

Abstract
MHC mismatched DBA/2 renal allografts are spontaneously accepted by C57BL/6 mice by poorly understood mechanisms, but both immune regulation and graft acceptance develop without exogenous immune modulation. Previous studies have shown that this model of spontaneous renal allograft acceptance is associated with TGF-β dependent immune regulation, suggesting a role for T-regulatory (Treg) cells. The current study shows that TGF-β immune regulation develops 30 days post-transplant, but is lost by 150 days post-transplant. Despite loss of detectable TGF-β immune regulation, renal allografts continue to function normally for >200 days post-transplantation. Because of its recently described immunoregulatory capabilities, we studied indoleamine 2,3-dioxygenase (IDO) expression in this model, and found that intragraft IDO gene expression progressively increases over time, and that IDO in “regulatory” dendritic cells (RDC) may contribute to regulation associated with long-term maintenance of renal allografts. Immunohistochemistry evaluation confirms presence of both Foxp3+ T-cells and IDO+ DCs in accepted renal allografts, and localization of both cell types within accepted allografts suggests the possibility of synergistic involvement in allograft acceptance. Interestingly, at the time when RDCs become detectable in spleens of allograft acceptors, ~30% of these mice challenged with donor-matched skin allografts accept these skin grafts, demonstrating progression to “true” tolerance. Together, these data suggest that spontaneous renal allograft acceptance evolves through a series of transient mechanisms, beginning with TGF-β and Treg cells, which together may stimulate development of more robust regulation associated with RDC and IDO.
Introduction
It has been known for many years that transplantation of fully mismatched renal allografts in non-immunosuppressed mice results in spontaneous acceptance for some strain combinations. This curiosity was first reported by Russell et al (1, 2) who demonstrated immune alteration during the first 30 days post-transplantation in renal allograft acceptors. Despite these initial observations from almost 30 years ago, little progress has been made to fully explain them. Many believe Russell et al’s initial speculation, that these models, which are not dependent upon immunosuppressive strategies, might hold important clues to development of immune tolerance. Our group has therefore studied spontaneous acceptance of DBA/2 renal allografts by C57BL/6 mice, hoping to elucidate mechanisms that lead to long term renal allograft acceptance, and to gain further insight into development of tolerance.
We have reported that like cardiac allografts in this strain combination, renal allograft acceptance occurs despite presence of alloreactive T and B cells. Interestingly, spontaneous renal allograft acceptors display regulation of splenocyte alloresponse to donor antigen. Presence of alloreactive T-cells and regulation are demonstrated as DTH-detectable immune regulation mediated by splenic T cells involving TGF-β (3, 4). In addition, these allograft acceptors also develop donor reactive antibodies (3, 4). Given its known immunoregulatory functions, and its association with T-regulatory cells (Tregs), involvement of TGF-β suggested that regulation associated with spontaneous renal allograft acceptance involved Treg cells.
Although we assumed involvement of Treg cells, subsequent data suggested that Tregs alone are not robust enough to explain allograft acceptance and regulation in this model (5). Immunoregulatory mechanisms involving TGF-β and Tregs have been identified in rodent recipients that have spontaneously accepted renal allografts, but their association with graft acceptance is poorly understood (3, 6). Additionally, TGF-β associated immune regulation is only transiently expressed and not observed in late renal allograft acceptor splenocytes (>150 days post-transplant) (5). These observations led us to explore several possibilities regarding immune regulation in our model: 1) graft acceptance might not require Tregs or TGF-β mediated regulation, which only develop as epiphenomena; 2) persistence of allograft acceptance beyond 150 days involves replacement by a subsequent regulatory mechanism, or alternatively clonal anergy or deletion; or 3) regulatory T cells relocate or establish themselves locally in accepted grafts, and are not maintained in the periphery. Even more fundamentally, it is unknown whether spontaneous renal allograft acceptance represents true immunologic tolerance, or merely prolonged or delayed rejection, given our previous observations that early renal allograft acceptance is associated with development of alloantibodies and mild fibrosis (3). We therefore studied the long term fate of spontaneously accepted renal allografts, to determine if graft survival persists over time, and what mechanisms might contribute to long term allograft acceptance and tolerance.
In the current report, we present evidence suggesting that early renal allograft acceptance is associated with TGF-β induced DTH immune regulation, both peripherally by splenocytes as well as locally by graft infiltrating cells. Curiously, during the late transplant period (>150 days), TGF-β associated mechanisms no longer appear to be operative in GICs or splenocytes, and even more interesting, some renal allograft acceptors are able to accept donor matched skin allografts at this time. Our data suggest that early TGF-β associated mechanisms may be involved in induction, but not long term maintenance of allograft acceptance. To preserve late graft function in the absence of TGF-β associated mechanisms, presumably some other mechanism must be operational. We demonstrate that IDO expression is significantly increased in accepted renal allografts 150 days after transplantation, and further, that immune regulation at day 150 may be mediated by IDO. Perhaps most importantly, we demonstrate that regulatory dendritic cells (RDC) are one likely source of IDO in late allograft acceptors, and that these cells are capable of controlling alloresponses. Both RDCs and Tregs localize in accepted allografts, suggesting that their synergism might contribute to maintenance of tolerance. Taken together, these data suggest that spontaneous renal allograft acceptance is a complex immune response that evolves through different mechanistic steps over time, and that acceptance and tolerance may ultimately be mediated by RDCs.
Methods
Mice
C57Bl/6 (H-2b), DBA/2 (H-2d), and FVBN (H-2q) mice were obtained from Taconic (Germantown, NY). All mice were housed and treated in accordance with Animal Care Guidelines established by the National Institute of Health and The Ohio State University.
Murine Kidney Transplantation
Murine kidney transplantation was performed as described by Zhang et al (7). Briefly, donor left kidneys were isolated by ligating and dividing the adrenal and testicular vessels with micro suture. The aorta and inferior vena cava (IVC) were mobilized at their junction, with the left renal artery and vein. The aorta was ligated below the renal vessel. An elliptical patch of bladder containing the left ureterovesical junction was excised. Grafts were perfused in situ with 0.2 to 0.4ml of cold, heparinized Ringer’s lactate. Finally, kidneys with their vascular supply and ureter attached to the bladder patch were harvested en bloc. Recipient right native kidneys were removed immediately prior to transplantation. The infra-renal aorta and IVC were carefully isolated and cross-clamped. An end-to-side anastomosis between donor renal vein and the recipient IVC was performed. Following successful anastomosis the kidney graft perfused instantly. Urinary reconstruction was then performed by a bladder-to-bladder anastomosis. The left native kidney was removed one week post-transplantation. Kidney graft survival was followed by daily examinations of overall animal health and weekly serum creatinine checks. All transplanted mice received a single subcutaneous injection of penicillin (500U/10g) just after surgery in addition to drinking water containing sulfatrim (100mg/kg) until the experiment was terminated.
Serum Creatinine Determination
Quantitative serum creatinine levels were determined using kits from Roche Molecular Biochemicals (Indianapolis, IN), formerly Boehringer Mannheim. Creatinine reagents and a Boehringer Mannheim/Hitachi analyzer were used to perform the analysis. Conventional units (mg/dl) were converted to SI units by multiplying conventional units by 88.4. Concentrations of creatinine in the serum are expressed in μmol/L.
Murine skin transplantation
Skin allografts were performed using abdominal skin from donor mice. Square full-thickness grafts (approximately 8x10 mm) were placed on graft beds prepared on each recipient’s flank. Grafts were covered with protective bandages for 7 days. Rejection was considered to occur when grafts exhibited dark discoloration, scabbing and necrotic degeneration.
Graft infiltrate isolation
Renal graft tissues were excised and suspended in 1mg/ml collagenase (Sigma, St. Louis, MO). Grafts were cut into small pieces in a petri dish using a scalpel and small plunger and incubated for 40 minutes at 37 C. Cells were washed and removed from the debris followed by RBC lysis. After three washes in PBS, cells were used in transfer DTH assays.
Transfer DTH responses
Graft infiltrating cells or splenocytes were isolated from kidney acceptor mice at 60 and 150 days post-transplant and were tested in transfer DTH assays. For this assay, pinnae of naive B6 mice were injected using a 30-gauge insulin syringe, with 35μl containing cells (either 1–3 x106 graft infiltrating cells (GIC), 8 x106 renal allograft acceptor unfractionated splenocytes, 8 x 106 renal allograft acceptor DC depleted splenocytes, 8 x106 skin allograft rejector splenocytes, or 0.25 x 106 DC+ renal allograft acceptor splenocytes) plus subcellular donor alloantigens +/− 25μg of neutralizing TGF-β or IDO antibodies. Changes in ear thickness were measured both before injection and 24 hours after injection using a dial thickness gauge (Swiss Precision Instruments, Carlstadt, NJ). For reference, changes ranging 0–30 x10−4 inches represent background swelling due to injection trauma, changes ranging 40–60 x10−4 inches represent moderate DTH responses, and changes ranging 70–100 x10−4 inches represent strong DTH responses. It was also noted that in 33% of experiments performed, injections of GIC alone from day 60 and 150 renal allograft acceptors resulted in swelling responses ranging 50–90 x10−4 inches. We have previously observed this “hot prep” phenomenon when cell isolates are contaminated with red blood cells or platelets (unpublished observations). Although it might also be the nature of these cells, these “hot” results were seen equally in day 60 and 150 graft acceptors. Thus, only experiments for which an acceptable background swelling response was obtained for GIC alone (<50 x10−4 inches) were included in data presented in this paper. Renal allograft acceptor splenocytes contain normal numbers of T-cells when compared to non-transplanted control mice (data not shown)
Subcellular alloantigen
Subcellular alloantigen was prepared according to published methods of Engers et al (8). Briefly, fresh RBC-depleted DBA/2 splenocytes suspended in PBS were subjected to three rapid freeze/thaw cycles, using liquid nitrogen, and spun at 13,000 rpm for 30 minutes to remove residual debris. Supernatants were adjusted to 3–5 mg protein/ml and used as subcellular alloantigen. For DTH challenge, 25 μl (75–125 μg protein) of this solution was injected into murine pinnae.
