

Abstract
We review development of evidence and current perceptions of the multiple and significant functions of cardiac troponin I in regulation and modulation of cardiac function. Our emphasis is on the unique structure function relations of the cardiac isoform of troponin I, especially regions containing sites of phosphorylation. The data indicate that modifications of specific regions cardiac troponin I by phosphorylations either promote or reduce cardiac contractility. Thus, a homeostatic balance in these phosphorylations is an important aspect of control of cardiac function. A new concept is the idea that the homeostatic mechanisms may involve modifications of intra-molecular interactions in cardiac troponin I.
Keywords: Cardiac, Sarcomeres, Adrenergic stimulation, Mechanics, Kinases, Phosphatases
In the time since the troponin discovery and naming by the laboratory of Setsuro Ebashi [1], there has been a constant stream of evidence indicating the substantial role of modifications in cardiac troponin (cTn) as a critical control element in the body’s response to physiological stressors such as the hemodynamic demands of exercise. It is now widely recognized that regulatory processes at the level of the sarcomeres and the hetero–trimeric Tn complex involve not only the reception of Ca2+ by cTnC, but also transduction of this Ca-binding signal by cTnI and cTnT [2,3]. Multiple interactions of cTnI and cTnT with actin and tropomyosin (Tm) control the number and kinetics of interactions of cross-bridges with the thin filament. Our focus here on cTnI serves to highlight the significant impact of modifications in the function of Tn in working cardiac myocytes and in the integrated physiological responses to exercise, which we have reviewed previously [4,5]. Evidence summarized below indicates that phosphorylation of cTnI by adrenergic signaling cascades is an indispensable control mechanism in matching cardiac output to venous return and myocyte dynamics to heart rate.
Specific modifications in troponin I affect the dynamics and intensity of the heart beat
Elucidation of the function of an N-terminal extension of ~30 amino acids with sites (Ser-23, Ser-24) of phosphorylation by protein kinase A (PKA), not present in the fast skeletal (fsTnI) or slow skeletal isoforms (ssTnI), provided the first evidence that cTnI may have a special role in the control of cardiac contractility [6,7]. Although it had been reported that phosphorylation of this region depressed sarcomeric response to Ca2+ and increased cross-bridge kinetics [2,3], studies with transgenic models have provided the strongest evidence for a critical role of Tn, especially cTnI, in cardiac function. The first series of studies were carried out with a transgenic mouse model expressing ssTnI (TG-ssTnI) to the exclusion of cTnI in the cardiac compartment [8]. We chose the slow skeletal isoform for these studies inasmuch as it is naturally expressed in the embryonic/neonatal period. Hearts of TG-ssTnI mice had slower relaxation kinetics, depressed effects of β-adrenergic stimulation [8], and protection against acidosis [9,10] and ischemia/reperfusion injury [11]. All of these properties are properties of preparations from neonatal rat hearts, and indicated that the special cardiac function in the neonatal period is importantly controlled by the isoform of TnI. More recent studies using mutant forms of cTnI with either pseudo-phosphorylated PKA sites (cTnI-S23D, S24D) or non-phosphorylatable mutant cTnI-S23A, S24A strongly support a critical role of cTnI phosphorylation in the control of cardiac contractility. Yasuda et al. [12] concluded from their studies of hearts of mice expressing these mutant forms of cTnI that phosphorylation of cTnI is a significant factor in cardiac relaxation with an importance similar to that of phospholamban phosphorylation. Takimoto et al. [13] also reported that hearts of mice expressing cTnI-S23D/S24D had a constitutive enhancement of rate-dependent increases in systolic and diastolic function in vivo. Along these lines, Varian and Janssen [14] reported a frequency dependent decrease in myofilament sensitivity to Ca2+ associated with increasing heart rates and most likely attributable to cTnI phosphorylation. The role of cTnI phosphorylation in the relaxant effect of adrenergic stimulation has also been emphasized in work reported by Stelzer et al. [15] in studies of mice expressing cTnI-S23D/S24D against a cTnI and myosin binding protein C null background. These studies together with others provide compelling evidence for a prominent role of cTnI phosphorylation in the maintenance of power and frequency response in ejecting ventricles [13,16–19]. Apart from their significant effects on cardiac dynamics, cTnI phosphorylation at the PKA sites affects length dependence of activation (LDA) [20]. Cooperative mechanisms involving feedback effects of strongly bound cross-bridges are important for LDA and are also sensitive to isoform specific structure in cardiac TnI [21].
