

Abstract
Understanding the mechanism for sucrose-induced protein stabilization is important in many diverse fields, ranging from biochemistry and environmental physiology to pharmaceutical science. Timasheff and Lee [Lee, J. C. & Timasheff, S. N. (1981) J. Biol. Chem. 256, 7193–7201] have established that thermodynamic stabilization of proteins by sucrose is due to preferential exclusion of the sugar from the protein’s surface, which increases protein chemical potential. The current study measures the preferential exclusion of 1 M sucrose from a protein drug, recombinant interleukin 1 receptor antagonist (rhIL-1ra). It is proposed that the degree of preferential exclusion and increase in chemical potential are directly proportional to the protein surface area and that, hence, the system will favor the protein state with the smallest surface area. This mechanism explains the observed sucrose-induced restriction of rhIL-1ra conformational fluctuations, which were studied by hydrogen–deuterium exchange and cysteine reactivity measurements. Furthermore, infrared spectroscopy of rhlL-1ra suggested that a more ordered native conformation is induced by sucrose. Electron paramagnetic resonance spectroscopy demonstrated that in the presence of sucrose, spin-labeled cysteine 116 becomes more buried in the protein’s interior and that the hydrodynamic diameter of the protein is reduced. The preferential exclusion of sucrose from the protein and the resulting shift in the equilibrium between protein states toward the most compact conformation account for sucrose-induced effects on rhIL-1ra.
Understanding sucrose interactions with proteins and their consequences on structural stability is critical for many research areas, as evidenced by the numerous effects documented, and the various terms applied to the sugar, by different disciplines. For example, sucrose has long been known in biochemistry as a “solute” that enhances thermal stability of the native protein (1–3). This effect has been exploited in pharmaceutical science, where sucrose has been used as a protective “excipient” during high-temperature processing steps (4). In environmental physiology, sucrose is known as an “osmolyte” and “compatible solute” because it protects organisms against osmotic stress (5). In cryobiology, sucrose is referred to as a “cryoprotectant” because it protects proteins during freezing (6).
Fortunately, a single thermodynamic mechanism, developed by Timasheff and colleagues (for a review, see ref. 3), can explain all of these stabilizing effects of sucrose in aqueous solution. Lee and Timasheff (2) found that sucrose is preferentially excluded from the surface of proteins, which increases protein chemical potential. The degree of preferential exclusion and the increase in chemical potential are directly proportional to the surface area of protein exposed to solvent (2). By the LeChatelier Principle, the system will minimize the thermodynamically unfavorable effect of preferential sucrose exclusion by favoring the state with the smallest surface area. Furthermore, the corresponding shift in the equilibrium toward the native state can be ascribed to the Wyman relationship between ligand binding and state equilibria (3). For example, for the equilibrium between native and denatured states, the increase in protein chemical potential is greatest for the denatured state, which has a greater surface area (2). Hence, with preferentially excluded sucrose the free energy of denaturation is increased and the native state is stabilized.
Based on the Timasheff mechanism, sucrose should also favor the most compact protein conformation, even under nondenaturing conditions. It is well documented with hydrogen exchange studies that the native conformation is flexible and does not exist as discreet, single structure (7–10). Rather, fluctuations from the most compact form of the protein will transiently expose portions of the protein backbone to solvent. Again by the LeChatelier Principle these fluctuations should be more unfavorable in preferentially excluded cosolvents than in water. In fact, Bolen and colleagues (9) documented that dynamic structural fluctuations of ribonuclease, which lead to transient increase in protein surface area and allow hydrogen–deuterium exchange, are attenuated in the presence of sucrose. They attribute this effect to the known preferential exclusion of sucrose from ribonuclease (2) and the concomitant shift in the equilibrium toward the state with smallest surface area, and to the unfavorability of exposing the protein backbone to sucrose (11). Sucrose-mediated restriction of conformational mobility would be expected for any protein, but this hypothesis has not been tested.
Finally, it is also important to consider the potential effects of sucrose exclusion on the “static” (e.g., that which would be measured with a spectroscopic method) conformation of the native protein. As assessed by CD spectroscopy, the native conformations of α-chymotrypsin, chymotrypsinogen, and tubulin were not altered in the presence of preferentially excluded sucrose (2, 12). A small change in the spectra of ribonuclease in sucrose was noted, but it was not indicative of any major structural change (2). Most likely sucrose does not alter the structure of these proteins, because the native conformation, in the absence of sucrose, is already representative of the most compact conformation. In other words, there is not a detectable capacity for further structural ordering and increased packing density of residues.
However, theoretically, if a protein’s native structure has the capacity for further ordering and a concomitant reduction in surface area, then this effect should be manifested in the presence of sucrose. We reported previously (13) that the infrared spectrum of rhIL-1ra in the conformationally sensitive amide I region exhibits narrowing of the dominant β-sheet band in the presence of 60% (wt/vol) sucrose. We suggested that this change reflected an increase in the structural order of the protein and that this effect could be due to the preferential exclusion of sucrose from the protein (13).