Neutralizing reagents
Polyclonal rabbit anti-TGF-β and control rabbit Ig were obtained from R&D Systems (Minneapolis, MN). Polyclonal rabbit anti-IDO Ig was obtained from Alexis Biochemicals (San Diego. CA). In addition to anti-IDO antibody, IDO activity was neutralized using 1-methyl D tryptophan (1-MT) obtained from Sigma-Aldrich (St Louis, MO).
Alloantibody analysis
Presence of donor-reactive IgG antibody was determined by the ability of sera to bind to DBA/2 thymocytes. Binding was detected by flow cytometry, using fluorescein isothiocyanate-conjugated goat anti-mouse IgG ( chain specific) (Pierce Chemical Co., Rockford, IL). Results are shown as percentage of DBA/2 thymocytes that bound detectable alloantibody. Treatment of DBA/2 thymocytes with sera from naive C57Bl/6 mice resulted in less than 5% binding, and sera from allosensitized C57BL6 mice react with >98% of thymocytes.
Histological examination of renal tissue
Renal tissues were excised and fixed in 10% neutral buffered formalin, dehydrated in upgraded ethanol (70%, 95%, and 100%) and embedded in paraffin. For histological analysis, 4 μ sections were mounted on slides and stained with hematoxylin and eosin (H&E) or trichrome
Immunohistochemistry techniques
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue from representative accepted renal DBA to B6 allografts (n=12, days 28–158) and B6 to B6 isografts (n=9, days 30–153) with a double marker technique optimized for the simultaneous identification of Foxp3 and a surface differentiation molecule CD3. Sections were baked for 30 min in oven, deparaffinized in xylene, rehydrated in absolute and 95% ethanol, incubated for 5 minutes in 3% H2O2 in methanol to block endogenous peroxidase, and washed in TBS/Tween 20. Antigen retrieval was done with Borg Decloaker™ solution pH 9.5 (Biocare Medical, Walnut Creek, CA) in pressure cooker. Blocking normal goat serum 1:50 and avidin D 1:10 were employed, followed by CD3 polyclonal antibody (DAKO, Carpinteria, CA) diluted at 1:400 in 1% TBS/BSA and incubated overnight at 4°C. After biotin blockade, slides were incubated with biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories, Burlingame, CA) for 35 min, followed by Streptavidin peroxidase (Biogenex) for 60 minutes, and developed with 3,3′-diaminobenzidine. All steps included washing with TBS/Tween 20 in between. Slides were then incubated in blocking normal goat serum 1:50 and avidin D 1:10 for 20 min. An affinity purified rat anti-FOXP3 (clone FJK-16s) antibody (eBoioscience) diluted at 1:50 in 1% TBS/BSA was incubated overnight at 4°C. After biotin blockade, a biotinylated rabbit anti-rat (mouse ads) IgG secondary antibody (Vector Laboratories, Burlingame, CA) was used for 35 min followed by avidin-biotinylated-alkaline phosphatase complex (ABC-AP, Vector Laboratories, Burlingame, CA) for 60 min. All steps included washing slides in between with TBS/Tween 20 and with TBS only before developing FOXP3 staining. Tissue sections were developed with Vector Blue™ alkaline phosphatase substrate (Vector Laboratories, Burlingame, CA) and mounted with Faramount (DAKO, Carpinteria, CA).
For IDO, after the Decloaker and blocking steps above, polyclonal rabbit anti IDO antibody at a 1:2000 dilution (Alexis Biochemical) was added, and the sections were incubated overnight at 4°C. Sections were then rinsed in PBS and treated with biotin (10ug/liter PBS), washed three times in PBS, and incubated in the biotinylated goat anti rabbit IgG for 35 minutes. Slides were washed and incubated for 1 hour in Streptavidin Peroxidase (Biogenex). Sections were washed and developed with Romulin AEC Chromogen (Biocare Medical), counterstained with hematoxylin, dehydrated in ethanol, cleared in Xylene, and coverslipped with permanent mounting media. Sections stained for Foxp3 and CD3 were scored for positive cells per 10x fields in coded sections and converted to cells/mm2 (10x field = 2.14 mm2). Sections stained for IDO were scored qualitatively according to the type and frequency of positive cells.
Foxp3 and IDO RT-PCR
Total RNA was isolated from kidney tissue using Trizol™ (Invitrogen). 2ug of total RNA was reverse transcribed using Qiagen Omniscript RT kit™ and amplified using Superarrays gene specific RT2 End-point™ Foxp3 and IDO PCR kits. Samples were separated on a 2% ethidium bromide containing agarose gel and bands visualized through UV transillumination. Images were captured using a Kodak DC120 camera and analyzed using Kodak 1D gel Analysis Software™. Band intensity was normalized using a mouse GAPDH internal control and reported as relative units.
MACs Microbead Cell Separations
Splenocytes were depleted of dendritic cells utilizing MACs microbead technology developed by Miltenyi Biotec (Auburn, CA). Cells were labeled with pan DC microbeads, and passed through a separation column, LS+/VS+, in a magnetic field. The positive and negative fractions of cells were separately collected and used for further analysis.
Results
Long-term Renal Allograft Acceptor Function
We have previously reported that ~70% of C57Bl/6 mice spontaneously accept heterotopic DBA/2 renal allografts for at least 60 days (3). Longitudinal graft function in renal allograft acceptors was evaluated for deterioration over time. Baseline serum creatinine levels were maintained in renal allograft acceptors when measured either at day 60 (30+/−15 μmol/l), > day 150 (31+/−10), or at day 200 (36+/−13). Note that day 200 was following a donor skin graft challenge at d150. These levels were essentially identical to levels that were measured in normal, non-transplanted animals and in C57Bl/6->C57Bl/6 renal isograft acceptors during the same time periods (data not shown). Mice driven to reject renal allografts by priming with skin allografts (14 days prior to renal grafting) develop much higher creatinine levels (102 +/− 12 μmol/L) within 14 days of kidney transplantation (Table 1).
Relocation of Active Immune Regulation
We have reported that renal allograft acceptors gain DTH-detectable immune regulation by 60 days post-transplant, but maintain the ability to respond to third party antigens such as tetanus toxin (3). In those studies recipient splenocytes were tested for donor-reactive immune regulation in transfer DTH assays. Other previous studies have suggested that development of donor-reactive immune regulation in spleens of cardiac allograft acceptor and renal allograft acceptor mice is transient (5). In the present study, we extended DTH testing of splenocytes to 150 days post-transplant to determine the immune status of the recipient at this later time point. As shown in Figure 1A, splenocytes show DTH-detectable, TGF-β-mediated, graft-reactive immune regulation on day 60, that becomes non-TGF-β mediated by day 150 post-transplant. Consistent with our previous studies (3), IL-10 was also unable to restore DTH responses (data not shown). Despite losing peripheral immune regulation mediated by TGF-β by day 150, allografts maintained normal function. Because graft function was maintained, we hypothesized that the site of graft-reactive immune regulation migrates over time from peripheral lymphoid organs to the graft itself.
Figure 1. Changes in mechanism of regulation over time in renal allograft acceptors.

Splenocytes (A) and graft infiltrating cells (B) were isolated from accepted renal allografts at d60 and d150 post-transplant. Cells from each mouse were combined with alloantigen alone or alloantigen + polyclonal antibodies to TGF-β, then injected into pinnae of naïve C57BL/6 mice. Day 60 acceptors demonstrated prominent donor-reactive DTH regulation in both splenocytes (A) and graft infiltrating cells (B) that appeared to be mediated by TGF-β (*swelling significantly greater when TGF-β blocked p<0.002). Day 150 acceptors continued to demonstrate donor-reactive regulation in both sites, but TGF-β antibodies no longer restored donor reactive responses in either site, suggesting a change in mechanism of regulation. DTH responses were measured after 24 hours as change in ear thickness (mean ± SD x 10-4 inches). DTH responses (73 +/− 12 x 10-4 inches) to tetanus toxoid, a pre-sensitized antigen, were displayed by splenocytes from day 150 renal acceptors demonstrating their ability to respond by DTH at this later time point (data not shown). Similar positive control testing with GIC from day 150 renal acceptors was not possible due to limited cells.