Sites of phosphorylation on cTnI distinct from the PKA sites have different effects on cardiac dynamics and myofilament sensitivity to Ca2+ and thus have led to the concept of “yin-yang” regulation of the function of cTnI dependent on the relative balance of phosphorylation of these sites [22,23]. Sites at Ser-42, and Ser-44 are substrates mainly for protein kinase C, and when phosphorylated induce a depression in maximum tension, and a decrease in cross-bridge kinetics [24–26], as reflected in a depression of thin filament sliding in the motility assay and ATPase rate. On the other hand, it is apparent that phosphorylation of Thr-143 induces an increase in sensitivity to Ca2+ [27] and an apparent depression in cross-bridge kinetics [25]. As reviewed elsewhere, phosphorylation of sites at Ser-42, Ser-44, and Thr-143 may be considered maladaptive, when and if they predominate as may occur in disorders of the heart [28]. cTnI-Ser-150, a highly conserved and strategically located amino acid, is also a substrate for phosphorylation by P21 activated kinase [29], and when phosphorylated induces an increase in Ca2+-sensitivity. However, at this time the physiological significance of cTnI-S150 phosphorylation is poorly understood.
Molecular mechanisms of cTnI function in Ca2+ and crossbridge dependent activation of cardiac sarcomeres
Structure–function analysis of cTnI reveals mechanisms of the ability of this single protein to control and modulate sarcomeric activation and dynamics [2,3]. As illustrated in Fig. 1, cTnI is a rod-like and flexible protein with the following distinct regions: a cardiac specific N-terminal extension region, an IT-arm region, the inhibitory region (Ip), the switch region (H3), and the C-terminal mobile domain. The IT-arm region consists of two α-helices that interact with the C-lobe of cTnC and the α-helix of the C-terminal domain of TnT (T2). Apart from IT-arm region, the rest of TnI molecule appears flexible and, as described below, the switch region and the C-terminal mobile domain undergo disordered–ordered structural transitions depending on the Ca-bound state of the regulatory Ca2+-binding site in the N-lobe of cTnC.
Fig. 1.
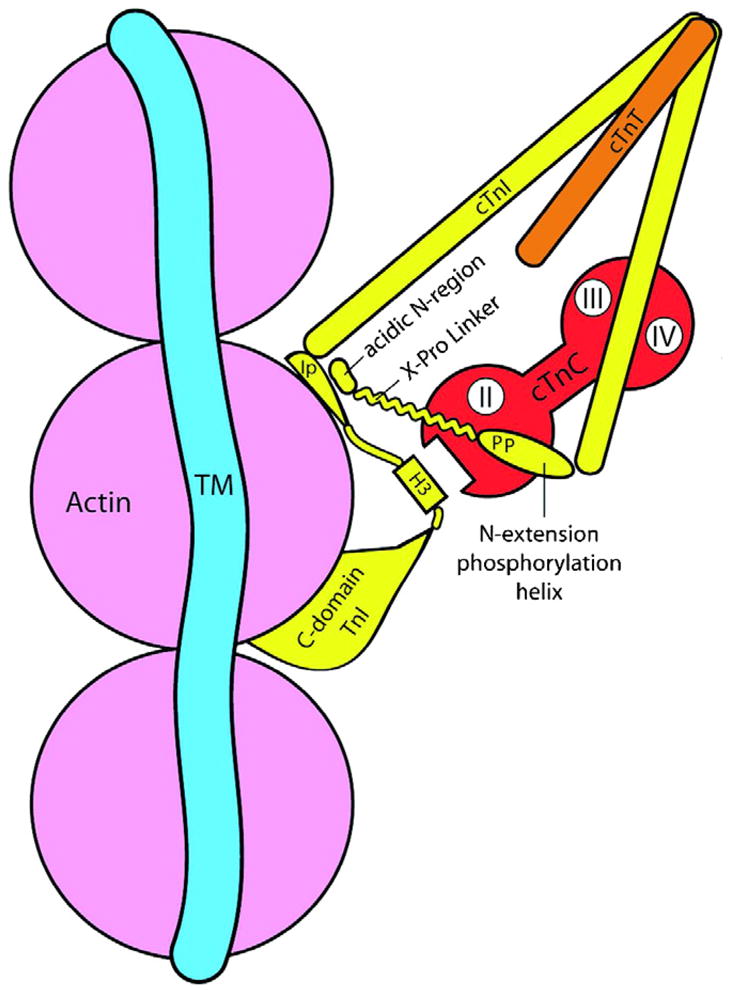
Model illustrating interactions of troponin (Tn) components in the diastolic state and with phosphorylation of Ser-23, Ser-24 in the unique N-terminal extension containing a phosphorylation helix, a proline helix linker and an acidic region. Tm, tropomyosin, cTnI, cardiac troponin I, cTnT, cardiac TnT, H3, the switch peptide that binds to a hydrophobic patch on cTnC as indicated, Ip, a basic inhibitory peptide. cTnC is illustrated in apo state and indicates the N-lobe regulatory Ca-binding domain (II) and C-lobe slowly exchanging Ca/Mg sites in the mobile C-domain demonstrates a second actin binding site. The model emphasizes the potential for the phosphorylated N-extension of cTnI to interact with the inhibitory peptide. See text and Ref. [47] for details.