The purpose of the current study is to address more rigorously the mechanistic link between sucrose effects on equilibria between states, conformational dynamics, and structure of the native conformation by: (i) measuring directly the preferential interaction of sucrose with rhIL-1ra; (ii) measuring the effect of sucrose on the H–D exchange and cysteine reactivity of rhIL-1ra; and (iii) addressing in more detail, with both infrared and electron paramagnetic resonance spectroscopies, the apparent increased ordering of the native structure of rhIL-1ra in the presence of sucrose.
MATERIALS AND METHODS
Protein and Reagents.
Pharmaceutical quality rhIL-1ra was produced and purified at Amgen Biologicals. The protein was greater than 99.5% homogeneity based on size-exclusion chromatography and approximately 98% pure based on cation-exchange chromatography. Sucrose was purchased from Pfanstiehl Laboratories, and other chemicals were purchased from Sigma. All chemicals were of reagent grade or higher quality.
Formulation Buffer and Sucrose Solutions.
The formulation buffer was 140 mM NaCl/10 mM sodium citrate, pH 6.5, at room temperature. All sucrose solutions were prepared to achieve the same final concentrations of buffer components as in the formulation buffer and were adjusted to pH 6.5.
Preferential Interaction Measurements.
The preferential interaction parameter, (∂m3/∂m2)T,μ1,μ3, where mi is the molal concentration of component i, and μi is the chemical potential of component i, was determined at 1 M sucrose by high-precision densimetry, according to previously established methods (1, 2, 14). Components are defined as: water, component 1; protein, component 2; sucrose, component 3 (1, 2, 14). A Precision Density Meter DMA 602 (Anton Parr, Gratz, Austria) was used to measure the densities of the solvents and protein solutions. All measurements were done at 25°C. The partial specific volume of sucrose in a 1 M sucrose solution was taken from ref. 2. The apparent partial specific volume, φ, of rhIL-1ra was calculated from density measurements as a function of protein concentration, under conditions of constant molality and constant chemical potential of sucrose. Constant chemical potential was attained by exhaustive dialysis of rhIL-1ra against 1 M sucrose in the formulation buffer. Irreversible aggregation upon lyophilization of rhIL-1ra in the absence of sucrose precluded measurement of the isomolal φ2 in the usual way, in which a dried protein is rehydrated in the various molal concentrations of sucrose (2). Instead, φ2 (isomolal 1 M sucrose) was approximated by substituting φ2 obtained in the absence of sucrose. This type of approximation should not lead to large errors in the calculation of the preferential interaction parameter, because in most cases φ2 shows no dependence on sucrose concentration (see ref. 2). Calculations of (∂g3/∂g2)T,μ1,μ3, (∂m3/∂m2)T,μ1,μ3, and (∂μ2/∂m3)T,P,m2, where gi is the weight concentration of component i, were made using the approaches and sucrose data given in (2).
The calculated interaction parameter based on sucrose-induced surface free energy perturbations was determined from a form of the Gibbs adsorption isotherm (2),
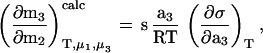 |
1 |
as previously described (1, 2, 15). An interpolated value of rhIL-1ra’s surface area, s, was obtained based on a plot of surface area vs. molecular weight for several globular proteins (data taken from ref. 1). The surface tension/sucrose activity increment, (∂σ/∂a3)T, was taken from (2).
Extinction Coefficient Measurements.
The extinction coefficient of 1 mg/ml rhIL-1ra at 280 nm was determined in the presence of various sucrose concentrations by the method of Lee and Timasheff (14). Three separate protein solutions were prepared for each sucrose concentration. The absorbencies were corrected for light scattering based on the method of Leach and Scheraga (16). Values (mean ± SD, n = 3) were 0.770, 0.774 ± 0.002, 0.775 ± 0.003, 0.782 ± 0.002, and 0.783 ± 0.003 ml/(mg⋅cm) for rhIL-1ra in 0, 10, 20, 30, 40, and 50% wt/vol sucrose solutions, respectively.
Dynamic Light Scattering.
The measurement of the preferential interaction parameter assumes the system is at thermodynamic equilibrium (14). Therefore, it first was necessary to determine whether solution conditions might cause polymerization of rhIL-1ra, over the hours-long time scale needed to reach dialysis equilibrium and make the densimetry measurements. The hydrodynamic radius of rhIL-1ra was measured by dynamic light scattering on a DynaPro-801 (Protein Solutions, Charlottesville, VA) molecular size detector, with appropriate viscosity and refractive index corrections (17). The protein was found to be monomeric at sucrose concentrations of 0 and 50% wt/vol (data not shown), and the respective calculated hydrodynamic radii were 2.42 ± 0.2 and 2.09 ± 0.4 nm (mean ± SD) (17).