To test this hypothesis, we performed transfer DTH assays using allograft infiltrating lymphocytes derived from renal allograft acceptor mice. Accepted DBA/2 renal allografts were obtained from C57Bl/6 recipients either 60 or 150 days post transplant. Graft infiltrating cells (GICs) were isolated by collagenase digestion and were transferred into pinnae of naïve C57Bl/6 mice along with subcellular donor alloantigen. Donor-reactive DTH responses were measured as ear swelling 24 hours later. As shown in Figure 1B, 60 day kidney GICs did not elicit ear swelling when challenged with donor alloantigen unless TGF-β was neutralized at DTH sites. Similarly, 150 day kidney GICs were unable to promote donor alloantigen-induced ear swelling. Interestingly, TGF-β-neutralizing antibodies did not restore donor alloantigen-induced ear swelling for 150 day kidney GICs. Thus, within 60 days post-transplant, cells present within accepted renal allografts demonstrate DTH-detectable, TGF-β-mediated, graft-reactive immune regulation. By 150 days after transplantation, however, graft-reactive immune regulation no longer appears to be dependent upon TGF-β.
Together, these findings suggest that both spleen and graft serve as concurrent active sites for expression of graft-reactive immune regulation in renal allograft acceptors. Furthermore, it appears that loss of TGF-β mediated splenic immune regulation does not reflect late migration of this regulatory center from peripheral lymphoid organs to the graft itself.
Donor-reactive Alloantibody
Previous studies have demonstrated that donor-reactive alloantibodies are generated by renal allograft acceptor mice at levels comparable to rejecting heart graft recipients (3). In the current study, we tested for alloantibody persistence. Donor-reactive humoral immune responses were tested in renal allograft acceptors by flow cytometric analysis of serum reactivity with donor thymocytes. All kidney allograft acceptors demonstrate donor reactive antibodies, and as shown in Figure 2, these antibodies persist 150 post-transplant. Additional analyses demonstrated that alloantibody titers were 50-fold lower in renal allograft acceptors compared to skin allograft rejectors (data not shown). These findings confirm that donor-reactive humoral immune responses in renal allograft recipients develop, and persist under conditions in which donor-reactive cellular immune responses are subject to immunoregulation.
Figure 2. Untreated renal allograft acceptors generate donor-reactive alloantibodies.
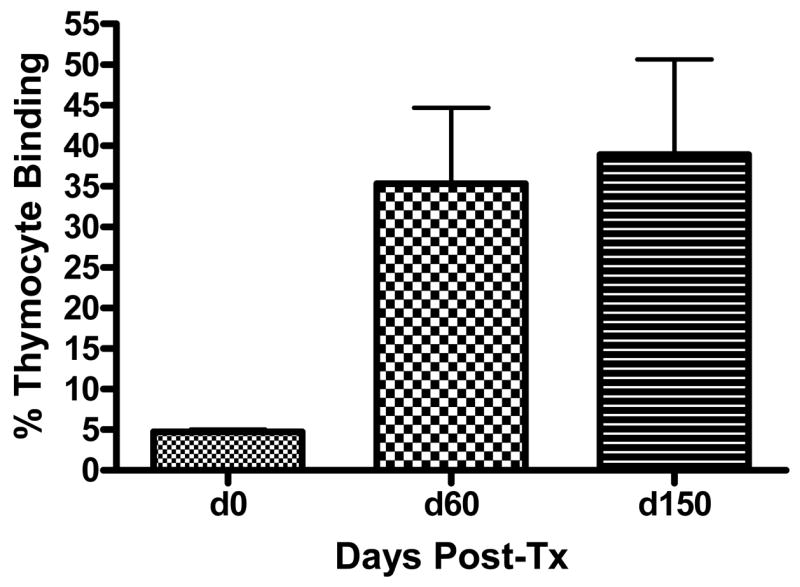
Sera from C57BL/6 mice were collected pre-transplant on day 0 (n=8), day 60 post-transplant (n=13), day 150 post-transplant (n=8) and were tested for reactivity to DBA/2 (donor) thymocytes by flow cytometric analysis. Donor reactive antibodies were thus detected by day 60 and persisted until day 150 post-transplant. Results are expressed as mean percentage binding for each time point.
Challenge with Donor Skin Allografts
Renal allografts induce long-lasting, donor reactive alloantibodies, and yet continue to function after apparent loss of DTH-detectable immune regulation, raising the possibility that immune tolerance to donor antigen develops in renal allograft acceptors. Currently, the most stringent functional test for alloantigen tolerance in murine organ transplant models is acceptance of donor-matched skin allografts. To test this hypothesis, cohorts of DBA/2 ->C57BL/6 renal allograft acceptor mice received DBA/2 skin allografts either 60, 150, or 180 days post-transplant. As shown in Figure 3A, 100% of 60-day kidney allograft acceptors reject DBA/2 skin grafts within 24 days post-transplant, and 6 of 8 acceptors retain functional kidney allografts for more than 50 days post-skin transplant. In surviving mice, serum creatinine levels remain normal after skin grafting, indicating that renal allograft function does not deteriorate following skin allograft rejection (Table 1).
Figure 3. Graft survival of renal allograft recipients following skin allograft challenge.
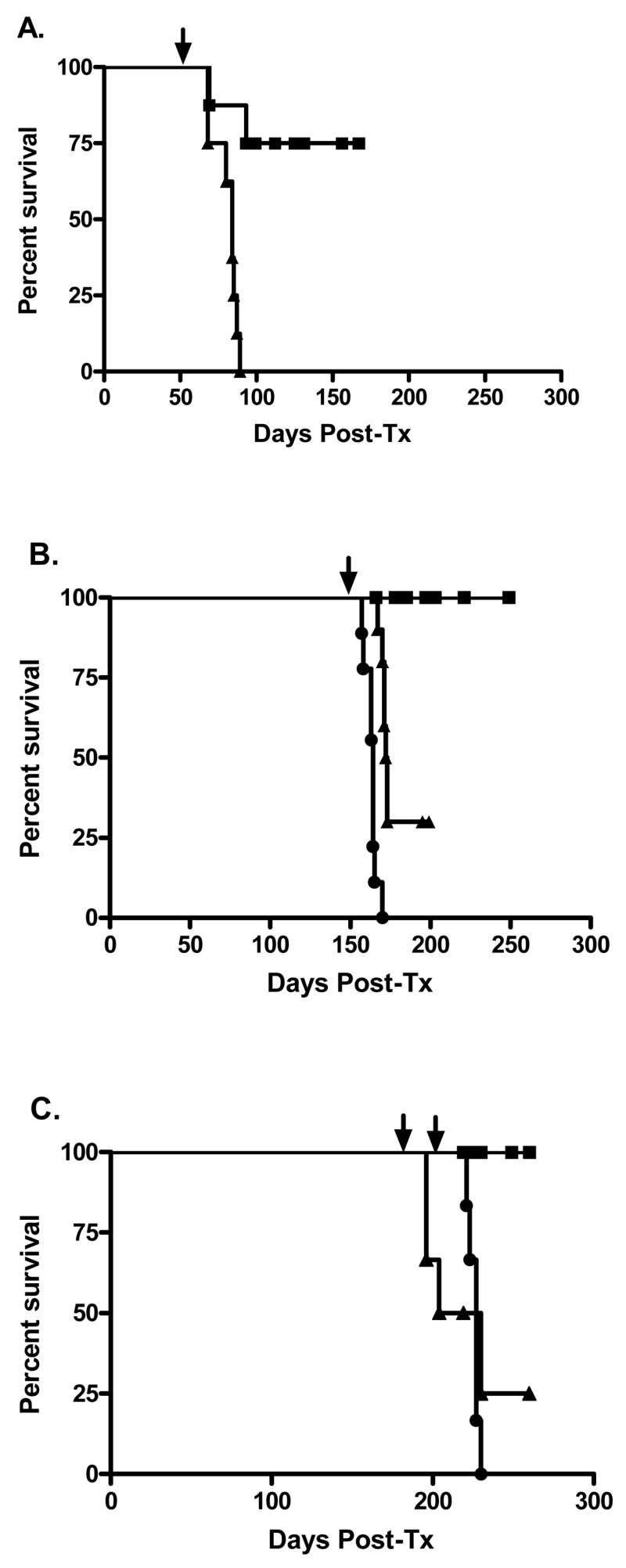
A. When challenged at 60 days post-transplantation, all renal allograft recipients (n=8) rejected donor matched skin grafts (▲) with 25% of recipients rejecting their renal allografts (■). B. When renal allograft acceptors were challenged at 150 days post-transplantation with concomitant donor matched and 3rd party skin allografts, 30% of renal allograft recipients (n=10) accepted donor matched skin grafts (▲) despite all of them rejecting their 3rd party grafts (●) suggesting further progression of donor specific regulation. None of the 150 day skin graft recipients rejected their renal allografts (■). C. When renal allograft acceptors were challenged at 180 days post-transplantation with concomitant donor matched skin allografts, 33% of renal allograft recipients (n=6) accepted donor matched skin grafts (▲) despite all of them rejecting their 3rd party grafts (●) placed 30 days later. None of the 180 day skin graft recipients rejected their renal allografts (■). Renal graft function was assessed by serum creatinine analysis and animal survival. Skin graft survival was assessed by tissue necrosis. Arrows indicate time of skin transplantation.