In the resting state (diastole), which is depicted in Fig. 1, the inhibitory region and the second actin-binding site in the mobile domain interact with actin to prevent strong, force generating reactions with myosin heads. Zero-length chemical cross-linking experiments investigating interactions between fsTnI and actin indicated that the inhibitory region interacts with the N-terminus of actin, whereas photo-active cross-linking experiments with BP-mal indicated the inhibitory region, as well as Cys-133 of fsTnI, which corresponds to Cys-166 of cTnI near the second-actin binding site, are close to Met-47 of actin in the reconstituted thin filaments. The structure of the inhibitory region when it binds to actin is not known. From sequence comparisons with profilin, which interacts with the same of region of actin as TnI, and the crystal structure of the actin–profilin complex, Tung et al. suggested that the inhibitory region may form a β-hairpin conformation when actin bound [30]. The atomic model for the interaction between the mobile domain, which includes the second actin binding site and actin-Tm filament, was proposed based on the NMR structure of the mobile domain determined by Murakami et al. [31]. Yet ESR experiments with reconstituted thin filaments indicate that most of the mobile domain remains flexible even in the absence of Ca2+ [32]. Investigation [33] of the effects of the inhibitory region showed that actin-binding peptides derived from the myosin head were released from actin when a peptide corresponding to the inhibitory region was added to actin or actin-Tm. On the other hand, when the peptide corresponding to the second actin-binding site was added to actin or actin-Tm, the myosin peptides were not released from the actin surface. These observations indicated that the binding of the inhibitory region, but not the second actin binding site, induces a structural change in actin and prevents actin–myosin strong interaction. On the basis of results of experiments with cardiomyopathy-linked mutations of cTnI, we proposed that the functional role of the second actin-binding site may be to increase the local concentration of the cTnI molecule for actin to facilitate the interaction of the inhibitory region and actin [34].
Upon Ca2+-binding to the regulatory site of cTnC, the switch region of TnI binds to a newly exposed hydrophobic patch in the N-lobe of TnC [35]. This interaction releases both the inhibitory region and the second actin-binding site from actin. Based on their crystal structures of fsTn complex in the presence and absence of Ca2+, Vinogradova et al. [36] proposed a model for Tn activation and relaxation, in which the central linker region of TnC undergoes disordered to helical structure upon activation, whereas the inhibitory region undergoes short helix to flexible extended conformation. Consistent with their proposal, experimental results suggest that the inhibitory region undergoes a conformational switch from a β-hairpin type structure when bound to actin to a more elongated structure having a propensity to form a helix [36–38]. The elongated inhibitory region interacts with the central helical region of TnC to stabilize the active form of the Tn complex. It remains unclear whether this structural transition of the linker region of the Tn complex is essential or not for regulation. An interesting aspect of the specialization of the cTnI isoform is an alanine at position 164 near the mobile domain, which is replaced by His at the homologous position in ssTnI. Dargis et al. [39] reported that this substitution is of significance in studies in which reconstitution with the mutant, cTnI (A164H), rendered cardiac myofilament preparations relatively more sensitive to Ca2+ and relatively less sensitive to deactivation by acidic pH. Incorporation of cTnI (A164H) into hearts of transgenic mice recapitulated many of the effects of ssTnI described above, while retaining phosphorylation sites [40].
