

Abstract
A number of antipsychotic and antidepressant drugs are known to increase the risk of ventricular arrhythmias and sudden cardiac death. Based largely on a concern over QT prolongation and the development of life-threatening arrhythmias, a number of antipsychotic drugs have been temporarily or permanently withdrawn from the market or their use restricted. Some antidepressants and antipsychotics have been linked to QT prolongation and the development of Torsade de pointes arrhythmias, whereas others have been associated with a Brugada syndrome phenotype and the development of polymorphic ventricular arrhythmias. This review examines the mechanisms and predisposing factors underlying the development of cardiac arrhythmias, and sudden cardiac death, associated with antidepressant and antipsychotic drugs in clinical use.
Keywords: antidepressants, antipsychotics, arrhythmias, Brugada syndrome, long QT syndrome, sudden cardiac death
1. Introduction
Sudden unexplained death in individuals with mental health problems was first described in 1849 and a link with antipsychotic drugs was postulated over 40 years ago [1,2]. Starting in early 1990, a clear relationship has been established between antipsychotics, prolongation of the QT interval of the electrocardiogram (ECG), an atypical polymorphic tachycardia known as Torsade de pointes (TdP), and sudden cardiac death (SCD). Based on a concern over QT and rate-corrected QT intervals (QTc) (QT corrected for heart rate) prolongation, a number of antipsychotic drugs have been temporarily or permanently withdrawn from the market or their use restricted. In other cases, close follow-up with an ECG has been recommended or a modification of label imposed. The list of antipsychotic drugs implicated includes pimozide, sertindole, thioridazine, mesoridazine, droperidol, sultopride and ziprasidone.
A link between antidepressants and arrhythmias was first suggested following the Cardiac Arrhythmia Suppression Trial (CAST) trial, based on the sodium channel-blocking properties of a number of antidepressant drugs including imipramine. Tricyclic antidepressants were subsequently shown to block potassium channels, and thus to prolong the QT interval and induce TdP arrhythmias. These effects of tricyclic antidepressants were generally observed when combined with other QT-prolonging agents, or in cases of overdose. In addition, a number of case reports have linked tricyclic antidepressants as well as antipsychotic drugs to an ECG pattern of right bundle branch and ST segment elevation in right precordial leads resembling the Brugada syndrome, in some cases associated with syncope secondary to the development of rapid polymorphic ventricular tachycardia and fibrillation (VT/VF).
This review examines the mechanisms and predisposing factors underlying the development of cardiac arrhythmias and sudden cardiac death among antidepressant and antipsychotic drugs in clinical use.
2. Methods
The bibliography used in this review extended from 2000 to 2007. The most relevant papers related to the theme of sudden death, antipsychotics and antidepressants were selected and used as references. References were accessed in the PubMed database. The following terms were used for the search: sudden death, antidepressants, antipsychotics, long QT syndrome, Torsade de pointes, and Brugada syndrome.
3. Antidepressant and antipsychotic agent-induced long QT syndrome
The QT interval provides a measure of the time interval between the start and the end of the electrical activation of the ventricles of the heart. QTc are usually around 400 ms and values < 450 ms are considered normal. QTc values > 450 ms in men and 460 ms in women are generally considered prolonged. TdP, from the French for ‘twisting of the points,’ is an atypical ventricular tachycardia characterized by oscillations of the points or peaks (‘pointes’) around the main axis of the ECG, giving rise to a unique morphology. Since the original work of Dessertenne [3], it has been well recognized that many conditions can cause prolonged or abnormal repolarization, giving rise to QT prolongation, abnormal T/U wave morphologies, and the development of TdP. Often self-limited, TdP arrhythmias can degenerate into ventricular fibrillation, thus leading to sudden cardiac death (Figure 1).
Figure 1. A. Self limiting TdP episode. B. TdP leading to ventricular fibrillation.
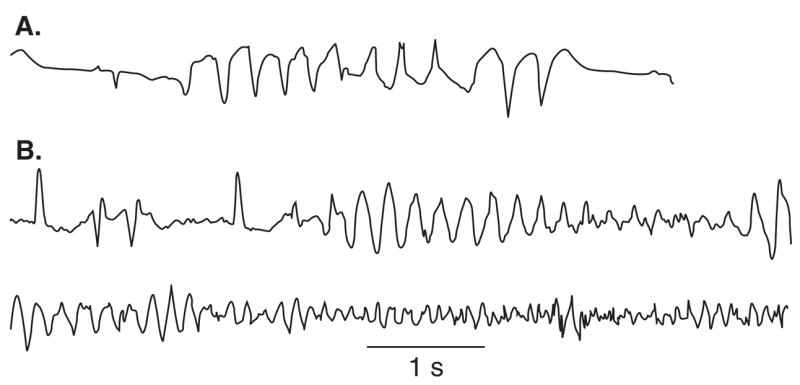
TdP is preceded by a short-long-short sequence.