Infrared Spectroscopy.
A 200-mg/ml rhIL-1ra solution was diluted with the appropriate sucrose solution buffer to achieve the desired final sucrose concentration and a 50-mg/ml final protein concentration. Identical aliquots of the formulation buffer were diluted similarly with the stock sucrose solutions for use as buffer blanks. IR spectra were recorded at 25°C with a Nicolet Magna 550 Fourier transform IR spectrometer equipped with a deuterated tungsten gallium selenium detector as previously described in ref. 18. For each spectrum, a 256-scan interferogram was collected in single-beam mode, with a 4 cm−1 resolution. The spectra for the appropriate buffer blank and gaseous water were subtracted from the protein spectra, according to previously established criteria (19). The final protein spectra were smoothed with a 7-point function to remove white noise. Second derivative spectra were calculated with Nicolet omnic software. All second-derivative spectra were baseline-corrected, using Galactic’s grams 386 software, based on the method of Dong and Caughey (19), and area-normalized under the second-derivative amide I region, 1600–1700 cm−1 (20).
Hydrogen–Deuterium (H–D) Exchange.
Deuterated formulation buffer and stock sucrose solutions were prepared by lyophilizing (Labconco Freeze Dryer 4.5, Kansas City, MO) solutions and rehydrating them to the original volumes with D2O at least three times. Between lyophilization cycles, the samples were freeze-thawed at least three times to promote H–D exchange. Fifty microliters of rhIL-1ra (200 mg/ml) was added to 150 μl deuterated formulation buffer or deuterated sucrose solution at time = 0 min. The solution was placed in an IR cell (15-μm spacer), which was thermostated at 25°C. From time = 0, the above process took ca. 3 min. The macro function of Omnic (Nicolet) was used to collect 20 interferograms at various time points. Spectra were processed as described in the IR spectroscopy methods. Additional processing included area-normalizing amide I peaks to account for slight path-length differences in the IR cell between samples.
Electron Paramagnetic Resonance Spectroscopy.
The protein was spin-labeled with the nitroxide radical 4-maleimido-2,2,6,6-tetramethyl-1-piperidinyloxy. The reaction mixture contained a 1:1 mole ratio of spin label to rhIL-1ra (rhIL-1ra concentration = 50 mg/ml). The reaction was performed in 50% (wt/vol) sucrose buffer solution, which minimized irreversible aggregation losses during the labeling and resulted in labeling of only cysteine 116, based on tryptic peptide mapping and mass spectroscopy (see below). The reaction proceeded for 19 hr and was quenched by exhaustive dialysis against the formulation buffer without sucrose. The dialyzed protein sample was concentrated to 65 mg/ml with a Centricon-10 (Beverly, MA) centrifuge concentrator. The sample was then diluted with buffer alone or buffer containing sucrose to achieve desired final sucrose concentrations and a 16.3-mg/ml protein concentration.
X-band EPR spectra of samples were recorded on a Bruker ESP 300 spectrometer (Billerica, MA) at a field modulation of 100 kHz. The spectrometer settings were maintained at a modulation amplitude of 1.0 Gauss, a scan width of 150 Gauss divided into 1,024 intervals, and a frequency of approximately 9.75 GHz. Microwave power was set at 10 mW. Thirty-two consecutive scans were co-added. Digitized spectra were analyzed on a Hewlett–Packard series 755 workstation, using a spectral fitting program (21), based on EPR simulation code (22). The nitrogen hyperfine coupling constant, An, was obtained directly from the fitting routine (21). For a measure of rotational dynamics, the protein’s hydrodynamic diameter, Dh, was calculated from measured values of rotational diffusivity, Drot, also obtained from the spectral fitting routine (21), and the Debye–Stokes–Einstein formula for isotropic spherical rotation under no-slip boundary conditions: DH = 3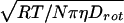 (23), where R is the gas constant, T is temperature (K), N is Avogadro’s number, and η is viscosity.
(23), where R is the gas constant, T is temperature (K), N is Avogadro’s number, and η is viscosity.
CD Spectroscopy.
CD (Aviv 62DS) spectra were collected by co-adding 15 scans in the near and far UV regions using 0.01 and 0.001 cm path-length cells, respectively. Protein concentration was 15 mg/ml, and measurements were performed at 25°C in a thermostated cell holder.
Reactivity of rhIL-1ra Cysteines.
The reaction of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NbdCl, stock concentration in 100% ethanol = 16.4 mg/ml) with free thiol groups of rhIL-1ra (final concentration = 0.085 mg/ml) was initiated by pipetting 3 μl of the stock NbdCl into 1 ml of the appropriate reaction solution. The reaction was followed by monitoring absorbance at 420 nm over time as described by Yancey and Somero (24). As a control for the potential direct effect of sucrose on thiol reactivity, the rates of reaction of NbdCl with glutathione (final concentration = 1.5 μg/ml) were determined as a function of sucrose concentration. In addition, for all reactions, the absorbance change noted with just the appropriate sucrose solution was used to correct the sample absorbencies obtained at each time point.