As shown in Figure 3B, all 150 day renal allograft acceptors retain normal renal function after DBA/2 skin allograft implantation, and 30% (3/10) have skin allograft acceptance for more than 50 days post-skin transplant. These 150 day acceptors simultaneously reject all concurrently placed, third party FVBN (H-2q) skin allografts within about 15 days (Figure 3B). Thus some renal allograft acceptors gain the ability to accept donor-matched skin grafts by day 150 post-transplant, despite loss of detectable TGF-β-mediated DTH immune regulation. These data suggest progression to true tolerance in 30% of renal allograft acceptors between 60 and 150 days post-transplant. To determine if this progression continues to evolve over time we similarly studied 180 day renal allograft acceptors. As shown in Figure 3C, 33% (2/6) of 180 day renal allograft acceptors have donor matched skin graft acceptance and all retain normal renal function and their ability to reject third party FVBN skin allografts. This indicates no further progression in development of tolerance over time.
Allograft Histopathology
Recipient renal function is maintained solely by allograft kidneys in this model, thus serum creatinine levels were used to monitor allograft function throughout all experiments. Because renal tissues can show major histological injury before physiologic function is impaired to a detectable degree, accepted allografts were also examined histologically after staining with H&E. As shown in Figure 4, day 60 (panel A) and day 150 (panel B and C) accepted renal allografts exhibit abnormal histology. Both undergo prominent leukocytic cuffing of larger vessels, as well as mild fibrosis, while most renal parenchyma is unremarkable. Interestingly, diffuse interstitial leukocyte migration that is characteristic of acute cardiac allograft rejection was not observed in accepted renal allografts. Observed histopathology in accepted renal allografts is alloantigen-dependent, as no similar leukocytic infiltration is seen in accepted, d150 renal isografts (panel D) which maintain normal tissue histology throughout this time period.
Figure 4. Histologic features in accepted renal allografts.
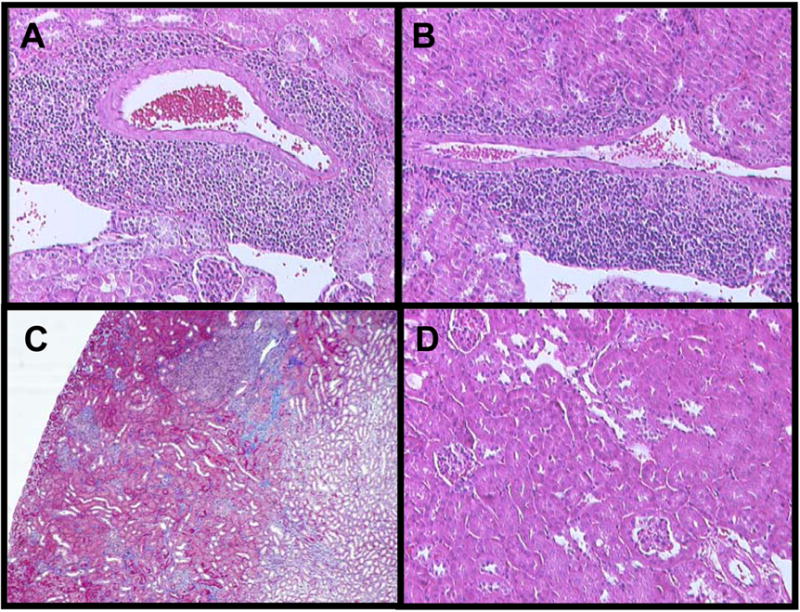
Renal grafts from C57BL/6 recipients were collected and formalin fixed sections were prepared and stained with H&E or trichrome. Magnification X100. Prominent perivascular mononuclear cell infiltration is observed in accepted renal allografts collected at 60 days post-transplant (A) and at 150 days post-transplant (B). Mild interstitial fibrosis is observed in renal allografts at 150 days post-transplant (C). The renal parenchyma of accepted renal isografts at day 150 post-transplant is normal (D).
As suggested in Figure 2, most renal allografts endure prolonged exposure to circulating, donor-reactive antibodies. The immunologic and histologic impact of these alloantibodies remains unclear. We have confirmed by immunofluorescence that C3d is not depositing in accepted allografts (data not shown). In cardiac allografts, alloantibodies have been associated with development of neo-intimal formation and tissue fibrosis (9). Neointimal formation is not observed in arteries of the renal allografts, but mild interstitial fibrosis is present (Figure 4C). Based on these observations, both day 60 and day 150 renal allografts appear to be the focus of an active, but unusual immune process.
Allograft Foxp3 Expression
Foxp3 expression is unique to regulatory T cells (10–13), thus presence of Treg cells in renal allografts should be mirrored by expression of Foxp3. To determine presence of active Treg cells, total RNA was isolated from accepted renal allografts at 30, 60, and 150 days post-transplant to evaluate for presence or absence of Foxp3. Controls included normal, non-transplanted DBA/2 and C57BL/6 kidneys and renal isografts collected at 30, 60, and 150 days post-transplant. These RNA were analyzed by RT-PCR for expression of Foxp3. As shown in Figure 5, Foxp3 mRNA is significantly elevated in renal allografts by day 30 post-transplant, but decreases to control kidney levels by day 150. Foxp3 expression in renal isografts is comparable to background with only a slight increase at day 150. Increased expression of Foxp3 in 30–60 day renal allografts occurs concomitantly with expression of TGF-β-mediated DTH immune regulation. It is intriguing that loss of TGF-β mediated immune regulation between day 60 and 150 (Figure 1) occurs simultaneous to decreases in Foxp3 transcription. We therefore conclude that diminishing levels of Foxp3 suggest decreased presence or activity of Treg cells, and further, that alternate mechanisms must be involved in subsequent maintenance of renal allograft acceptance.
Figure 5. Foxp3 mRNA expression associated with early but not late renal allograft acceptance.
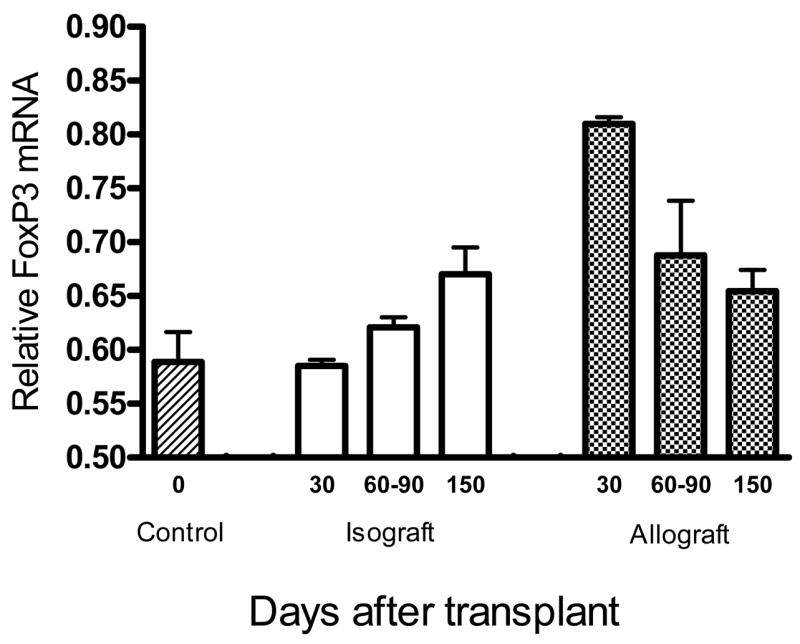
Tissue homogenates from accepted renal allografts and isografts were evaluated by real time RT-PCR for presence of Foxp3 mRNA before (control), 30, 60–90, and 150 days after transplant. There was significant elevation of Foxp3 expression in day 30 allografts (Students two sided t-test, p=0.005), but Foxp3 expression returned to baseline by day 150 post-transplant.
Allograft IDO Expression
IDO, which is known to be expressed by dendritic cells (DC) (14), is known to suppress alloreactive T cell responses (15–18). Because the TGF-β and Foxp3 data together suggested a diminishing role for Treg cells, we hypothesized that late tolerance might be maintained by transition from Treg mediated to DC mediated mechanisms in accepted allografts. Accepted allografts were therefore evaluated by RT-PCR for presence of IDO transcripts. As shown in Figure 6, IDO mRNA expression increases over time in accepted renal allografts, becoming significantly elevated by day 150 post transplant. In contrast, low IDO mRNA levels are found in normal non-transplanted kidneys, and renal isografts show only slight elevation by day 150. Thus increasing presence of IDO over time in accepted renal allografts suggests possible development of an alternate mechanism of allograft acceptance dependent upon immunoregulatory effects of IDO.
Figure 6. Renal allograft acceptance associated with progressive IDO expression at the graft site.
