation of MMPs (Alexander and Elrod, 2002). Previous studies showed that increased activation of MMP-9 leads to BBB disruption after focal cerebral ischemia (Gasche et al., 2001; Rosenberg et al., 1998). A recent study provides direct evidence that MMPs open the BBB by degrading tight junction proteins (Yang et al., 2007). It has been shown in a humanized dynamic in vitro BBB model, which was based on an endothelial cell line, that BBB failure is related to proinflammatory factors (interleukin-6 and interleukin-1β) and MMP-9 (Cucullo et al., 2007). In another BBB model where endothelial cells grew on porous membranes with basement membrane matrix, increased MMP was linked to degradation of the basement membrane protein and BBB dysfunction (Haorah et al., 2007). It has also been shown that MMP-9 expression is increased and results in BBB disruption after SCI (Noble et al., 2002). We demonstrated a similar pattern of MMP-9 activity increase and BBB disruption in this study. We also showed the colocalization of MMP gelatinolytic activity with endothelial cells, which indicates the role of endothelial damage in BBB disruption.
Another report showed that decreased expression of MMP-9 in knockout mice reduces infarct size after cerebral ischemia (Asahi et al., 2001). Similarly, decreased expression of MMP-9 in MMP-9–null mice or treatment with an MMP inhibitor improves functional outcome after SCI (Noble et al., 2002). Inhibition of MMP-9 has been linked to attenuation of neuronal apoptosis and a decrease in infarct volume after cerebral ischemia (Gu et al., 2005). We showed in this study that inhibition of MMP-9 expression with the specific inhibitor SB-3CT decreased BBB disruption and apoptosis. Active MMP-9 has been linked to neuronal apoptosis by disruption of supporting extracellular matrix in a cell culture study and a 3-nitropropionic acid injury model (Gu et al., 2002; Kim et al., 2003). We demonstrated that MMP gelatinolytic activity colocalized with neurons and speculate that MMP-9 activation and endothelial damage contribute to neuronal apoptosis by disruption of the BBB and surrounding environment of neurons after SCI.
Neutrophils begin to appear in the lesion site at 4-6 h, peak at 12-24 h, and disappear within 5 days after SCI in the rat (Taoka et al., 1997). Neutrophils also infiltrate the lesion site in human SCI patients (Fleming et al., 2006). Treatment with the MMP inhibitor decreases the ability of monocytes to migrate through endothelium (Reijerkerk et al., 2006). It has been demonstrated that MMP-mediated endothelial change is partly responsible for dendrite cell migration through endothelium (Zozulya et al., 2007). Neutrophils could be beneficial for tissue repair, but they can also cause tissue damage if there is overwhelming neutrophil infiltration (Taoka et al., 1997). We demonstrated that gelatinolytic activity-positive neutrophils present in the lesion site after SCI and suggest that neutrophil infiltration after BBB disruption may contribute to tissue damage.
It has been reported that low concentrations of ROS, like superoxide anions and hydrogen peroxide, can activate pro-MMPs by oxidation of a sulfide bond (Rajagopalan et al., 1996). Previous studies provided evidence that superoxide anion overproduction contributes to MMP-9 activation and BBB disruption after cerebral ischemia (Gasche et al., 2001; Kamada et al., 2007). We showed that gelatinolytic activity colocalized with superoxide anion production, which is further evidence that ROS may cause an increase in gelatinolytic activity. ROS can damage cellular macromolecules leading to cell death after central nervous system injury (Chan, 2001). Overexpression of SOD1 in Tg rodents decreases oxidative stress and further decreases neuronal death after central nervous system injury (Chan, 2001). Decreased oxidative stress in SOD1 Tg rodents attenuates MMP activation after cold injury-induced brain trauma, 3-nitropropionic acid injury, and focal cerebral ischemia (Kamada et al., 2007; Kim et al., 2003; Morita-Fujimura et al., 2000). In contrast, decreased expression of SOD1 in knockout mice results in more intense MMP-9 expression and gelatinolytic activity after cerebral ischemia (Gasche et al., 2001). Similarly, knockout mice with 50% manganese-superoxide dismutase activity demonstrated delayed BBB breakdown associated with MMP activation and brain hemorrhage while it was absent in their Wt littermates (Maier et al., 2006). One report showed that oxidative stress increases after SCI (Azbill et al., 1997). Overexpression of SOD1 in Tg rats decreases oxidative stress and is neuroprotective after SCI (Sugawara et al., 2002). Here we demonstrated that overexpression of SOD1 in Tg rats attenuates MMP-9 activation and BBB disruption after SCI, which may further decrease tissue and neuronal damage.
We found in our zymography study that MMP-2 and active MMP-2 increased 1 day after SCI and continued to increase until 7 days after SCI. In the SOD1 Tg rats, there was a similar pattern, but with a significantly greater increase compared with the Wt rats 3 and 7 days after SCI. However, the cellular source of MMP-2 expression is unclear. Since microglia/macrophages are also activated at these time points after SCI, it is tempting to suggest that these inflammatory cells may be a source of MMP-2 overexpression. However, more work is needed to confirm this speculation. It has been shown that MMP-2 is upregulated 7 to 14 days after SCI and contributes to wound healing and functional recovery after SCI (Hsu et al., 2006). Also, delayed upregulation of MMP-9 7 to 14 days after cerebral ischemia has been shown to play a beneficial role (Zhao et al., 2006). Thus, the neuroprotective role and repair mechanisms of MMPs (MMP-9 and -2) at later time points after SCI warrant further studies.