To determine the extent of labeling of the various cysteine residues in rhIL-1ra, identical reactions were carried out separately, for 4 hr, quenched with 100-fold mole ratio excess of glutathione relative to NbdCl, dialyzed exhaustively against the formulation buffer, and analyzed with tryptic peptide mapping, as described below.
Identification of Labeled Cysteine.
Tryptic mapping of 4-maleimido-2,2,6,6-tetramethyl-1-piperidinyloxy-labelled rhIL-1ra was carried out as previously described (25). Peptides containing the labeled cysteine were further digested with Glu-C in 25 mM ammonium carbonate (pH 7.8) for 18 hr at 25°C. The digested sample was analyzed by reversed-phase HPLC method using a Vydac C4 column. Mass spectrometry was performed to confirm the identity of the labeled peptides by using a Kompact matrix-assisted laser desorption isotherm–time of flight mass spectrometer calibrated with gentisic acid matrix and substance P. The mass determination was done by the addition of 0.2 μg of the rhIL-1ra digest to the slide, which was allowed to dry, followed by the addition of 0.5 μl of gentisic acid. N-terminal sequence analysis was done as previously described (26) to identify the position of labeled cysteine.
RESULTS AND DISCUSSION
Thermodynamic Interaction of Sucrose with rhIL-1ra.
Sucrose has been shown to be excluded preferentially from the surface of α-chymotrypsin, chymotrypsinogen, ribonuclease, and tubulin and is expected to have the same interaction with most proteins (2, 27, 28). However, to invoke rigorously the consequences of preferential sucrose exclusion on stability of a given protein, it is necessary to make the interaction measurements for that protein.
The preferential interaction of 1 M sucrose in terms of weight concentrations is (∂g3/∂g2)T,μ1,μ3 = −0.164 (g sucrose/g rhIL-1ra), documenting that sucrose is preferentially excluded from the surface of rhIL-1ra. This value is comparable to the values determined in 1 M sucrose for chymotrypsinogen A (−0.138 g/g), α-chymotrypsin (−0.104 g/g), and ribonuclease (−0.190 g/g) (2). The preferential interaction parameter on a mole basis (3) is (∂m3/∂m2)T,μ1,μ3 = −8.290 (mol sucrose/mol rhIL-1ra). Furthermore, in terms of chemical potential perturbation it becomes (∂μ2/∂m3)T,P,m2 = 19.4 [kJ/(mol rhIL-1ra)/(mol sucrose)]. The preferential exclusion of sucrose and the increased protein chemical potential indicate that any state of the protein that has an increased surface area should be thermodynamically less favorable than more compact states.
A critical assumption for this conclusion is that the degree of preferential exclusion varies directly with protein surface area and is not altered by changes in the chemical properties of the exposed surface (e.g., exposure of hydrophobic residues). In support of this assumption, preferential exclusion of sucrose from α-chymotrypsin, chymotrypsinogen, ribonuclease, and tubulin has been shown to be due to the increase in the surface tension of water by sucrose and not to any specific property of individual proteins (1–3, 15, 28). Furthermore, it is primarily the unfavorable interactions of the peptide backbone, and not side chains, with sucrose that gives rise to the increased protein chemical potential in sucrose solutions (11).
If the surface tension effect is also the dominant factor in preferential exclusion of sucrose from rhIL-1ra, then the preferential interaction parameter approximated from the Gibbs adsorption isotherm (see Materials and Methods) should agree well with the experimentally determined value. For this calculation, it must be assumed that the surface tension increment of sucrose for the air–water interface is roughly equivalent to that at the protein–water interface (2). Furthermore, the surface area of rhIL-1ra must be approximated, based on its molecular weight (see Materials and Methods). Given these assumptions (2), the calculated value of (∂m3/∂m2)T,μ1,μ3calc. = −10.9 (mol sucrose/mol rhIL-1ra) agrees reasonably well with the experimental value of −8.3 (mol sucrose/mol rhIL-1ra). Thus, it appears that the exclusion of sucrose from the surface of rhIL-1ra is due to the increase of water surface tension by sucrose and should vary directly with protein surface area. Furthermore, the stabilizing effect of sucrose may be related to the surface free energy of cavity formation in the solvent. Because it takes free energy to expand the cavity occupied by rhIL-1ra in the presence of sucrose, the transition to a more expanded state is more unfavorable in sucrose than in water (2).