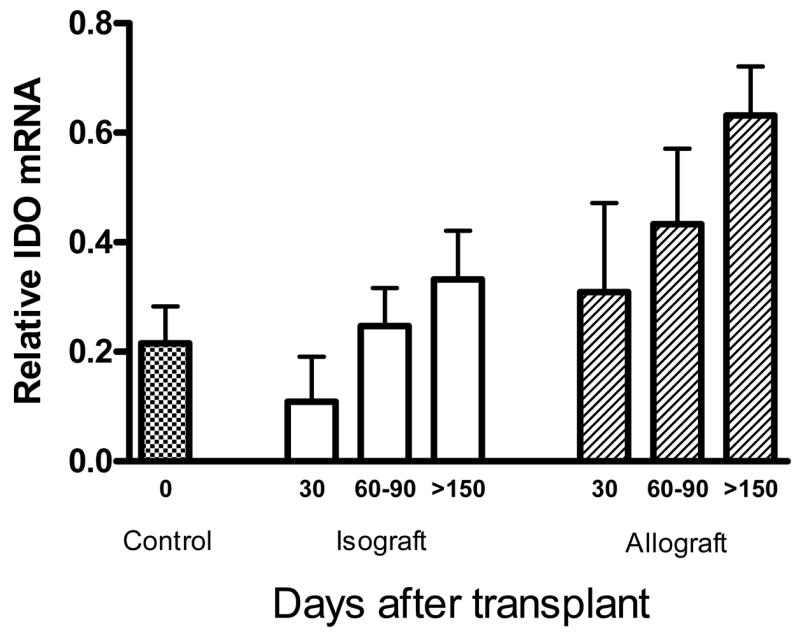
Tissue homogenates from accepted renal allografts and isografts were evaluated by real time RT-PCR for enzyme indoleamine 2,3-dioxygenase (IDO) mRNA before (control), 30, 60–90, and 150 days after transplant. Allografts showed increased levels of IDO transcription over time that were significantly higher than controls and isografts by day 150 post-transplant (p=0.009). Each bar represents data from at least 2 mice.
Evidence of IDO-mediated immune regulation of alloresponse
IDO has been shown to inhibit alloreactive responses in vitro, yet there are few in vivo data to support this (16–19). As shown in Figure 2, 150 days after transplantation, splenic donor-reactive DTH responses remain suppressed, but no longer appear to be associated with TGF-β. Because IDO transcription is elevated at this time point (Figure 6), we hypothesized that alloreactive immune responses were suppressed by IDO. To test this, splenocytes from 60 or 150 day renal allograft acceptors were combined with donor alloantigens with or without neutralizing antibodies to IDO and used in our transfer DTH assay. As shown in Figure 7, day 150 allograft acceptor splenocytes show IDO-mediated, donor-reactive immune regulation. In contrast, IDO regulation does not appear to be active at day 60, when TGF-β appears to be active. We initially chose to neutralize IDO in our DTH assay with antibodies because of our experience with other neutralizing antibodies, but in retrospect it was somewhat surprising that these neutralized IDO function, since IDO is thought to be an intracellular enzyme. Accordingly, this experiment was repeated using 1-MT, a known inhibitor of IDO and tryptophan metabolism (20, 21). Addition of 1-MT at DTH challenge sites restored DTH reactivity with results identical to those observed with anti-IDO antibody (data not shown). Anti-IDO antibody or 1-MT mixed with splenocytes (without donor antigen) showed DTH responses consistent with negative controls (not shown). These results suggest that regulation of donor-reactive DTH responses transitions from TGF-β mediated early (60 days) to IDO- mediated later (150 days).
Figure 7. IDO associated regulation of donor-reactive immune responses.
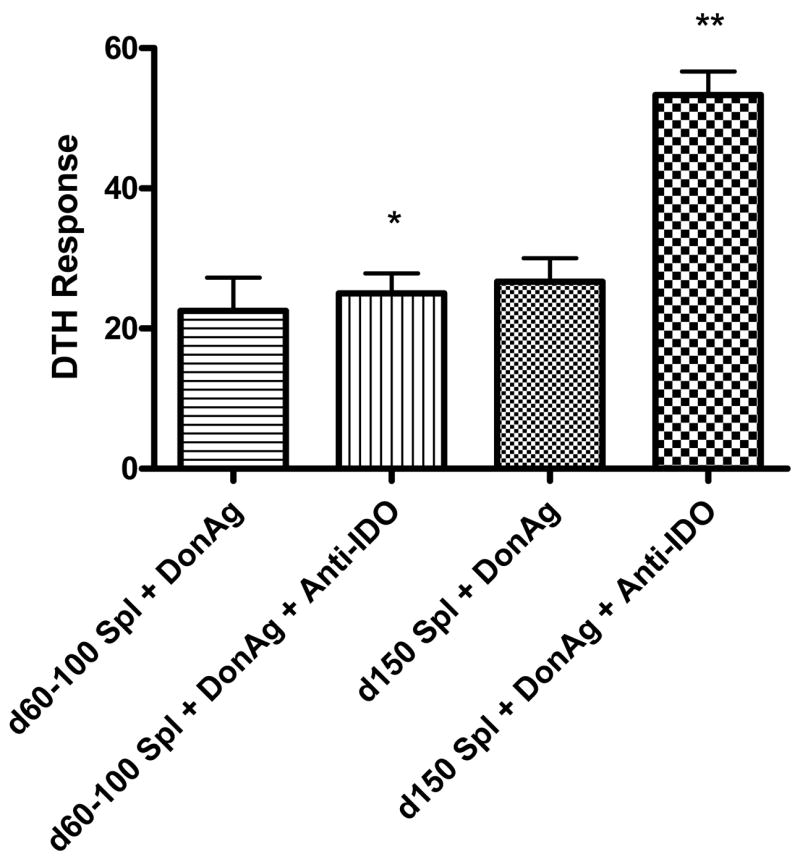
Splenocytes from renal allograft acceptors early (day 60) or late (day 150) post-transplant were combined with alloantigen alone or alloantigen + polyclonal antibodies to IDO. These combinations were injected into pinnae of naïve C57BL/6 mice for trans-vivo DTH measurements. Early allograft acceptors showed no evidence of IDO regulation (*p=0.76), but late renal allograft acceptors demonstrated prominent donor-reactive DTH regulation by IDO (**p=0.005). Anti-IDO antibody and splenocytes without donor antigen showed DTH responses consistent with controls (not shown). DTH responses were measured after 24 hours as change in ear thickness (mean ± SD x 10-4 inches).
Evidence of immune regulation by dendritic cells
Dendritic cells are known to be an important source of IDO (14, 22, 23). Given the compelling IDO data from unfractionated splenocytes, we hypothesized that DCs might be the source of IDO associated with regulation in spontaneous renal allograft acceptors (Figure 7). To test this hypothesis, DCs were isolated from splenocytes of late renal allograft acceptors and combined with alloreactive splenocytes from sensitized (alloreactive) mice. DC regulation was then tested using trans-vivo DTH. As shown in Figure 8, when DCs from allograft acceptors were added to alloreactive splenocytes, tolerance to alloantigen was induced. Further, blocking IDO restores alloresponses, suggesting that this conferred tolerance is mediated by IDO. DCs from naïve mice were similarly isolated, and showed no regulation of alloreactive splenocytes (data not shown). These results thus suggest that “regulatory” DCs (RDCs) are present in spontaneous renal allograft acceptors, and that these cells have the capacity to regulate alloresponse in our DTH model.
Figure 8. Regulatory dendritic cells (RDC) can regulate donor-reactive DTH responses.
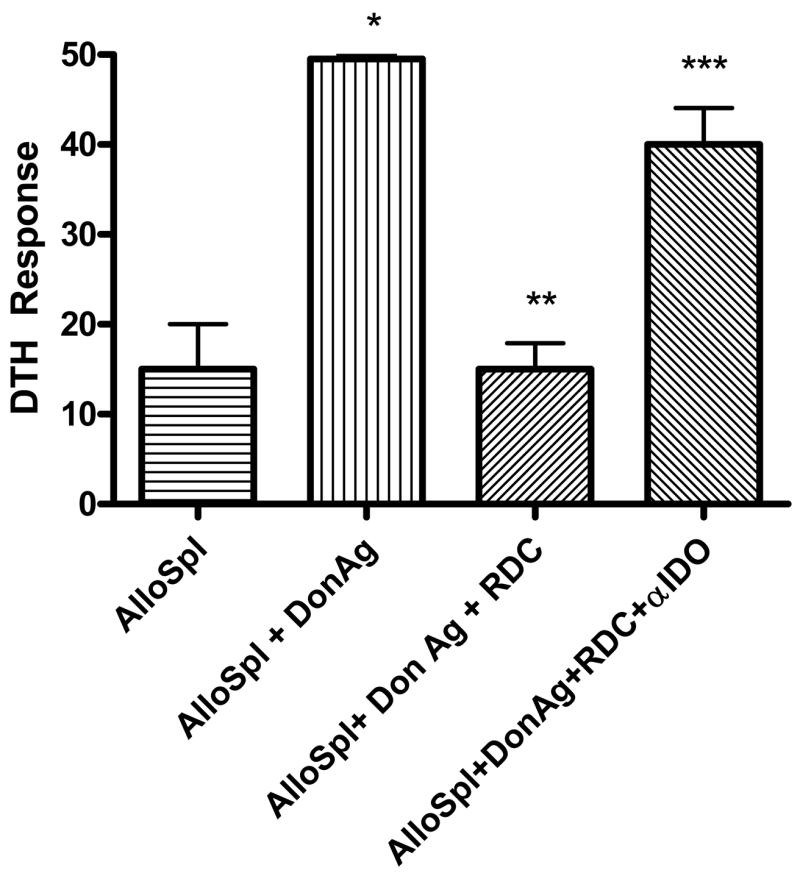
Dendritic cells (DC) isolated from late (day 150) renal allograft acceptor splenocytes were combined with alloreactive splenocytes and donor antigen alone or alloantigen + polyclonal antibodies to IDO. These combinations were injected into pinnae of naïve C57BL/6 mice for trans-vivo DTH measurements. Addition of donor antigen to allosplenocytes caused significant swelling (*p=0.02). RDCs regulated responses of alloreactive splenocytes to donor antigen (**p=0.001), and blocking secreted indoleamine 2, 3 dioxygenase (IDO) restored alloreactivity (***p=0.003). DCs from normal mice did not regulate alloreactive splenocytes with or without IDO antibody (data not shown). DTH responses were measured after 24 hours as change in ear thickness (mean ± SD x 10-4 inches).