Modified from [111] with permission.
TdP: Torsade de pointes.
Torsade de pointes can be caused by either congenital or acquired long QT syndrome (LQTS). Congenital LQTS is subdivided into 10 genotypes distinguished by mutations in at least seven different ion genes and a structural anchoring protein located on chromosomes 3, 4, 6, 7, 11, 17 and 21 [4–11]. The prevalence of this disorder is estimated at 1 – 2:10,000. The ECG diagnosis is based on the presence of prolonged repolarization (QT interval) and abnormal T-wave morphology [12]. In the different genotypes, cardiac events may be precipitated by physical or emotional stress (LQT1) or a startle (LQT2), or may occur at rest or during sleep (LQT3). Antiadrenergic intervention with β-blockers is the mainstay of therapy. For patients unresponsive to this approach, implantable cardiac defibrillator (ICD) and/or cardiac sympathetic denervation may be therapeutic alternatives [13,14].
Acquired LQTS refers to a syndrome similar to the congenital form but caused by exposure to drugs that prolong the duration of the ventricular action potential [15] or secondary to cardiomyopathies such as dilated or hypertrophic cardiomyopathy, as well as to abnormal QT prolongation associated with bradycardia or electrolyte imbalance [16–21]. The acquired form of the disease is far more prevalent than the congenital form, and in some cases may have a genetic predisposition.
Table 1, modified from [22], lists the antidepressant and antipsychotic drugs that have been shown to prolong QT interval and induce TdP. Among antidepressant agents, aminotryptyline, imipramine, and maprotiline are the agents most commonly associated with TdP; the greatest prolongation of QT interval is observed with maprotiline [23]. Table 1 shows the relative risk of TdP for distinct tricyclic antidepressants, by classifying the drugs into TdP risk categories ranging from the highest risk of TdP (Category 1, drugs generally accepted by authorities to have a risk of causing TdP) to the lowest risk (Category 4, drugs weakly associated with TdP). As shown in Table 1, most anti-depressants are classified as Category 4, with a low risk of TdP. Vieweg and Wood [23] reported 13 cases of TdP induced by antidepressants. As with most cases of acquired LQTS, most cases (12 of 13) involved women. One case involved a child. In addition to female gender, risk factors include age, bradycardia, metabolic inhibitors, hypokalemia, hypomagnesemia, drug overdose and co-administration of QT-prolonging drugs (Table 2). QRS duration of the ECG, measured in 5 of 13 cases, showed prolongation in 2 and no change in 3, suggesting that QT prolongation with anti-depressants may be due to sodium as well as potassium channel blockade. Antipsychotic drugs, especially those in the phenothiazine group, can also induce QT prolongation and TdP. The first compelling evidence associating sudden death and antipsychotics was reported by Mehtonen et al. in 1991 [24]. A higher incidence of sudden death was observed in otherwise healthy patients receiving anti-psychotic medication. The authors reported 49 cases of sudden death (31 women and 18 men) associated with the use of these agents. A therapeutic dose of phenothiazine was involved in 46 of the 49 cases. Thioridazine was the only antipsychotic drug administered in 15 cases.
Table 1.
Drugs that prolong the QT interval and/or induce Torsade de pointes.
| Drug | Clinical use | TdP category | Ref. |
|---|---|---|---|
| Antidepressants | [22,59] | ||
| Tricyclic antidepressants | |||
| Amitryptyline | Depression | 4 | [105–107] |
| Amoxapine | Depression | 4 | |
| Clomipramine | Depression | 4 | [108] |
| Desipramine | Depression | 4 | |
| Citalopram | Depression | 4 | |
| Doxepin | Depression | 4 | [109,110] |
| Imipramine | Depression | 4 | |
| Nortryptyline | Depression | 4 | |
| Protryptyline | Depression | 4 | |
| Trimipramine | Depression | 4 | |
| Other antidepressants | |||
| Fluoxetine | Depression | 4 | |
| Sertraline | Depression | 4 | |
| Venlafaxine | Depression | 2 | |
| Antipsychotics | [24–26] | ||
| Chlorpromazine | Schizophrenia | 1 | |
| Clozapine | Schizophrenia | 2 | |
| Haloperidol | Schizophrenia | 1 | |
| Pimozide | Tourette’s tick Females > Males | 1 | |
| Quetiapine | Schizophrenia | 2 | |
| Risperidone | Schizophrenia | 2 | |
| Sertindole* | Schizophrenia | 1 | |
| Thioridazine | Schizophrenia | 1 | |
| Ziprasidone | Schizophrenia | 2 | |
Drug withdrawn.
Females > males: substantial evidence indicates a greater risk (usually > twofold) of TdP in women.
Risk category
Drug List 1: Drugs generally accepted by authorities to have a risk of causing TdP.
Drug List 2: Drugs that in some reports may be associated with TdP but at this time lack substantial evidence for causing TdP.