This mechanism has also been invoked for sucrose-induced inhibition of certain irreversible denaturation processes. For example, sucrose slowed the irreversible loss of lactate dehydrogenase activity under denaturing conditions of acidic pH (29). An intermediate state with a volume approximately 4% greater than, and in equilibrium with, the native state was on the inactivation pathway. Sucrose slowed irreversible inactivation because the expanded state was more thermodynamically unfavorable than the native state. Similarly, for stabilization of rhIL-1ra by sucrose under nondenaturing conditions, it has been proposed that the formation of an intermediate state on the degradation pathway is less favorable in sucrose than in water (25). The intermediate state is thought to have a larger volume than, and to be in equilibrium with, the native state. Thus, preferential exclusion of sucrose from rhIL-1ra should make formation of the intermediate state less favorable and stabilize the native protein.
Hydrogen Exchange Kinetics and Cysteine Reactivity.
To determine the effects of sucrose on the conformational dynamics of rhIL-1ra, we first used infrared spectroscopy to study H–D exchange kinetics in the presence of 0, 34.2% (1 M), and 50% (wt/vol) sucrose. Typically, exchange is initiated by dissolving a lyophilized protein directly into a D2O buffer (8). However, many proteins, including rhIL-1ra, are unfolded in the dried solid (13, 30). Thus, residues that would normally be buried in the protein’s interior are exposed. During rehydration and refolding, H–D exchange may proceed more rapidly than expected for a native protein. To avoid this potential complication, H–D exchange was initiated by combining a 200-mg/ml rhIL-1ra solution in the H2O formulation buffer with the appropriate D2O buffer (with or without deuterated sucrose), to give a final protein concentration of 50 mg/ml. The potential direct effects of sucrose on the H–D exchange reaction have been examined by Wang et al. (9), who found that exchange kinetics for the model peptide poly-d,l-alanine, are not affected by 1 M sucrose.
The amide II infrared band for proteins is predominantly due to N–H stretching and, hence, is very sensitive to H–D exchange (8). As exchange of rhIL-1ra proceeds with time, absorbance is shifted from around 1,550 cm−1 to around 1,450 cm−1, which is the position assigned to amide II′ (Fig. 1 Inset). The rate of H–D exchange is reflected in the change in the amide II′/II peak height ratio versus time (Fig. 1). By visual inspection of these plots (Fig. 1), it is apparent that the exchange rate is dramatically reduced by increasing sucrose concentrations, suggesting that sucrose inhibits fluctuations that increase solvent accessibility of the protein backbone.
Figure 1.

Amide II′/II peak height ratio as a function of time of deuteration for rhIL-1ra in 0 M (circles), 1 M (squares), and 50% wt/vol (triangles) sucrose. (Inset) Representative IR absorbance spectra of rhIL-1ra at various times during deuteration (arrows represent direction of peak increase/decrease over time).
Deuteration of proteins also affects the component bands of the amide I infrared absorbance (18). By following specific bands in the second derivative amide I region (Figs. 2), the solvent accessibility of different regions of the protein can be inferred (18). The rate of change in the peak height of the β-sheet band at 1,627 cm−1 is rapid relative to that for the turn band at 1,678 cm−1. This result suggests that the amide groups corresponding to the 1,627 cm−1 frequency are relatively more solvent exposed than those corresponding the 1,678 cm−1 band, most likely because the latter groups are more deeply buried in the protein’s interior.
Figure 2.

Second derivative peak value: 1,627 cm−1 (a); 1,678 cm−1 (b). d2I/dx2 (I is IR absorbance; x is wave number cm−1) is plotted as a function of time of deuteration. Symbols are the same as in Fig. 1. (Insets) Representative second-derivative IR absorbance spectra of rhIL-1ra at various times of the deuteration reaction (arrows represent direction of peak increase/decrease over time).
Large-scale structural changes are needed to allow H–D exchange of residues deep in the protein’s interior relative to those needed for exchange of residues closer to the surface (9). Because the large-scale structural transitions should increase protein surface more, these should be most sensitive to attenuation by sucrose. Thus, sucrose should have a greater effect on exchange kinetics for the H–D kinetics monitored at the 1,678 cm−1 than those noted with the 1,627 cm−1 band. This is indeed the case (Fig. 2).
Next we employed a complementary method to study further the effects of sucrose on rhIL-1ra conformational mobility. The native protein contains four free cysteine residues and no disulfide bonds (25). Reactivity of thiol groups with NbdCl is due to increased solvent accessibility around the reactive cysteines, when the protein is expanded from the native state (24, 31). The overall rate of cysteine reactivity with NbdCl was determined by monitoring absorbance at 420 nm as a function of time for 90 min. The slopes of the linear plots (R2 > 0.99 in all cases) are 8.8, 7.0, and 3.6 a.u./min × 10−4 in 0, 10%, and 50% wt/vol sucrose, respectively. The largest standard error for the rate measurements is 0.5 × 10−4 a.u./min (n = 3). Thus, sucrose significantly reduces cysteine reactivity. As a control for the direct effects of sucrose on reactivity of NbdCl, the reaction of the reagent with glutathione (GSH) was studied. Sucrose does not alter the reaction kinetics (data not shown). Thus, consistent with the H–D exchange results, sucrose appears to be attenuating the labeling of cysteine residues by making the structural fluctuations needed to expose the residues to solvent thermodynamically unfavorable (24, 31).