Tregs and RDCs in accepted allografts Allograft
Although our RT-PCR and DTH data were suggestive that there is transition from Treg regulated immune responses to RDC regulated responses in allograft acceptors, it remained unclear whether Treg or RDCs were present in accepted allografts. IHC analyses of late accepted allografts show prominent infiltration of CD3+ cells becoming less diffuse and more nodular with time (mean infiltrate 101.9±44.4 cells/mm2). Foxp3+ cells initially infiltrate diffusely in the cortex, eventually developing nodular aggregates around small arteries or tubules (35.2±40.1 cells/mm2) (Figure 9A). Inconsistent with our quantitative RT-PCR results, allograft Foxp3 staining density seems to increase over time (Foxp3+ cells days 30–90 = 18.4±11.1/mm2; days 150+ = 60.2±56.1/mm2). Of all CD3+ cells, 21.9+10.8% were Foxp3+, a fraction that increases over time from 17+10% (days 30–90) to 28+10% (days >150). Conversely, almost all (98.8%) Foxp3+ cells express CD3. Cells expressing IDO are prominent in allografts at all times studied. IDO positive DCs (Figure 9B&E) are identified as larger angular cells within nodular aggregates. In addition, IDO positive arterial endothelium and tubules are often associated with nodular infiltrates of Foxp3 cells (Figure 9E).
Figure 9. Foxp3 and IDO staining of accepted renal grafts.

Allograft day 158 (A&B) A. Nodule of lymphoid cells stained for CD3 and Foxp3 shows many CD3+ cells (brown) with numerous Foxp3+ cells (blue). B. The same aggregate shows scattered DC (arrowheads) that are IDO positive (red). Isograft day 153 (C&D): C. Sections stained for Foxp3 (blue) and CD3 (brown) show very few CD3+ or Foxp3+ staining cells present in the cortex. D. Sections from the same area show no IDO positive cells (brown). E. Allograft day 28 shows a nodular infiltrate with IDO positive arteriolar endothelium (arrow) as well as DC (arrowhead) and tubular epithelial cells (asterisk).
In contrast, isografts show scattered CD3+ cells (35.4±18.2/mm2) and rare Foxp3+ cells (0.5±0.4/mm2) (Figure 9C). Foxp3+ cells comprise 1.9±2.0% of CD3+ cells. Density of these cells does not change appreciably between days 30 and 150 (not shown). Rare IDO+ tubular cells are found in the cortex in early samples (day ~30). No IDO+ DC or endothelial cells are identified in any isografts at any time studied (Figure 9D).
These data thus confirm coexistence of both Tregs and RDCs in spontaneously accepted renal allografts.
Discussion
This study demonstrates for the first time the presence of DCs with a regulatory phenotype (RDCs) in mice that spontaneously accept MHC mismatched renal allografts. It appears that development of spontaneous long-term renal allograft acceptance in mice is associated with multiple immunoregulatory mechanisms that evolve over time. Early escape from cellular rejection is associated with regulation of graft-reactive T-cells by a transient TGF-β-mediated mechanism, while later escape is associated with continued regulation of graft reactive T-cells by IDO and not TGF-β. Our data further suggest that clonal deletion is not required for spontaneous renal allograft acceptance, and that persisting alloreactive cells may be regulated by IDO secreted by RDCs. Even more intriguing, spontaneous development of true tolerance occurs in ~30% of these graft acceptors at late time points, when RDCs can be isolated from their spleens. While these data are far from conclusive, they support the very exciting possibility that development of immune regulation and tolerance may involve development of RDCs.
We have previously demonstrated that allosensitization occurs in renal allograft acceptors evidenced by presence of donor reactive T-cells and alloantibody (5, 24). Despite this allosensitization to donor antigen, data from the current study confirm that accepted grafts continue to function indefinitely (Figure 3), confirming that this model is not one of delayed or slow rejection. Spontaneous renal allograft acceptors display immune regulation of cellular responses, and our data suggest that regulation of cellular responses appears to be divided into at least two functionally distinct phases: early and late (arbitrarily defined as <90 and >150 days respectively) after transplant. Early on, donor reactive cells may be regulated at least in part by TGF-β, as anti-TGF-β antibody restores reactivity to donor antigen in DTH studies (Figure 1). This regulation occurs both systemically in splenocytes, and locally in graft infiltrating cells (Figure 1A & 1B). Because TGF-β is known to stimulate maturation of CD4+CD25+ Treg cells (13, 25–27), it is perhaps not surprising that Foxp3, a marker of active Treg cells (10, 28–30) is highly expressed in accepted allografts when TGF-β appears to be contributing to regulation. Because functional tolerance must depend at least in part upon regulation of cellular or humoral responses to donor antigen, we postulate that early expression of TGF-β in accepted renal allografts stimulates or activates Treg cells, contributing to induction of early tolerance.
Since their first description by Sakaguchi et al in 1995 (31), we have been enamored with the possibility that Tregs contribute to our models of immune regulation and allograft tolerance. These unique cells are now known to regulate both CD4 and CD8 T-cell responses, and there are accumulating data that support the role of regulatory T-cells in long-term allograft acceptance (3, 27, 32–35). Recently, Waldmann and colleagues have shown that Tregs are both highly enriched in accepted allografts (36) and dependent on TGF-β (27), and that TGF-β appears to be an essential intermediary in establishing long term tolerance (37) We too have shown a role for CD4+CD25+ regulatory cells in our heart transplant model (24), and in the current study confirm presence of Foxp3+ cells within our accepted renal allografts (Figure 9A). Based upon these results and those of others, we therefore hypothesized that TGF-β and Tregs might contribute to long term regulation.
Despite our convictions, however, Tregs alone are not robust enough to explain all of our experimental findings. We have previously observed that when Treg cells are depleted from allograft acceptor splenocytes, tolerance to alloantigen still persists in remaining splenocytes, and this tolerance is transferable to alloreactive cells (5). In addition, current data shows loss of TGF-β mediated regulation in renal allograft acceptors over time. As Figure 1 clearly demonstrates, anti-TGF-β antibody no longer restores reactivity to alloantigen in late allograft acceptors. Three possible conclusions could initially be drawn from these observations. First, there could be a switch from TGF-β mediated regulation, to some other subsequent mechanism that maintains regulation of alloreactive cells. Second there could be clonal deletion or anergy of alloreactive cells. Third, transient TGF-β regulatory mechanisms might be an epiphenomenon, associated with, but not responsible for acceptance of renal allografts.
Our data strongly suggest that there is transition from TGF-β to IDO mediated regulation by day 150 post-transplant. Concomitant with loss of TGF-β mediated regulation, there is increasing IDO expression in accepted allografts (Figure 6). More importantly, IDO appears to mediate suppression of donor-reactive immune responses of day 150 renal allograft acceptor splenocytes (Figure 7). One source of IDO in our model appears to be RDCs localized both in accepted allografts (Figure 9) and in spleens of late allograft acceptors (Figure 8). Perhaps most compelling is the observation that RDCs isolated from allograft acceptors are capable of regulating fully alloreactive cells (Figure 8). Taken together, these findings confirm persistence of alloreactive T-cells in our model, and suggest transition to RDC mediated IDO immune regulation by day 150.
There is substantial and growing interest in IDO and its tolerogenic properties, which are thought to occur primarily through profound influences upon T-cell mediated responses (15–17, 38, 39). Functionally, IDO is known to prevent rejection of fetuses during pregnancy by inhibiting alloreactive T cells (20), modulate immune resistance of tumors (40), and regulate alloreactive T-cell responsiveness (15–18). Other investigators have recently identified more specific roles for IDO in transplantation tolerance. Using a rat model, Degauque and colleagues have shown that accepted renal allografts accumulate Foxp3 and IDO 100 days post-transplant, and that IDO plays an important role in controlling tolerant T-cell responses to donor alloantigens (6). Yet other studies have demonstrated prolongation of islet graft survival attributed to IDO function (41). Our current data suggest that IDO may indeed play a regulatory role in spontaneous renal allograft acceptance.