The role of the IT-arm region of the Tn complex has been considered to be structural rather than functional in that it is apparent that the structure of the IT-arm region of the Tn complex remains unchanged during diastole-systole cycle. However, the relative orientation of the IT-arm to actin filament axis is more sensitive to strong cross-bridge attachment than to Ca2+-binding to the regulatory sites [41]. Moreover, recent data indicate that the IT-arm region is involved in the cross-bridge dependent activation of myofilaments [21]. These studies demonstrated that a region of cTnI in the near N-terminal region (surrounding Ala-65), which interacts with C-terminal regions of cTnC and cTnT, is of particular significance in transducing signaling of thin filament activation by strong cross-bridges.
Molecular mechanisms of the effects of cTnI phosphorylation on cardiac function
Phosphorylation of cTnI at sites in the unique N-terminus may exert their functional effects by both altered intermolecular and intra-molecular interactions. The major inter-molecular interaction is with the N-lobe, regulatory domain of cTnC, which occurs in the absence of phosphorylation. Although structural and biochemical evidence indicate that this interaction is relatively weak [42–45], it establishes a relatively high affinity Ca2+-binding state to the N-lobe of cTnC [46]. With phosphorylation of cTnI Ser-23/Ser-24, interaction with the N-lobe is further weakened and Ca2+-affinity of the N-lobe is reduced in association with enhanced off rate for Ca2+ exchange.
Recent evidence has provided insights into the disposition of the now free and flexible N-extension and predicted an intra-molecular interaction between the N-extension and the inhibitory region [47]. As mentioned above, Ca2+ dissociation from N-lobe or regulatory domain of cTnC is associated with a helical to β-hairpin type conformational change in the inhibitory region. PKA phosphorylation at Ser-23/Ser-24 in the cardiac N-extension of cTnI decreases Ca2+-sensitivity and increases the conformational transition rate of the inhibitory region [38]. These results demonstrate a structural linkage between the inhibitory region and the cardiac N-extension of cTnI. Both regions of cTnI have unique cardiac isoforms. Further support for a linkage between the cardiac N-extension and inhibitory region comes from the human mutation Arg-145 to Gly resulting in the loss of a basic charge in the inhibitory region linked to development of familial hypertrophic cardiomyopathy (see [48] for review). This mutation leads to a reduced ability to inhibit the actin-activated ATPase and an increase in Ca2+-sensitivity [49–51]. These studies suggest that the R145G mutation prevents the inhibitory region of cTnI from properly interacting with actin-tropomyosin in the thin filament. In addition, cTnI carrying the R145G mutation is insensitive to phosphorylation of the two adjacent Ser residues in the cardiac N-extension [52]. Thus, replacement of a basic Arg residue in the inhibitory region of cTnI with Gly appears to disrupt the effect of PKA phosphorylation at Ser-23/Ser-24 in the cardiac N-extension.