Drug List 3: Drugs to be avoided for use in patient with diagnosed or suspected congenital long QT syndrome. (Drugs on Lists 1, 2 and 4 should also be avoided by patients with congenital long QT syndrome.)
Drug List 4: Drugs weakly associated with TdP and/or QT prolongation but that are unlikely to pose a risk for TdP when used at recommended dosages and in the absence of other risk factors.
Modified from [22].
TdP: Torsade de pointes.
Table 2.
Risk factors for TdP and Brugada syndrome by antidepressant and antipsychotic drugs.
| Risk factor | Increased risk for LQTS and TdP | Increased risk for BrS |
|---|---|---|
| Gender | Female | male |
| Bradycardia | + | + |
| Hypokalemia | + | + |
| Hypomagnesemia | + | − |
| Drug interaction (QT prolonging agents) | + | − |
| Drug interaction (sodium or calcium channel blockers, parasympathetic agonists) | − | + |
| Drug interaction (slow metabolism by CYP inhibitors 2D6, 1A2, 3A4) | + | + |
| Hepatic dysfunction (increased drug concentration) | + | + |
| Genetic predisposition | Congenital LQTS | Congenital BrS |
BrS: Brugada syndrome; LQTS: Long QT syndrome; TdP: Torsade de pointes.
Antipsychotic drugs generally have a higher torsadogenic potential (Category 1 – 2) than antidepressants (Category 4). As a consequence, antidepressant-induced TdP is more typically observed in the presence of drug combinations. In 2001, Ray et al.[25] conducted a retrospective cohort study of half a million Medicaid patients between 1988 and 1993, before the introduction of atypical antipsychotics, and observed that the risk for sudden death increased 2.39 times in individuals receiving antipsychotic drugs compared with those who did not receive these agents. Although the study did not demonstrate causality, it suggested that the potential adverse cardiac effects of antipsychotics should be considered in clinical practice, particularly for patients with cardiovascular disease. Hennessy et al.[26], in a study of 90,000 patients, observed that patients treated for schizophrenia had a higher incidence of cardiac arrest and ventricular arrhythmias than non-schizophrenia patients. The drugs used were clozapine, haloperidol, risperidone, and thioridazine. Liperoti et al.[27], in a study of nursing home residents in six states, observed that the use of conventional antipsychotics led to a twofold increase in risk of hospitalization for ventricular arrhythmias and cardiac arrest, especially in patients with pre-existing cardiac disease.
4. Antidepressant and antipsychotic drug-induced Brugada syndrome
The Brugada syndrome (BS) was introduced as a new clinical entity by Pedro and Josep Brugada in 1992 [28]. The syndrome has attracted much interest because of its high incidence in many parts of the world and its association with high risk of sudden death, especially in males as they enter their third and fourth decade of life. A consensus report published in 2002 delineated diagnostic criteria for the syndrome [29,30]. A second consensus conference report published in 2005 focused on risk stratification schemes and approaches to therapy [31,32].
BS is characterized by an ECG pattern of right bundle branch in right precordial leads, ST segment elevation in the right precordial leads, relatively normal QTc interval, coupled with syncope and sudden death due to VT/VF. About 15% of probands with BS have gene mutations in the SCN5A gene. Over 100 mutations in SCN5A have been linked to the syndrome in recent years (see [33] for references; also see [34]). A second locus on chromosome 3, close to but distinct from SCN5A, has recently been linked to the syndrome [35] in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively benign prognosis. The third and fourth genes associated with BS encode the α1 (CACNA1C) and β(CACNB2b) subunits of the L-type cardiac calcium channel. Mutations in the α1 and β subunits of the calcium channel also lead to a shorter than normal QT interval, in some cases creating a combined Brugada/short QT syndrome [36].
A number of factors modulate the ECG and arrhythmic manifestations of BS. ST segment elevation in BS is often dynamic. The Brugada ECG may often be concealed, but can be unmasked or modulated by sodium channel blockers, a febrile state, vagotonic agents, α-adrenergic agonists, β-adrenergic blockers, tricyclic or tetracyclic antidepressants, first-generation antihistaminics (dimenhydrinate), a combination of glucose and insulin, hyperkalemia, hypokalemia, hypercalcemia, and by alcohol and cocaine toxicity [37–47]. These agents may also induce acquired forms of BS. Lithium, a widely used antidepressant agent, has recently been added to the list of drugs to avoid in patients with BS. Lithium is a potent blocker of cardiac sodium channels and can unmask patients with BS [48].