As an alternative method to spectroscopy, tryptic digest peptide mapping was performed on samples that were exposed to NbdCl (see Materials and Methods). Reaction of the cysteines in a given peptide with the reagent reduces the peak area in the native peptide position on the chromatogram, because the retention time of the reacted peptide is altered (data not shown). Peptide fragment T8, which contains residues 65–71 and two cysteines (25), is 3, 7, and 23% unreacted in 0, 10, and 50% (wt/vol) sucrose, respectively. For peptide fragment T13, which contains residues 114–145 and two cysteines (25), the unreacted amount is 4, 23, and 46% in 0, 10, and 50% sucrose, respectively. These results are consistent with the spectroscopic reaction NbdCl data and the H–D exchange data, all of which document that sucrose restricts the conformational mobility of rhIL-1ra.
Sucrose-Induced Ordering and Compaction of the Native Conformation.
To examine the effect of sucrose on the native “static” conformation, first we compared the effect of 0–50% (wt/vol) sucrose on the amide I region of the IR spectra of rhIL-1ra in H2O. In the presence of increasing amounts of sucrose, there is an increased narrowing of the dominant β-sheet band at 1,641 cm−1 and increased resolution of the β-sheet band at 1,630 cm−1 (Fig. 3). Band narrowing can be visualized more clearly by examining inverted second derivative spectra to which curves have been fit to the component bands (Fig. 4). Also, the degree of band narrowing at 1,641 cm−1 can be quantified by measuring the band width at half-height, which results in values of 17.1, 17.0, 16.8, 15.3, 15.0, and 13.7 cm−1, in 0, 10, 20, 30, 40, and 50% (wt/vol) sucrose, respectively.
Figure 3.

Second-derivative IR absorbance spectra of rhIL-1ra in various H2O sucrose solutions. Zero percent sucrose is indicated by solid line. (a) rhIL-1ra in 10 (dotted line) and 20% (dashed line) wt/vol sucrose. (b) rhIL-1ra in 30 (dotted line), 40 (dashed line), and 50% (dot–dashed line) wt/vol sucrose.
Figure 4.

Inverted second-derivative IR absorbance spectra with curve–fit component bands. (a) Zero percent sucrose. (b) Fifty percent wt/vol sucrose.
The sucrose-induced IR band narrowing arises because the dynamic distribution of the various β-sheet structural microenvironments is decreased; i.e., these structural elements become more homogeneous (32). This decreased dynamic distribution may reflect an increased packing density. Thus, we conclude that the relatively broad band noted at 1,641 cm−1 in 0% sucrose indicates some degree of heterogeneity in environment of the representative β-sheet structure, reflecting a capacity to have an increase in structural order induced by sucrose.
By assigning bands to secondary structural types and quantitating relative absorbencies of these bands with curve fitting (Fig. 4), the effects of sucrose on the overall secondary structural composition can be calculated with an approximate error of 5% (19, 33). The secondary structure of rhIL-1ra in the absence of sucrose is 66% β-sheet, 6% helix, and 28% β-turn. Even in the presence of 50% sucrose, where the most extreme changes in band widths are noted, the structure is 65% β-sheet, 8% helix, and 27% β-turn. Thus, the overall secondary structure is not altered by sucrose. Consistent with this finding, there are no detectable differences in near- and far-UV CD spectra of rhIL-1ra in the presence of up to 50% (wt/vol) sucrose (data not shown). Therefore, the increased ordering in protein structure is not due to any large-scale redistribution between secondary structural elements.
EPR spectroscopy was also used to investigate sucrose-induced conformational changes in native rhIL-1ra. Any conformational change that alters the local environment around a spin-labeled protein residue will be reflected in an altered EPR spectrum (34). With our protocol for attaching the spin label, only cysteine 116 is labeled (see Materials and Methods). Thus, our EPR experiments report structural information about this single residue, which is located near the surface of the protein (35).
EPR spectra for spin-labeled proteins reflect both local polarity of the environment around the spin label as well as the spin label’s rotational dynamics (34). For a covalently attached spin label located well away from the end of the polypeptide, as is the case here, the label is strongly immobilized. Thus, the rotational dynamics of the spin label reflect the dynamics of the protein as a whole and provide a measure of the protein’s hydrodynamic size. A measure of the local polarity surrounding the spin probe is the nitrogen hyperfine-coupling constant, AN, which varies directly with environmental polarity.