These observations suggested that interactions between the inhibitory region of cTnI and actin could be modulated by the bisphosphorylated cardiac N-extension. While the inhibitory region in both skeletal and cardiac TnI has been shown to be helical in the absence of Ca2+-binding to the regulatory domain [36], bioinformatics analyses supports a β-turn type conformation for this region when bound to actin [29], as mentioned. Recently, the determined structure for the cardiac N-extension bisphosphorylated Ser-23/Ser-24 was found to contain a C-terminal helix (residues 21–30) containing the phosphorylation motif, an extended poly(L-proline)II helix (residues 11–19), and an acidic N-terminus with some propensity for helical structure [47]. Using this structure, the X-ray crystal structure of the cTn core, and uniform density models of the cTn subunits derived from neutron contrast variation data, atomic models were built that show the conformational transition induced by PKA phosphorylation at Ser-23/Ser-24 of cTnI and suggest a molecular linkage between the cardiac N-extension and the inhibitory region of cTnI [47]. In the absence of phosphorylation, structural and biochemical studies show that the cardiac N-extension interacts weakly with the N-lobe or regulatory domain of cTnC [42–44]. Binding to the N-lobe is further weakened by phosphorylation of Ser-23/Ser-24 in the N-extension. In addition, neutron contrast variation data demonstrated that bisphosphorylation resulted in a dramatic bending of the rod-like cTnI at the N-terminal end that interacts with cTnC, resulting in a broader, shorter structure [53]. The phosphorylation-induced bending of cTnI suggests an alternative binding mode for the N-extension of cTnI. A model maximizing electrostatic interactions generated by constrained docking yielded a single conformation requiring a hinge bending motion between residues 33–42 of cTnI [47]. This resulted in a sharp bend in the cTnI similar to that observed in the molecular envelops for cTn components derived from small-angle scattering with deuterium labeling. In the model, the acidic amino terminus of the N-extension interacts with basic residues within the inhibitory region of cTnI. The phosphorylation–induced bending of cTnI is consistent with biochemical studies showing that the axial ratio decreases upon phosphorylation and an asymmetrical to a more symmetrical change in shape consistent with a shorter, broader structure [54–56].
A two-step molecular mechanism was proposed to account for the observed physiological consequences of PKA phosphorylation at Ser-23/Ser-24 of cTnI. Interactions between the cardiac N-extension of cTnI and the N-lobe of cTnC form the first step of the phosphorylation switch and affect myofilament Ca2+-sensitivity. The second step of the phosphorylation switch results from bisphosphorylation at Ser-23/Ser-24 of cTnI and weakening of interactions with the N-lobe of cTnC. This induces a bend in cTnI such that the acidic N-terminus of the cardiac N-extension is positioned for interaction with the inhibitory region of cTnI. As a result, weakening the interaction of the inhibitory region with actin and altering cross-bridge reactions. This model can be extended to provide an enhanced molecular understanding of phosphorylation at Ser-42, Ser-44, and Thr-143 in cTnI.
Cardiac TnI also can be phosphorylated by PKC at Ser-42 and Ser-44 in the N-domain and Thr-143 in the inhibitory region of cTnI [57]. Phosphorylation at Ser-42/Ser-44 is known to decrease maximal actomyosin Mg2+ATPase activity and Ca2+-sensitivity by stabilizing the inactive state of the thin filament [24–26,58]. Residues 42 and 44 are located in the N-domain of cTnI near the interaction site with the C-lobe hydrophobic crevice of cTnC. Comparison of the cTnC bound structures for the N-domain of cTnI having Ser-42/Ser-44 mutated to Asp, to mimic the phosphorylated state, with the N-domain of cTnI demonstrated that negative charge at residues 42 and 44 stabilized and extended the amino terminus of the N-domain helix (Howarth, Finley, and Rosevear, unpublished). In agreement with the structural results, bioinformatics analysis predicted that introducing negative charge at positions 42 and 44 of cTnI by mutation to Glu extends the amino terminus of the N-domain helix [25]. Extension and stabilization of the N-domain helix would alter the dynamical properties at the neighboring hinge region, residues 32–41, of cTnI required for PKA phosphorylation-dependent bending of the cardiac specific N-extension of cTnI. Thus, PKC phosphorylation at Ser-42/Ser-44 of cTnI would decrease mobility of the hinge region of cTnI and thus have opposite effects on Ca2+-sensitivity and cross-bridge formation.