Three types of repolarization patterns in the right precordial leads are recognized [29,30]. Type 1 ST segment elevation is diagnostic of BS and is characterized by a coved ST-segment elevation ≥ 2 mm (0.2 mV) followed by a negative T-wave. Type 2 ST segment elevation has a saddleback appearance with a high take-off ST-segment elevation of ≥ 2 mm, followed by a trough displaying ≥ 1 mm ST elevation, followed by either a positive or biphasic T-wave. Type 3 ST segment elevation has either a saddleback or coved appearance with an ST-segment elevation of < 1 mm. These three patterns may be observed sequentially in the same patient or following the introduction of specific drugs, particularly sodium channel blockers. Type 2 and Type 3 ST segment elevation are not considered to be diagnostic of BS. BS is definitively diagnosed only when a Type 1 ST-segment elevation (Brugada ECG) is observed in more than one right-precordial lead (V1 – V3), in the presence or absence of a sodium channel blocking agent, and in conjunction with one or more of the following: documented ventricular fibrillation; polymorphic ventricular tachycardia; a family history of sudden cardiac death (< 45 years old); coved-type ECGs in family members; inducibility of VT with programmed electrical stimulation; syncope; or nocturnal agonal respiration [29–32].
Table 3 lists the antidepressants and antipsychotic agents that can induce the Brugada pattern in the ECG. It should be noted that all cases of BS induced by antidepressants and antipsychotics were of type 1 phenotype (Figure 2). A study of 98 patients experiencing an overdose of tricyclic anti-depressants reported that 15 of them displayed an ECG consistent with BS [41]. The overall mortality was 3% among all patients but 6.7% among patients who displayed a Brugada phenotype. Rouleau et al. described three cases of psychotropic drug-induced BS ECG [49], occurring during concomitant administration of amitryptyline and a phenothiazine (case 1), overdose of fluoxetine (case 2), and co-administration of trifluoperazine and loxapine (case 3). Babaliaros and Hurst [40] described a Brugada pattern in patients receiving increasing doses of imipramine. Akhtar and Goldschlager [47] recently reported a case of BS following massive ingestion of desipramine and clonazepam. Chow et al.[50] reported a similar case following desipramine. Bolognesi et al. described a Brugada ECG pattern following overdose of amitryptyline and with maprotiline [51]. Two additional cases of BS have been reported following overdose with nortryptyline [42,52] or lithium [48].
Table 3.
Drugs that induce ECG Brugada pattern (type 1 ST segment elevation).
| Drug | Major clinical indication | Ref. | |
|---|---|---|---|
| Antidepressants | |||
| Tricyclic antidepressant | Amitryptyline | Depression | [49,51] |
| Desipramine | Depression | [40,50] | |
| Nortryptyline | Depression | [42,52] | |
| Tetracyclic antidepressant | Maprotiline | Depression Anxiety | [51] |
| Other | Lithium | Depression | [48] |
| Antipsychotics | Trifl uoperazine | Anxiety Psychotic disorders | [49] |
| Loxapine | Psychotic conditions, including hallucinations, delusions, and confusion | [111] | |
Figure 2. Brugada syndrome phenotype induced by an overdose of desipramine and clonazepam in a 44-year-old man previously medicated with desipramine, clonazepam and trazodone.
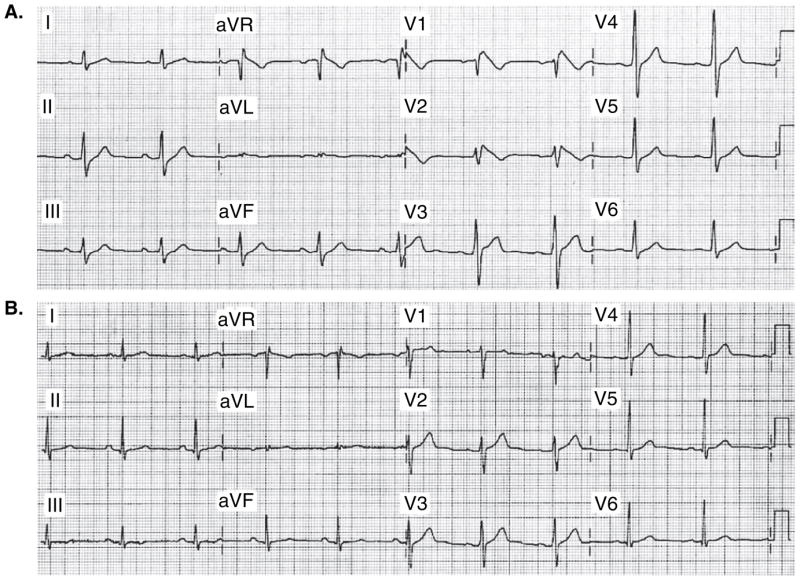
A. ECG shows sinus bradycardia, first-degree AV block (228 ms), prolonged QRS interval (132 ms), and downsloping ST elevation (Type 1) in leads V1 – V2, ST elevation in lead V3, upsloping ST depression in leads II, III, and aVF. B. Baseline ECG from 1 year ago is normal.
Modified from [42], with permission.
ECG: Electrocardiogram.
The available data suggest that most cases of antidepressant and antipsychotic-induced BS phenotype occurs as a consequence of drug overdose or drug combination.