As shown in Fig. 5, AN decreases gradually with increasing concentrations of sucrose. Thus, the spin label at cysteine 116 on rhIL-1ra is exposed to a more hydrophobic environment, most likely because the spin label becomes progressively more buried in the protein’s interior. Consistent with this interpretation, the overall changes in the spectrum noted in the presence of 50% (wt/vol) sucrose relative to that noted in the absence of sugar (Fig. 5 Inset) visually correlate well with those noted when a spin label becomes increasingly buried in the protein’s structure (34).
Figure 5.

Results of EPR spectral simulation in terms of the apparent hydrodynamic diameter, DH,apparent (solid circles), of labeled rhIL-1ra and the nitrogen hyperfine-coupling constant, AN (open triangles). (Inset) First-derivative EPR spectra (arbitrary units) of rhIL-1ra in 0 (solid line) and 50% wt/vol (dotted line) sucrose. Width of spectra is 150 Gauss, centered at 3,472 Gauss.
In addition, the apparent hydrodynamic diameter decreases gradually with increased sucrose concentration. The value decreases from 24 to 14 Å as sucrose concentration is increased from 0–50% (wt/vol). For reference, the estimated hydrodynamic diameter of rhIL-1ra, from an extrapolation based on molecular weight, is 18 Å. The magnitude of the sucrose-induced reduction in apparent hydrodynamic diameter reflects a qualitative trend and not an absolute change in the protein’s hydrodynamic diameter; i.e., there is not a 42% reduction in diameter in the presence of 50% sucrose. The effects of sucrose most likely are exaggerated because the Debye–Stokes–Einstein equation DH = 3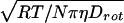 assumes an isotropic spherical rotation. A deviation toward a more rod-like shape results in a transition from a cube-root function to a square-root function for this equation (23). Thus, an increase in deviation from the spherical mode of rotation as a function of sucrose concentration would exaggerate the apparent reduction in hydrodynamic diameter, relative to the actual change induced by sucrose. The magnitude of this exaggeration is not known.
assumes an isotropic spherical rotation. A deviation toward a more rod-like shape results in a transition from a cube-root function to a square-root function for this equation (23). Thus, an increase in deviation from the spherical mode of rotation as a function of sucrose concentration would exaggerate the apparent reduction in hydrodynamic diameter, relative to the actual change induced by sucrose. The magnitude of this exaggeration is not known.
In conclusion, the EPR spectroscopic results are consistent with the sucrose-induced increased ordering of the protein structure noted with infrared spectroscopy, which occurs concomitant with a compaction of the native state.
CONCLUSIONS
Sucrose is preferentially excluded from the surface of rhIL-1ra. Thus, in the presence of sucrose, increases in protein surface area are more thermodynamically unfavorable than in water and the equilibrium between states is shifted toward that with the smallest surface area. These effects account for: (i) the inhibition H–D exchange and cysteine reactivity, which indicate restriction of the protein conformation mobility and (ii) an increase in the structural order and a compaction of the native state. The formation of a more compact and ordered native state could contribute to the inhibition of H–D exchange and cysteine reactivity. However, just the sucrose-induced shift in the equilibrium toward the native state (even if it has not been compacted) is all that is necessary for the thermodynamic effects of sucrose to restrict rhIL-1ra conformational mobility.
Acknowledgments
We gratefully acknowledge Shinichi Hara and Scott Lauren for peptide sequencing work. These studies were supported by National Science Foundation Grant BES9505301 and predoctoral fellowships for B.S.K. from the American Foundation for Pharmaceutical Education and the Colorado Institute for Research in Biotechnology.
References
- 1.Arakawa T, Timasheff S N. Biochemistry. 1982;21:6536–6544. doi: 10.1021/bi00268a033. [DOI] [PubMed] [Google Scholar]
- 2.Lee J C, Timasheff S N. J Biol Chem. 1981;256:7193–7201. [PubMed] [Google Scholar]
- 3.Timasheff S N. Annu Rev Biophys Biomol Struct. 1993;22:67–97. doi: 10.1146/annurev.bb.22.060193.000435. [DOI] [PubMed] [Google Scholar]