PKC is also responsible for phosphorylation at Thr-143 located in the inhibitory region of cTnI. This region is flexible and not observed in the core crystal structure of cardiac troponin [59]. While the structural role of Thr-143 phosphorylation remains to be elucidated, a model can be suggested based on our current understanding of the effects of PKA and PKC phosphorylation of cTnI. Introduction of negative charge at Thr-143, via phosphorylation, would be predicted to decrease electrostatic interactions between the inhibitory region and both the acidic amino terminus of the cardiac N-extension of cTnI and actin. The end result would be stabilization of a more extended structure resulting in an increase in cross-bridge kinetics. Consistent with this hypothesis, introduction of negative charge at Thr-143 appeared to potentate the effects of PKA phosphorylation at Ser-23/Ser-24 in a transgenic mouse model [60].
Acknowledgments
This chapter is dedicated to the memory of Professor Setsuro Ebashi. We are grateful to our many colleagues for collaborations permitting the study of troponin at many levels of organization in the heart. Work described here was supported in part by grants from the NIH NHLBI and AHA.
References
- 1.Ebashi S, Kodama A. A new protein factor promoting aggregation of tropomyosin. J Biochem. 1965;58:107–108. doi: 10.1093/oxfordjournals.jbchem.a128157. [DOI] [PubMed] [Google Scholar]
- 2.Solaro RJ. Modulation of cardiac myofilament activity by protein phosphorylation. In: Page E, Fozzard H, Solaro RJ, editors. Handbook of Physiology: Section 2. The Cardiovascular System. Vol. 1. Oxford University Press; New York: 2001. pp. 264–300. [Google Scholar]
- 3.Kobayashi T, Solaro RJ. Calcium, thin filaments, and integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 4.Solaro RJ, Westfall MV. Physiology of the myocardium. In: Sellke FW, del Nido PJ, Swanson SJ, editors. Surgery of the Chest. Elsevier Sanders; Philadelphia: 2005. pp. 767–779. [Google Scholar]
- 5.Solaro RJ. Integration of myofilament response to Ca2+ with cardiac pump regulation and pump dynamics. Am J Physiol. 1999;277:S155–S163. doi: 10.1152/advances.1999.277.6.S155. [DOI] [PubMed] [Google Scholar]
- 6.Solaro RJ, Moir AJG, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenalin in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 7.Ray KP, England PJ. Phosphorylation of the inhibitory subunit of troponin and its effect on the calcium dependence of cardiac myofibril adenosine triphosphatase. FEBS Lett. 1976;70:11–16. doi: 10.1016/0014-5793(76)80716-8. [DOI] [PubMed] [Google Scholar]
- 8.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AM, Moss RJ, Solaro RJ, Leiden JM. Impaired cardiomycyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol (London) 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolska BM, Vijayan K, Arteaga GM, Konhilas J, Phillips RM, Kim R, Naya R, Leiden T, Kim RM, Naya R, Leiden T, Martin JM, de Tombe AF, Solaro RJ. Expression of slow skeletal troponin I in adult heart muscle prevents force decline during acidic conditions. J Physiol (London) 2001;536(3):863–870. doi: 10.1111/j.1469-7793.2001.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urboniene D, Dias F, Peña JR, Walker LA, Solaro RJ, Wolska BM. Expression of slow skeletal troponin I in adult mouse heart helps to maintain the left ventricular systolic function during respiratory hypercapnia. Circ Res. 2005;97:70–77. doi: 10.1161/01.RES.0000173849.68636.1e. [DOI] [PubMed] [Google Scholar]
- 11.Arteaga GM, Warren CM, Milutinovic S, Martin AF, Martin RJS, Solaro RJ. Specific enhancement of sarcomeric response to Ca2+ protects murine myocardium against ischemia/reperfusion dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H2183–H2192. doi: 10.1152/ajpheart.00520.2005. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res. 2007;101:377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 13.Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 14.Varian KD, Janssen P. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–H2219. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]
- 15.Stelzer JE, Page JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 16.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and cross-bridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 17.Herron TJ, Korte FS, McDonald KS. Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ Res. 2001;89:1184–1190. doi: 10.1161/hh2401.101908. [DOI] [PubMed] [Google Scholar]