5. Cardiovascular risk of typical (conventional) versus atypical antipsychotics
Although a potential risk of arrhythmias exist for a number of antipsychotics, the newer currently available atypical antipsychotic medications are generally regarded as safer than the older typical antipsychotic medications [53]. Table 4 classifies the antipsychotic medication in terms of risk of arrhythmia and SCD. The older antipsychotic medications, particularly the phenothiazines are clearly associated with a risk of sudden death in schizophrenic patients [25]. The newer currently used atypical drugs have not been shown to increase the risk of sudden death in schizophrenic patients. Liperoti et al.[27] observed that the use of conventional antipsychotics (chlorpromazine, chlorprothixene, fluphenazine, haloperidol, loxapine, molindone, thioradizine, perphenazine, promazine, thiotixene and trifluoperazine) was associated with a twofold increase in the risk of hospitalization for cardiac arrhythmias or cardiac arrest, whereas no increased risk was associated with the use of atypical antipsychotics (olanzapine, clozapine, risperidone and quetiapine). Glassman and Bigger [54], in a review of the risk of TdP and SCD, established that the highest risk was with the typical antipsychotic thioridazine, followed by other typical agents, pimozide, sertindole, droperidol, and haloperidol; no association with TdP or SCD was observed with the atypical agents olanzapine, quetiapine, or risperidone. In the CATIE trial (Clinical Antipsychotic Trials of Intervention Effectiveness) [55], no increase in SCD was observed in schizophrenic patients randomly assigned to the currently used atypical antipsychotics olanzapine, risperidone, or quetiapine, following the suspension of the typical agent perphenazine. In addition, ziprasidone, the atypical agent with the greatest propensity to prolong QT, has not been linked to sudden death despite 6 years of availability. However, in 2005, the US FDA warned that the atypical antipsychotics olanzapine, aripriprazole, risperidone, and quetiapine increase in the risk of SCD in the elderly by 4.5% [56]. The advisory applied to these four drugs plus two other atypical agents, clozapine and ziprasidone.
Table 4.
Classification of antipsychotic medication in terms of risk of arrhythmia and sudden cardiac death.
| Antipsychotic drugs | Chemical structure | Risk of cardiac arrhythmias |
|---|---|---|
| Typical | ||
| Chlorpromazine | Aliphatic phenothiazine | Higher |
| Pimozide | Diphenylbutylpiperidine | Higher |
| Thioridazine | Piperidine | Higher |
| Trifl uoperazine | Piperazine | Lower |
| Haloperidol | Butyrophenone | Lower |
| Sulpiride* | Substitute benzamide | Lower |
| Atypical | ||
| Clozapine | Dibenzodiazepine | Higher |
| Quetiapine | Dibenzothiazepine | Lower |
| Risperidone | Benzisoxazole | Lower |
| Amisulpiride | Subsituted benzamide | Lower |
| Olanzapine | Thienobenzodiazepine | Lower |
| Zotepine | Dibenzothiepine | Lower |
Some regard sulpiride as an atypical antipsychotic.
From [2] with permission.
6. Effects of antidepressants and antipsychotics on ion channels
Antidepressants and antipsychotics have been reported to modulate the cardiac action potential by blocking cardiac ion channels present in ventricular myocytes, including the fast sodium inward current (INa), the inward slow calcium current (ICa), and one or more outward potassium currents (I K), particularly the rapidly activating delayed rectifier current (IKr). Drug-induced IKr block has attracted considerable attention in recent years due to the association of IKr block with QT interval prolongation in the ECG and life-threatening cardiac arrhythmias such as TdP. Drug-induced INa and ICa block is thought to underlie the development of the BS phenotype [57,58].
Table 5 illustrates the IC50 (concentration that produces a 50% inhibition of ion channel current) values for block of IKr, ICa and INa derived from expression systems and/or native myocytes for a number of antidepressants and antipsychotics that have been shown to induce arrhythmias [59]. The available studies suggest that most antidepressants inhibit both inward and outward currents; imipramine, amitryptyline, and fluoxetine block both IKr and ICa. Imipramine and amitryptyline also block INa. The ability of antidepressants to block both outward and inward currents is associated with lack of correlation between the degree of IKr block and QT prolongation, because calcium and/or sodium channel inhibition limits the effects of IKr block to prolong APD and thus to prolong the QT interval. In contrast to antidepressants, antipsychotic drugs produce more of an outward current inhibition, most commonly secondary to inhibition of the IKr channel. A 30-fold difference between the effective plasma concentration and the IC50 for inhibition of IKr has been suggested as an adequate margin of safety for avoiding the development of TdP as an adverse effect [60].
Table 5.