CONCLUSION
We conclude that increased oxidative stress after SCI leads to MMP-9 upregulation, BBB disruption, and apoptosis, and that overexpression of SOD1 in Tg rats decreases oxidative stress and further attenuates MMP-9–mediated BBB breakdown.
ACKNOWLEDGMENTS
We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for assistance with figure preparation. This work was supported by National Institutes of Health grants P50 NS14543, RO1 NS25372, RO1 NS36147, and RO1 NS38653.
REFERENCES
- ALEXANDER JS, ELROD JW. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J. Anat. 2002;200:561–574. doi: 10.1046/j.1469-7580.2002.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASAHI M, WANG X, MORI T, SUMII T, JUNG J-C, MOSKOWITZ MA, FINI ME, LO EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZBILL RD, MU X, BRUCE-KELLER AJ, MATTSON MP, SPRINGER JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- BINDOKAS VP, JORDÁN J, LEE CC, MILLER RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONFOCO E, KRAINC D, ANKARCRONA M, NICOTERA P, LIPTON SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- CHAN PH, KAWASE M, MURAKAMI K, CHEN SF, LI Y, CALAGUI B, REOLA L, CARLSON E, EPSTEIN CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J. Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUCULLO L, COURAUD P-O, WEKSLER B, ROMERO I-A, HOSSAIN M, RAPP E, JANIGRO D. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600525. [Epub ahead of print. doi:10.1038/sj.jcbfm.9600525] [DOI] [PubMed] [Google Scholar]
- DEEM TL, COOK-MILLS JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEMING JC, NORENBERG MD, RAMSAY DA, DEKABAN GA, MARCILLO AE, SAENZ AD, PASQUALE-STYLES M, DIETRICH WD, WEAVER LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- GASCHE Y, COPIN J-C, SUGAWARA T, FUJIMURA M, CHAN PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- GU Z, CUI J, BROWN S, FRIDMAN R, MOBASHERY S, STRONGIN AY, LIPTON SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J. Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU Z, KAUL M, YAN B, KRIDEL SJ, CUI J, STRONGIN A, SMITH JW, LIDDINGTON RC, LIPTON SA. S-Nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- HAORAH J, RAMIREZ SH, SCHALL K, SMITH D, PANDYA R, PERSIDSKY Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood–brain barrier dysfunction. J. Neurochem. 2007;101:566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- HSU J-YC, MCKEON R, GOUSSEV S, WERB Z, LEE J-U, TRIVEDI A, NOBLE-HAEUSSLEIN LJ. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J. Neurosci. 2006;26:9841–9850. doi: 10.1523/JNEUROSCI.1993-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMADA H, YU F, NITO C, CHAN PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats. Relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM GW, GASCHE Y, GRZESCHIK S, COPIN J-C, MAIER CM, CHAN PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J. Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAIER CM, HSIEH L, CRANDALL T, NARASIMHAN P, CHAN PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann. Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- MORITA-FUJIMURA Y, FUJIMURA M, GASCHE Y, COPIN J-C, CHAN PH. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J. Cereb. Blood Flow Metab. 2000;20:130–138. doi: 10.1097/00004647-200001000-00017. [DOI] [PubMed] [Google Scholar]
- NOBLE LJ, DONOVAN F, IGARASHI T, GOUSSEV S, WERB Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J. Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUSTOVRH MC, JAWERBAUM A, CAPOBIANCO E, WHITE V, MARTINEZ N, LOPEZ-COSTA JJ, GONZALEZ E. Oxidative stress promotes the increase of matrix metalloproteinases-2 and -9 activities in the feto-placental unit of diabetic rats. Free Radic. Res. 2005;39:1285–1293. doi: 10.1080/10715760500188796. [DOI] [PubMed] [Google Scholar]
- RAJAGOPALAN S, MENG XP, RAMASAMY S, HARRISON DG, GALIS ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. J. Clin. Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REIJERKERK A, KOOIJ G, VAN DER POL SMA, KHAZEN S, DIJKSTRA CD, DE VRIES HE. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. FASEB J. 2006;20:E1901–E1909. doi: 10.1096/fj.06-6099fje. [DOI] [PubMed] [Google Scholar]
- ROSENBERG GA, ESTRADA EY, DENCOFF JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- SUGAWARA T, LEWÉN A, GASCHE Y, YU F, CHAN PH. Overexpression of SOD1 protects vulnerable motor neurons after spinal cord injury by attenuating mitochondrial cytochrome c release. FASEB J. 2002;16:1997–1999. doi: 10.1096/fj.02-0251fje. express article 10.1096/fj.02-0251fje; summary: FASEB J. [DOI] [PubMed] [Google Scholar]
- TAOKA Y, OKAJIMA K, UCHIBA M, MURAKAMI K, KUSHIMOTO S, JOHNO M, NARUO M, OKABE H, TAKATSUKI K. Role of neutrophils in spinal cord injury in the rat. Neuroscience. 1997;79:1177–1182. doi: 10.1016/s0306-4522(97)00011-0. [DOI] [PubMed] [Google Scholar]
- YANG Y, ESTRADA EY, THOMPSON JF, LIU W, ROSENBERG GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- ZHAO B-Q, WANG S, KIM H-Y, STORRIE H, ROSEN BR, MOONEY DJ, WANG X, LO EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- ZOZULYA AL, REINKE E, BAIU DC, KARMAN J, SANDOR M, FABRY Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1α chemokine and matrix metalloproteinases. J. Immunol. 2007;178:520–529. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]