- 18.Pi Y, Zhang KR, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- 19.Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM. Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol. 2004;556:835–847. doi: 10.1113/jphysiol.2004.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konhilas JP, Irving TC, Wolska BM, Martin AF, Jweid E, Solaro RJ, deTombe PP. Troponin I in the heart: influence on length-dependent activation and inter-filament spacing. J Physiol (London) 2003;547(3):951–961. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel PL, Kobayashi T, Biesiadecki B, Davis J, Tikunova S, Wu S, Solaro RJ. Identification of a region of troponin I important in signaling cross-bridge dependent activation of cardiac myofilaments. J Biol Chem. 2007;282:183–193. doi: 10.1074/jbc.M512337200. [DOI] [PubMed] [Google Scholar]
- 22.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, van Buren P, Martin LA, Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 23.Roman BB, Goldspink PH, Spaite E, Urboniene D, McKinney R, Geenen DL, Solaro RJ, Buttrick PM. Inhibition of PKC phosphorylation of cTnI improves cardiac performance in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H2089–H2095. doi: 10.1152/ajpheart.00582.2003. [DOI] [PubMed] [Google Scholar]
- 24.Pyle WG, Sumandea MP, Solaro RJ, deTombe PP. Troponin I serines 43/45 and regulation of cardiac myofilament function. Am J Physiol Heart Circ Physiol. 2002;283:H1215–H1224. doi: 10.1152/ajpheart.00128.2002. [DOI] [PubMed] [Google Scholar]
- 25.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:1265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- 26.Mathur MC, Kobayashi T, Chalovich JM. Negative charges at protein kinase C sites of troponin I stabilize the inactive state of actin. Biophys J. 2008;94:542–549. doi: 10.1529/biophysj.107.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Grant JE, Doede CM, Sakthivel SJ, Walker JW. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J Mol Cell Cardiol. 2006;41:823–833. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 28.Solaro RJ, Wolska BM, Arteaga GM, Martin AF, Buttrick P, deTombe P. Modulation of thin filament activity in long and short term regulation of cardiac function. In: Solaro RJ, Moss RL, editors. Molecular Control Mechanisms in Striated Muscle Contraction. Kluwer Academic Publishers; The Netherlands: 2002. pp. 291–327. [Google Scholar]
- 29.Buscemi N, Foster DB, Neverova I, Van Eyk JE. p21-Activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ Res. 2002;91:509–516. doi: 10.1161/01.res.0000035246.27856.53. [DOI] [PubMed] [Google Scholar]
- 30.Tung CS, Wall ME, Gallagher SC, Trewhella J. A model of troponin-I in complex with troponin-C using hybrid experimental data: the inhibitory region is a beta-hairpin. Protein Sci. 2000;9:1312–1326. doi: 10.1110/ps.9.7.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami K, Yumoto F, Ohki S, Yasunaga T, Tanokura M, Wakabayashi T. Structural basis for Ca2+ regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 32.Aihara T, Ueki S, Nakamura M, Arata T. Calcium-dependent movement of troponin I between troponin C and actin as revealed by spin-labeling EPR. Biochem Biophys Res Commun. 2006;340:462–468. doi: 10.1016/j.bbrc.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Patchell VB, Gallon CE, Evans JS, Gao Y, Perry SV, Levine BA. The regulatory effects of tropomyosin and troponin-I on the interaction of myosin loop regions with F-actin. J Biol Chem. 2005;280:14469–14475. doi: 10.1074/jbc.M414202200. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi T, Solaro RJ. Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J Biol Chem. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 35.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 36.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca2+ regulated structural changes in troponin. Proc Natl Acad Sci USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong WJ, Robinson JM, Stagg S, Xing J, Cheung HC. Ca2+ induced conformational transition in the inhibitory and regulatory regions of cardiac troponin I. J Biol Chem. 2003;278:8686–8692. doi: 10.1074/jbc.M212886200. [DOI] [PubMed] [Google Scholar]
- 38.Dong WJ, An J, Xing J, Cheung HC. Structural transition of the inhibitory region of troponin I within the regulated cardiac thin filament. Arch Biochem Biophys. 2006;456:135–142. doi: 10.1016/j.abb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dargis R, Pearlstone JR, Barrette-Ng I, Edwards H, Smillie LB. Single mutation (A162H) in human cardiac troponin I corrects acid pH sensitivity of Ca2+ regulated actomyosin S1 ATPase. J Biol Chem. 2002;277:34662–34665. doi: 10.1074/jbc.C200419200. [DOI] [PubMed] [Google Scholar]