- 4.Vrkljan M, Foster T M, Powers M E, Henkin J, Porter W R, Staack H, Carpenter J F, Manning M C. Pharm Res. 1994;11:1004–1008. doi: 10.1023/a:1018935420680. [DOI] [PubMed] [Google Scholar]
- 5.Yancey P H, Clark M E, Hand S C, Bowlus R D, Somero G N. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter J F, Crowe J H. Cryobiology. 1988;25:244–255. doi: 10.1016/0011-2240(88)90032-6. [DOI] [PubMed] [Google Scholar]
- 7.Friedman J M. Methods Enzymol. 1994;232:205–231. doi: 10.1016/0076-6879(94)32049-7. [DOI] [PubMed] [Google Scholar]
- 8.Barksdale A D, Rosenberg A. Methods Biochem Anal. 1983;28:1–113. doi: 10.1002/9780470110485.ch1. [DOI] [PubMed] [Google Scholar]
- 9.Wang A, Robertson A D, Bolen D W. Biochemistry. 1995;34:15096–15104. doi: 10.1021/bi00046a016. [DOI] [PubMed] [Google Scholar]
- 10.Englander S W, Calhoun D B, Englander J J, Kallenbach N R, Liem R K, Malin E L, Mandal C, Rogero J R. Biophys J. 1980;32:577–589. doi: 10.1016/S0006-3495(80)84991-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Bolen D W. Biochemistry. 1995;34:12884–12891. doi: 10.1021/bi00039a051. [DOI] [PubMed] [Google Scholar]
- 12.Lee J C, Timasheff S N. Biochemistry. 1975;14:5183–5187. doi: 10.1021/bi00694a025. [DOI] [PubMed] [Google Scholar]
- 13.Chang B S, Beauvais R M, Dong A, Carpenter J F. Arch Biochem Biophys. 1996;331:249–258. doi: 10.1006/abbi.1996.0305. [DOI] [PubMed] [Google Scholar]
- 14.Lee J C, Gekko K, Timasheff S N. Methods Enzymol. 1979;61:26–49. doi: 10.1016/0076-6879(79)61005-4. [DOI] [PubMed] [Google Scholar]
- 15.Lin T Y, Timasheff S N. Protein Sci. 1996;5:372–381. doi: 10.1002/pro.5560050222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leach S J, Scheraga H A. J Am Chem Soc. 1960;82:4790–4792. [Google Scholar]
- 17.Protein Solutions, I. dynapro-801win Operators Manual. Inc., Charlottesville, VA: Protein Solutions; 1996. [Google Scholar]
- 18.Dong A, Matsuura J, Allison S D, Chrisman E, Manning M C, Carpenter J F. Biochemistry. 1996;35:1450–1457. doi: 10.1021/bi9518104. [DOI] [PubMed] [Google Scholar]
- 19.Dong A, Caughey W S. Methods Enzymol. 1994;232:139–175. doi: 10.1016/0076-6879(94)32047-0. [DOI] [PubMed] [Google Scholar]
- 20.Kendrick B S, Dong A, Allison S D, Manning M C, Carpenter J F. J Pharm Sci. 1996;85:155–158. doi: 10.1021/js950332f. [DOI] [PubMed] [Google Scholar]
- 21.Carlier C. Ph.D. Thesis. New Haven, CT: Yale University; 1994. [Google Scholar]
- 22.Schneider D H, Freed J H. In: Calculating Slow Motional Magnetic Resonance Spectra: A User’s Guide. Berliner L S, Reuben J, editors. NY: Plenum; 1989. [Google Scholar]
- 23.van Holde K E. Physical Biochemistry. Englewood Cliffs, NJ: Prentice–Hall; 1985. [Google Scholar]
- 24.Yancey P H, Somero G N. Biochem J. 1979;183:317–323. doi: 10.1042/bj1830317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang B S, Beauvais R M, Arakawa T, Narhi L O, Dong A, Aparisio D I, Carpenter J F. Biophys J. 1996;71:3399–3406. doi: 10.1016/S0006-3495(96)79534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu H S, Clogston C L, Wypych J, Fausset P R, Lauren S, Mendiaz E A, Zsebo K M, Langley K E. J Biol Chem. 1991;266:8102–8107. [PubMed] [Google Scholar]
- 27.Lee J C, Frigon R P, Timasheff S N. Ann N Y Acad Sci. 1975;253:284–291. doi: 10.1111/j.1749-6632.1975.tb19207.x. [DOI] [PubMed] [Google Scholar]
- 28.Kita Y, Arakawa T, Lin T Y, Timasheff S N. Biochemistry. 1994;33:15178–15189. doi: 10.1021/bi00254a029. [DOI] [PubMed] [Google Scholar]
- 29.Hall D R, Jacobsen M P, Winzor D J. Biophys Chem. 1995;57:47–54. doi: 10.1016/0301-4622(95)00044-x. [DOI] [PubMed] [Google Scholar]
- 30.Prestrelski S J, Tedeschi N, Arakawa T, Carpenter J F. Biophys J. 1993;65:661–71. doi: 10.1016/S0006-3495(93)81120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapoor M, Parfett C L. Arch Biochem Biophys. 1977;184:518–528. doi: 10.1016/0003-9861(77)90461-1. [DOI] [PubMed] [Google Scholar]
- 32.Byler D M, Susi H. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 33.Dong A, Huang P, Caughey W S. Biochemistry. 1990;29:3303–3308. doi: 10.1021/bi00465a022. [DOI] [PubMed] [Google Scholar]
- 34.Likhtenshtein G I. Spin Labeling Methods in Molecular Biology. New York: Wiley; 1976. [Google Scholar]
- 35.Vigers G P A, Caffes P, Evans R J, Thompson R C, Eisenberg S P, Brandhuber B J. J Biol Chem. 1994;269:12874–12879. [PubMed] [Google Scholar]