Fortunately, our Treg and RDC/IDO hypotheses are not mutually exclusive, and in fact published data and results from current experiments suggest that there is perhaps a synergistic relationship between Tregs and DCs in our model (42, 43). Mechanistically, Tregs have been demonstrated to “collaborate” with DCs to initiate IDO production (44, 45). Conversely, it has also been shown that IDO+ DCs are capable of stimulating development of Tregs (46–48). Our IHC results (Figure 9) demonstrate presence of lymphoid aggregates containing Foxp3+ CD3 cells in proximity to IDO+ DCs in accepted allografts, which is consistent with the hypothesis of synergism. In addition to DCs, we observe that other cells such as arterial endothelium and tubular epithelium in these lymphoid nodules also express IDO. Similar findings have been recently reported by Thebault et al. in a cardiac allograft model, whose results suggest that Foxp3 cells traffic to allografts and induce endothelial expression of IDO (43). Although we were initially surprised that late allografts contained Foxp3+ cells, given our quantitative RT-PCR data, Tregs are indisputably present in late allografts, and actually appear to increase numerically over time. The reason for this obvious discrepancy is unclear to us, but could include changes in mRNA stability, decreased Foxp3 protein expression per positive cell, or simple methodological differences from others who report Foxp3 mRNA expression relative to numbers of CD3+ cells (27). Whatever the cause, this early erroneous finding was fortuitous because it suggested to us the possibility of an alternate mechanism of regulation. Although not direct proof, our data taken together with that of others support the hypothesis that RDCs and Tregs might play complementary roles in tolerance induction, with Tregs possibly conditioning DCs to become RDCs capable of controlling alloresponses to donor antigen.,
Although the data presented most strongly support transition away from a TGF-β mechanism to an IDO mediated mechanism of regulation, an alternate early interpretation was that our findings might represent clonal deletion of alloreactive T-cells, or onset of anergy. Mechanisms such as clonal deletion and anergy have been documented in other transplant tolerance models (49, 50). Nonetheless in our model, blocking IDO restored late alloantigen responsiveness, confirming that alloreactive T-cells are still present, and not completely deleted from allograft acceptors. In addition, renal allograft acceptors maintain their ability to reject third party antigens at least 150 days post transplant (Figure 3), confirming specificity of this regulation, and further lack of general anergy. Although we have not ruled out the possibility that there is partial clonal deletion of alloreactive cells occurring by day 150, there are clearly alloreactive splenocytes that remain in these recipients that must be controlled by some regulatory mechanism.
In addition to regulation of cellular alloimmune responses, our data also suggest that spontaneous renal allograft acceptors might display regulation of humoral alloreactivity. We have found that cardiac (51) and renal (Figure 2) allograft acceptors display circulating donor-reactive IgG alloantibodies, but despite these antibodies, both demonstrate long term graft acceptance and function. It is also interesting that despite persistence of preformed antibody in these mice, 30% of renal allograft acceptors at day 150 do not reject donor-matched skin allografts (Figure 3). While one could argue that graft reactive antibodies induced in our model are of low affinity, of the wrong isotype, or of inadequate titer to disrupt tolerance, it is curious that this same antigenic challenge (skin) in naïve C57/BL6 recipients causes 50 fold higher antibody titers than those seen in our renal allograft acceptors challenged 60 days post-transplant. It has been previously shown that under certain circumstances, B-cell responses are dependent upon T-cell function (52, 53), so it is possible that spill-over of regulation from the cellular to the humoral immune response might be occurring in our model. While our data are inadequate to prove this hypothesis, and the argument for humoral regulation is admittedly not as strong as that for cellular regulation, our results do not dispute this possibility. Alternatively, there are growing data suggesting that under proper circumstances, B-cells are tolerogenic rather than immunogenic (54), and can in fact induce Tregs (55). Analogous to our changing understanding of the importance of alloantibody responses during human transplantation, which continues to evolve (56), the importance of antibody and B-cells in our model of allograft acceptance will clearly require further exploration.
Our model of renal allograft acceptance demonstrates a strong association with immunoregulatory mechanisms, but to fit the strictest definition of tolerance, animals should accept subsequent donor matched skin grafts. Interestingly, ~33% of late renal allograft acceptors became fully tolerant to donor antigen, compared to 0% of early renal allograft acceptors (Figure 3A). Additionally, no late renal allograft acceptors demonstrated renal allograft loss when challenged with donor skin, compared to 25% losses in early renal graft acceptors. Encouraged by our day 150 skin transplant acceptance, we performed day 180 donor matched skin transplants in another cohort, but saw no further improvement in tolerance (Figure 3C). It is possible that given more time, this tolerance might progress to involve a larger percentage of graft acceptors, and these studies are currently ongoing. Nonetheless, it is extremely exciting that true tolerance may be developing over time, despite presence of donor reactive T and B-cells. It is highly intriguing that at the time when some animals become tolerant to donor skin, there are DCs in their spleens capable of regulating donor responses. Although not proven here, we suspect that it is not simple coincidence that true tolerance occurs concomitant with peripheral (splenic) appearance of these RDCs. Thus, persistence of alloreactive cells in the setting of true tolerance suggests that regulation may be critically important to development of tolerance.
Based on data presented in this manuscript we thus propose a model of evolution of immune regulatory mechanisms associated with spontaneous renal allograft acceptance (Figure 10). In this model, donor-reactive immune responses are regulated by TGF-β and Tregs during early stages of renal allograft acceptance. TGF-β is known to down-regulate immune responses, and its presence in both grafts and peripherally may suppress early donor-reactive immune responses preventing graft destruction. This transient mechanism appears operative between 30 and 120 days post-transplant, and may be responsible for induction of allograft acceptance. Tregs are capable of inducing tryptophan catabolism in antigen presenting cells (44, 45, 57), and intragraft Tregs may thus facilitate development of RDCs and expression of the immunoregulatory molecule IDO. We posit that these different mechanisms are not mutually exclusive, and might in fact be complementary, as illustrated in Figure 10.
Figure 10. Model of Immune Regulatory Mechanisms Associated With Renal Allograft Acceptance.

In summary, metastable immunologic regulation involving TGF-β and Tregs might provide time and conditions that allow or promote development of immunoregulatory DCs. Presence of RDCs in both grafts and spleens of spontaneous allograft acceptors is extremely exciting, particularly given their apparent ability to modulate alloresponses. Although we feel that these evidence are compelling, we recognize that we have yet to directly prove that Tregs or RDCs are responsible for graft acceptance or tolerance in this model, and these studies are ongoing.
Acknowledgments
The authors would like to thank Kelly Nye, Misty Bear, Christina Sass, Bree Heestand, Nicholas DiPaola, and Jacob Iselin for their technical assistance with these studies.
Supported by NIH RO1 AI053094 (CHC) and a grant from the Roche Organ Transplant Foundation (RBC)
Footnotes
“This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org..”