- 40.Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, LaCross NC, D’Alecy LD, Ingwall JS, Metzger JM. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med. 2006;12:181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 41.Sun YB, Brandmeier B, Urving M. Structural changes in troponin in response to Ca2+ and myosin binding to thin filaments during activation of skeletal muscle. Proc Natl Acad Sci USA. 2006;103:17771–17776. doi: 10.1073/pnas.0605430103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaponenko V, Abusamhadneh E, Abbott B, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear Rosevear PR. Effects of TnI phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J Biol Chem. 1999;274:16681–16684. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- 43.Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, Cheung HC, Rosevear PR. NMR anaylsis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 1999;453:107–112. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- 44.Ward DB, Brewer SM, Cornes MP, Trayer IP. A cross-linking study of the N-terminal extension of human cardiac troponin I. Biochemistry. 2003;42:10324–10332. doi: 10.1021/bi034495r. [DOI] [PubMed] [Google Scholar]
- 45.Ward DG, Brewer SM, Calvert MJ, Gallon CE, Gao Y, Trayer IP. Characterization of the interaction between the N-terminal extension of human cardiac troponin I and troponin C. Biochemistry. 2004;43:4020–4027. doi: 10.1021/bi036128l. [DOI] [PubMed] [Google Scholar]
- 46.Robertson SP, Johnson JD, Holroyde MJ, Kranias E, Potter JD, Solaro RJ. The effect of TnI phosphorylation on static and kinetic Ca binding by cardiac TnC. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- 47.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J Mol Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 48.Gomes AV, Potter JD. Molecular and cellular aspects of troponin cardiomyopathies. Ann NY Acad Sci. 2004;1015:214–224. doi: 10.1196/annals.1302.018. [DOI] [PubMed] [Google Scholar]
- 49.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 50.Kruger M, Zittrich S, Redwood C, Blaudeck N, James J, Robbins J, Pfitzer G, Stehle R. Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. J Physiol. 2005;564:347–357. doi: 10.1113/jphysiol.2004.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott K, Watkins H, Redwood CS. Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2000;275:22069–22074. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 52.Deng Y, Schmidtmann A, Redlich A, Westerdorf B, Jaquet K, Thieleczek R. Effects of phosphorylation and mutation R145G on human cardiac troponin I function. Biochemistry. 2001;40:593–602. doi: 10.1021/bi0115232. [DOI] [PubMed] [Google Scholar]
- 53.Heller WT, Finley NL, Dong WJ, Timmins P, Cheung HC, Rosevear PR, Trewhella J. Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry. 2003;42:7790–7800. doi: 10.1021/bi0341509. [DOI] [PubMed] [Google Scholar]
- 54.Dong WJ, Chandra M, Xing J, She M, Solaro RJ, Cheung HC. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry. 1997;36:6754–6761. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- 55.Dong WJ, Robinson JM, Xing J, Umeda PK, Cheung HC. An interdomain distance in cardiac troponin C determined by fluorescence spectroscopy. Protein Sci. 2000;9:280–289. doi: 10.1110/ps.9.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reiffert SU, Jaquet K, Heilmeyer LM, Jr, Ritchie MD, Geeves MA. Bisphosphorylation of cardiac troponin I modulates the Ca(2+)-dependent binding of myosin subfragment S1 to reconstituted thin filaments. FEBS Lett. 1996;384:43–47. doi: 10.1016/0014-5793(96)00274-8. [DOI] [PubMed] [Google Scholar]
- 57.Noland TA, Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, Kuo JF. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem. 1995;270:25445–25454. doi: 10.1074/jbc.270.43.25445. [DOI] [PubMed] [Google Scholar]
- 58.Sumandea MP, Burkart EM, Kobayashi T, de Tombe PP, Solaro RJ. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann NY Acad Sci. 2004;1015:39–52. doi: 10.1196/annals.1302.004. [DOI] [PubMed] [Google Scholar]
- 59.Takeda S, Yamashita A, Maeda KM, Maeda Y. Structure of teh core domain of human cardiac tropoinin in the Ca(2+) saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 60.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]