IC50 values for block of IKr, ICa and INa.
| Drug | IKr IC50 | ICa IC50 | INa IC50 | Ref. |
|---|---|---|---|---|
| Antidepressants | ||||
| Amitryptyline* | 4.78 μm | 3.75 μm | > 1 μm | [59,112–115] |
| Imipramine* | 3.4 μm | 4 μm | 5 μm | [59,114,116] |
| Fluoxetine* | 1.5 – 3.1 μm | 2.8 μm | [59,114,117,118] | |
| Citalopram | 3.97 μm | [59] | ||
| Antipsychotics | ||||
| Chlorpromazine | 1.47 + 0.03 μm | [119,120] | ||
| Clozapine | 2.63 + 0.12 μm | [119,120] | ||
| Haloperidol | 1 μm | [121] | ||
| Sertindole ‡ | 2.9 μm | [121] | ||
| Thioridazine | 1.07 + 0.06 μm | [119,120] | ||
Drugs with mixed-ion channel block [59].
Drug withdrawn from the US market in 1998.
Reproduced with permission from The Thomson Corporation and Sala M, Coppa F, Cappucciati C, et al. Antidepressants: their effects on cardiac channels, QT prolongation and Torsade de pointes. Curr Opin Investig Drugs 2006;7:256–63. © 2006 The Thomson Corporation.
IC50: Concentration of antidepressants or antipsychotics that produces a 50% inhibition of a the ion-channel current (IKr, ICa and INa).
7. Mechanisms of arrhythmias in the long QT syndrome
Amplification of spatial dispersion of repolarization within the ventricular myocardium has been identified as the principal arrhythmogenic substrate in both acquired and congenital LQTS. The accentuation of spatial dispersion, typically secondary to an increase of transmural, trans-septal or apico-basal dispersion of repolarization, and the development of early after-depolarization (EAD)-induced triggered activity underlie the substrate and trigger for the development of TdP arrhythmias observed under LQTS conditions [61,62]. Models of the LQT1, LQT2, LQT3, LQT5, LQT6, LQT7, and LQT8 forms of the LQTS have been developed using the canine arterially perfused left ventricular wedge preparation [63–66]. These models suggest that in the first three forms of LQTS, preferential prolongation of the M cell action potential duration (APD) leads to an increase in the QT interval as well as an increase in transmural dispersion of repolarization (TDR), which contributes to the development of spontaneous as well as stimulation-induced TdP [67–69]. The unique characteristics of the M cells are at the heart of LQTS. The hallmark of the M cell is the ability of its action potential to prolong more than that of epicardium or endocardium in response to a slowing of rate [70–72]. This feature of the M cell is due to weaker repolarizing current during Phases II and III, secondary to a smaller IKs and a larger late INa and INa–Ca[73–75] compared to epicardial and endocardial cells.
These ionic distinctions also sensitize the M cells to a variety of pharmacological agents. Agents that block IKr (such as antidepressants and antipsychotics), slowly activating delayed rectifier potassium current (IKs) or increase ICa or late INa generally produce a much greater prolongation of the APD of the M cell than of epicardial or endocardial cells. The duration of the M cell action potential therefore determines the QT interval, whereas the duration of the epicardial action potential generally determines the QT peak interval.
Figure 3 presents a working hypothesis of the mechanisms underlying LQTS-related TdP based on available data. The hypothesis presumes the presence of electrical heterogeneity in the form of spatial dispersion of repolarization in the form of transmural and trans-septal dispersion of repolarization (TDR) under baseline conditions and the amplification of TDR by agents that reduce net repolarizing current via a reduction in IKr or IKs (or augmentation of ICa or late INa). Conditions leading to a reduction in IKr lead to a preferential prolongation of the M cell action potential. As a consequence, the QT interval prolongs and is accompanied by a dramatic increase in TDR, thus creating a vulnerable window for the development of re-entry. The reduction in net repolarizing current also predisposes to the development of EAD- and in rare cases delayed after-depolarization (DAD)-induced triggered activity in M and Purkinje cells, which provide the extrasystole that triggers TdP when it falls within the vulnerable period. β-Adrenergic agonists further amplify transmural heterogeneity in the case of IKs block as well as (transiently) in the case of IKr block, but reduce it in the case of INa agonists [69,76]. Inhibition of IKr is by far the most common cause of reduction in net outward current by antidepressant and antipsychotic drugs. The presence of other IKr blockers (combination of an anti-depressant and antipsychotic drug) or agents that reduced IKs or augment ICa or late INa can accentuate the reduction in repolarization forces and thus lead to an increase in the probability of arrhythmia.
Figure 3. Proposed mechanisms of arrhythmias in the long QT syndrome.