References
- 1.Russell PSCC, Colvin RB, Plate JM. Induced immune destruction of long-surviving, H-2 incompatible kidney transplants in mice. J Exp Med. 1978;147:1469–1486. doi: 10.1084/jem.147.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell PSCC, Colvin RB, Plate JM. Kidney transplants in mice. An analysis of the immune status of mice bearing long-term, H-2 incompatible transplants. J Exp Med. 1978;147:1449–1468. doi: 10.1084/jem.147.5.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bickerstaff AA, Wang JJ, Pelletier RP, Orosz CG. Murine renal allografts: spontaneous acceptance is associated with regulated T cell-mediated immunity. J Immunol. 2001;167:4821–4827. doi: 10.4049/jimmunol.167.9.4821. [DOI] [PubMed] [Google Scholar]
- 4.Bickerstaff AA, Wang JJ, Pelletier RP, Orosz CG. The graft helps to define the character of the alloimmune response. Transpl Immunol. 2002;9:137–141. doi: 10.1016/s0966-3274(02)00036-9. [DOI] [PubMed] [Google Scholar]
- 5.Bickerstaff A, Orosz C. Evidence for a limited contribution of immune regulation to cardiac allograft acceptance. Hum Immunol. 2002;63:935–947. doi: 10.1016/s0198-8859(02)00447-0. [DOI] [PubMed] [Google Scholar]
- 6.Degauque N, Lair D, Dupont A, Moreau A, Roussey G, Moizant F, Hubert FX, Louvet C, Hill M, Haspot F, Josien R, Usal C, Vanhove B, Soulillou JP, Brouard S. Dominant tolerance to kidney allografts induced by anti-donor MHC class II antibodies: cooperation between T and non-T CD103+ cells. J Immunol. 2006;176:3915–3922. doi: 10.4049/jimmunol.176.7.3915. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Schlachta C, Duff J, Stiller C, Grant D, Zhong R. Improved techniques for kidney transplantation in mice. Microsurgery. 1995;16(2):103–109. doi: 10.1002/micr.1920160212. [DOI] [PubMed] [Google Scholar]
- 8.Engers H, Thomas K, Cerottini JC, Brunner K. Generations of cytotoxic T lymphocytes in immune spleen cells to subcellular alloantigens. J Immunol. 1975;115:356–360. [PubMed] [Google Scholar]
- 9.Orosz CG. Local cellular immunology of experimental transplant vascular sclerosis. Clin Transpl. 1996;10:100–103. [PubMed] [Google Scholar]
- 10.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 11.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 12.Stassen M, Jonuleit H, Muller C, Klein M, Richter C, Bopp T, Schmitt S, Schmitt E. Differential regulatory capacity of CD25+ T regulatory cells and preactivated CD25+ T regulatory cells on development, functional activation, and proliferation of Th2 cells. J Immunol. 2004;173:267–274. doi: 10.4049/jimmunol.173.1.267. [DOI] [PubMed] [Google Scholar]
- 13.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-β, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 14.Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, Koni PA, Iwashima M, Munn DH. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 15.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL, Jr, Mellor AL. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 16.Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol. 2002;168:3771–3776. doi: 10.4049/jimmunol.168.8.3771. [DOI] [PubMed] [Google Scholar]
- 17.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20:469–473. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 18.Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beutelspacher SC, Tan PH, McClure MO, Larkin DF, Lechler RI, George AJ. Expression of indoleamine 2,3-dioxygenase (IDO) by endothelial cells: implications for the control of alloresponses. Am J Transplant. 2006;6:1320–1330. doi: 10.1111/j.1600-6143.2006.01324.x. [DOI] [PubMed] [Google Scholar]
- 20.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 21.Derks RA, Jankowska-Gan E, Xu Q, Burlingham WJ. Dendritic cell type determines the mechanism of bystander suppression by adaptive T regulatory cells specific for the minor antigen HA-1. J Immunol. 2007;179:3443–3451. doi: 10.4049/jimmunol.179.6.3443. [DOI] [PubMed] [Google Scholar]
- 22.Fallarino F, Vacca C, Orabona C, Belladonna ML, Bianchi R, Marshall B, Keskin DB, Mellor AL, Fioretti MC, Grohmann U, Puccetti P. Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol. 2002;14:65–68. doi: 10.1093/intimm/14.1.65. [DOI] [PubMed] [Google Scholar]
- 23.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 24.Orosz CG, Bickerstaff AA, Adams P, Hennessy P, Pelletier RP. Evidence that a similar range of alloimmune responses can develop in murine and human allograft recipients. Transpl Immunol. 2002;9:143–147. doi: 10.1016/s0966-3274(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 25.Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, Kroemer G, Martin F, Chauffert B, Zitvogel L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202:919–929. doi: 10.1084/jem.20050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 27.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 28.Adeegbe D, Bayer AL, Levy RB, Malek TR. Cutting edge: allogeneic CD4+CD25+Foxp3+ T regulatory cells suppress autoimmunity while establishing transplantation tolerance. J Immunol. 2006;176:7149–7153. doi: 10.4049/jimmunol.176.12.7149. [DOI] [PubMed] [Google Scholar]
- 29.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 31.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 32.Bickerstaff AA, Xia D, Pelletier RP, Orosz CG. Mechanisms of graft acceptance: evidence that plasminogen activator controls donor-reactive delayed-type hypersensitivity responses in cardiac allograft acceptor mice. J Immunol. 2000;164:5132–5139. doi: 10.4049/jimmunol.164.10.5132. [DOI] [PubMed] [Google Scholar]
- 33.Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol. 2004;172:923–928. doi: 10.4049/jimmunol.172.2.923. [DOI] [PubMed] [Google Scholar]
- 34.Graca L, Thompson S, Lin CY, Adams E, Cobbold SP, Waldmann H. Both CD4+CD25+ and CD4+CD25− regulatory cells mediate dominant transplantation tolerance. J Immunol. 2002;168:5558–5565. doi: 10.4049/jimmunol.168.11.5558. [DOI] [PubMed] [Google Scholar]
- 35.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-β induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 36.Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med. 2002;195:1641–1646. doi: 10.1084/jem.20012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daley SR, Ma J, Adams E, Cobbold SP, Waldmann H. A key role for TGF-beta signaling to T cells in the long-term acceptance of allografts. J Immunol. 2007;179:3648–3654. doi: 10.4049/jimmunol.179.6.3648. [DOI] [PubMed] [Google Scholar]
- 38.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 39.Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, Waldmann H, Cobbold S, Adams E, Munn DH. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–1401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 40.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 41.Alexander AM, Crawford M, Bertera S, Rudert WA, Takikawa O, Robbins PD, Trucco M. Indoleamine 2,3-dioxygenase expression in transplanted NOD Islets prolongs graft survival after adoptive transfer of diabetogenic splenocytes. Diabetes. 2002;51:356–365. doi: 10.2337/diabetes.51.2.356. [DOI] [PubMed] [Google Scholar]
- 42.Min WP, Zhou D, Ichim TE, Strejan GH, Xia X, Yang J, Huang X, Garcia B, White D, Dutartre P, Jevnikar AM, Zhong R. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J Immunol. 2003;170:1304–1312. doi: 10.4049/jimmunol.170.3.1304. [DOI] [PubMed] [Google Scholar]
- 43.Thebault P, Condamine T, Heslan M, Hill M, Bernard I, Saoudi A, Josien R, Anegon I, Cuturi MC, Chiffoleau E. Role of IFNgamma in allograft tolerance mediated by CD4+CD25+ regulatory T cells by induction of IDO in endothelial cells. Am J Transplant. 2007;7:2472–2482. doi: 10.1111/j.1600-6143.2007.01960.x. [DOI] [PubMed] [Google Scholar]
- 44.Munn DH, Sharma MD, Mellor AL. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol. 2004;172:4100–4110. doi: 10.4049/jimmunol.172.7.4100. [DOI] [PubMed] [Google Scholar]
- 45.Feunou P, Vanwetswinkel S, Gaudray F, Goldman M, Matthys P, Braun MY. Foxp3+CD25+ T Regulatory Cells Stimulate IFN-{gamma}-Independent CD152-Mediated Activation of Tryptophan Catabolism That Provides Dendritic Cells with Immune Regulatory Activity in Mice Unresponsive to Staphylococcal Enterotoxin B. J Immunol. 2007;179:910–917. doi: 10.4049/jimmunol.179.2.910. [DOI] [PubMed] [Google Scholar]
- 46.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–6761. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 47.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I, Horenstein AL, Fiore F, Massaia M, Colombo MP, Baccarani M, Lemoli RM. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood. 2007;109:2871–2877. doi: 10.1182/blood-2006-07-036863. [DOI] [PubMed] [Google Scholar]
- 49.Shirasugi N, Adams AB, Durham MM, Lukacher AE, Xu H, Rees P, Cowan SR, Williams MA, Pearson TC, Larsen CP. Prevention of chronic rejection in murine cardiac allografts: a comparison of chimerism- and nonchimerism-inducing costimulation blockade-based tolerance induction regimens. J Immunol. 2002;169:2677–2684. doi: 10.4049/jimmunol.169.5.2677. [DOI] [PubMed] [Google Scholar]
- 50.Kurtz J, Shaffer J, Lie A, Anosova N, Benichou G, Sykes M. Mechanisms of early peripheral CD4 T-cell tolerance induction by anti-CD154 monoclonal antibody and allogeneic bone marrow transplantation: evidence for anergy and deletion but not regulatory cells. Blood. 2004;103:4336–4343. doi: 10.1182/blood-2003-08-2642. Epub 2004 Feb 4312. [DOI] [PubMed] [Google Scholar]
- 51.VanBuskirk AM, Wakely ME, Sirak JH, Orosz CG. Patterns of allosensitization in allograft recipients: Long-term cardiac allograft acceptance is associated with active alloantibody production in conjunction with active inhibition of alloreactive delayed-type hypersensitivity. Transplantation. 1998;65(8):1115–1123. doi: 10.1097/00007890-199804270-00017. [DOI] [PubMed] [Google Scholar]
- 52.Gold MR, DeFranco AL. Biochemistry of B lymphocyte activation. Adv Immunol. 1994;55:221–295. doi: 10.1016/s0065-2776(08)60511-8. [DOI] [PubMed] [Google Scholar]
- 53.Jelinek DF, Lipsky PE. Regulation of human B lymphocyte activation, proliferation, and differentiation. Adv Immunol. 1987;40:1–59. doi: 10.1016/s0065-2776(08)60237-0. [DOI] [PubMed] [Google Scholar]
- 54.Raimondi MT. Engineered tissue as a model to study cell and tissue function from a biophysical perspective. Curr Drug Discov Technol. 2006;3:245–268. doi: 10.2174/157016306780368126. [DOI] [PubMed] [Google Scholar]
- 55.Chen X, Jensen PE. Cutting edge: primary B lymphocytes preferentially expand allogeneic FoxP3+ CD4 T cells. J Immunol. 2007;179:2046–2050. doi: 10.4049/jimmunol.179.4.2046. [DOI] [PubMed] [Google Scholar]
- 56.Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. 2005;5:807–817. doi: 10.1038/nri1702. [DOI] [PubMed] [Google Scholar]
- 57.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]