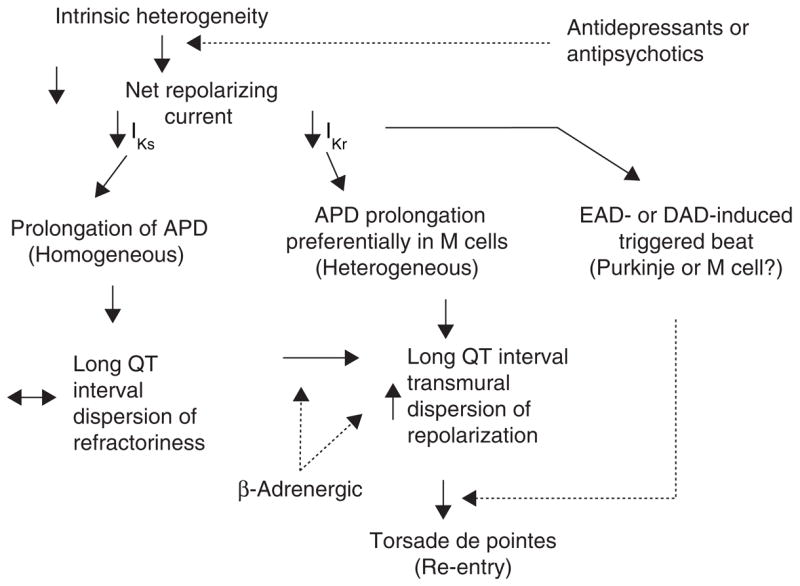
APD: Action potential duration; DAD: Delayed after-depolarization; EAD: Early after-depolarization.
8. Mechanisms of arrhythmia in the Brugada syndrome
The cellular basis for BS has been elucidated in recent years. The concept of Phase II re-entry, which is a trigger for BS, was described in the early 1990s and evolved in parallel with the clinical discovery of the syndrome [57,77–80]. Studies conducted over the past decade suggest that rebalancing of the currents active at the end of Phase I, leading to an accentuation of the action potential notch in right ventricular epicardium, is responsible for the accentuated J wave or ST segment elevation associated with BS [57,78–87]. The proposed cellular mechanism for BS is summarized in Figure 4. The available data support the hypothesis that BS results from amplification of heterogeneities intrinsic to the early phases of the action potential among the different transmural cell types. The amplification is secondary to a rebalancing of currents active during Phase I, including a decrease in INa orICa or augmentation of any one of a number of outward currents. ST segment elevation similar to that observed in patients with BS occurs as a consequence of the accentuation of the action potential notch, eventually leading to loss of the action potential dome in right ventricular epicardium, where transient outward current (Ito) is most prominent. Loss of the dome gives rise to a transmural as well as an epicardial dispersion of repolarization. The transmural dispersion is responsible for the development of ST segment elevation and the creation of a vulnerable window across the ventricular wall, whereas the epicardial dispersion gives rise to Phase II re-entry, which provides the extrasystole that captures the vulnerable window, thus precipitating VT/VF. Slowing of conduction within the right ventricular outflow tract can further exaggerate the epicardial and transmural dispersion of repolarization [88]. The VT generated is usually polymorphic, resembling a very rapid form of TdP.
Figure 4. Proposed mechanism for the Brugada syndrome.
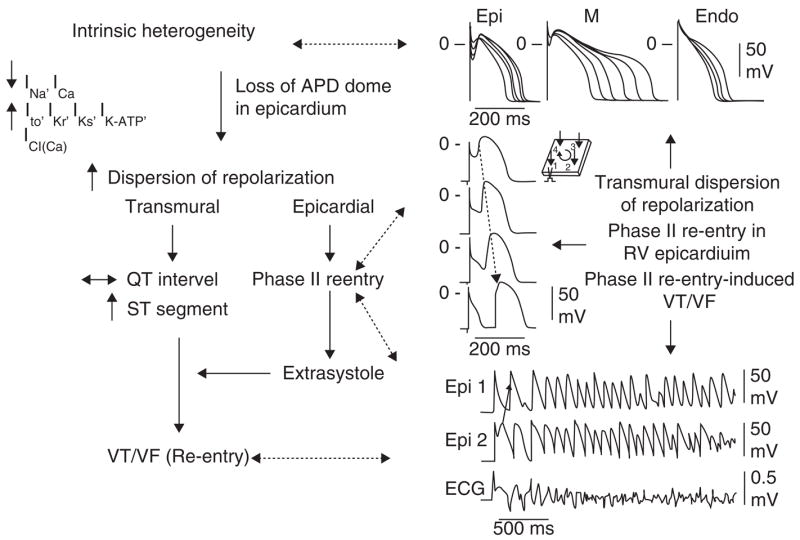
A shift in the balance of currents serves to amplify existing heterogeneities by causing loss of the action potential dome at some epicardial but not endocardial sites. A vulnerable window develops as a result of the dispersion of repolarization and refractoriness within epicardium as well as across the wall. Epicardial dispersion leads to the development of Phase II re-entry, which provides the extrasystole that captures the vulnerable window and initiates VT/VF via a circus movement re-entry mechanism.
Modified from [122], with permission.
APD: Action potential duration.
9. Genetic predisposition
The degree to which a genetic predisposition contributes to the clinical manifestation of antidepressant- and antipsychotic-induced arrhythmogenesis is not well defined. Available data suggest that up to 10 – 15% of individuals who develop TdP following exposure to QT-prolonging drugs possess mutations associated with the LQTS and may be considered to have a subclinical form of the congenital syndrome [89–93].
Abbott et al. were among the first to show that a polymorphism (a genetic variation that is present in greater than 1% of the population) in an ion channel gene is associated with a predisposition to drug-induced TdP [94]. They identified a polymorphism (T8A) of the KCNE2 gene encoding for MiRP, a β subunit of the IKr channel, that is present in 1.6% of the population and is associated with TdP related to quinidine and to sulfamethoxazole/trimethoprim administration. This finding suggests that common genetic variations may increase the risk for developmental of drug-related arrhythmias. Yang et al.[92] showed that DNA variants in the coding regions of genes predisposing to acquired LQTS can be identified in ~ 10 – 15% of affected subjects, predominantly in genes encoding ancillary subunits, providing further support for the hypothesis that subclinical mutations and polymorphisms may predispose to drug-induced TdP. Splawski et al.[95] further advanced this concept by identifying a heterozygous polymorphism involving substitution of serine with tyrosine in codon 1103 (S1103Y) in the sodium channel gene SCN5A (S1102Y in the shorter-splice variant of SCN5A) among Africans and African Americans, which increases the risk for acquired TdP. The polymorphism was present in 57% of 23 patients with proarrhythmic episodes, but in only 13% of controls. These findings suggest that carriers of such polymorphisms can be identified and excluded from treatment with drugs that are associated with a risk of proarrhythmia. It is conceivable that such testing could be applied widely, since accurate results could be made available rapidly at relatively low cost.
Another common polymorphism that has been associated with acquired forms of LQTS and TdP is K897T in KCNH2[96]. Most functional expression studies have reported that K897T reduces IKr[97–99], although one study has reported an increase in HERG current [100].
The action of antidepressants to precipitate BS may also have a genetic disposition. For example, the SCN5A promoter haplotype (so-called Hap B) has been shown to be associated with longer PR and QRS intervals as well as a more exaggerated response to sodium channel blockers [101].
Genetic defects can also contribute to drug-induced channelopathies by influencing the metabolism of drugs. In the case of relatively pure IKr blockers, there is a clear relationship between plasma levels of drug and the incidence of TdP. Genetic variants of the genes encoding for enzymes responsible for drug metabolism could alter pharmacokinetics so as to cause wide fluctuations in plasma levels, thus exerting a significant proarrhythmic influence [102,103]. For example, in the case of cytochrome CYP2D6, which is involved in the metabolism of some QT-prolonging drugs (terodiline, thioridazine), multiple polymorphisms have been reported that reduce or eliminate its function; 5 – 10% of Caucasians and African Americans lack a functional CYP2D6. Numerous proteins, including drug transport molecules and other drug-metabolizing enzymes, are involved in drug absorption, distribution, and elimination; and genetic variants of each of these have the potential to modulate drug concentrations and effects. Multiple substrates and inhibitors of the cytochrome P450 enzymes have been identified. A comprehensive database can be found at [104].
10. Summary and conclusion
Since the early 1990s, a clear relationship has been established between some antipsychotics, prolongation of the QT interval of the ECG, and the development of an atypical polymorphic tachycardia known as TdP and SCD. Antipsychotic drugs are more commonly associated with QT prolongation and TdP than are antidepressants. Not all the antipsychotics display the same risk: the newer atypical antipsychotics, olanzapine, risperidone, and quetiapine, display a much lower risk than the older typical antipsychotics, especially those in the phenothiazine group.
A growing number of case reports have linked tricyclic antidepressants as well as antipsychotic drugs to an ECG pattern of right bundle branch and ST segment elevation in right precordial leads resembling BS, in some cases associated with syncope secondary to the development of rapid polymorphic VT/VF.
Most cases of antidepressant-induced TdP occur following drug overdose or a combination with other QT-prolonging agents or conditions. Antidepressants, on the other hand, are more likely to predispose to BS phenotype. These proclivities are due to the fact that antipsychotic drugs generally exert a predominant effect to inhibit outward currents, IKr block in particular, whereas antidepressants exert a predominant effect to inhibit inward currents, such as INa and ICa.
The accentuation of spatial dispersion, typically secondary to an increase of transmural, trans-septal or apico-basal dispersion of repolarization, and the development of EAD-induced trigger activity underlie the substrate and trigger for the development of TdP arrhythmias observed under LQTS conditions, often observed with antipsychotics and/or antidepressant drugs. Available data point to an accentuation of the right ventricular action potential notch and eventual loss of the action potential dome as the basis of ST segment elevation and the development of the substrate for VT/VF observed in cases of antidepressant-induced acquired BS.
Available data suggest that at least 10 – 15% of individuals who develop TdP following exposure to QT-prolonging drugs possess mutations associated with the LQTS and may be considered to have a subclinical form of the congenital syndrome. The action of antidepressants to precipitate BS are also likely to have a genetic disposition, although little data are available at present. The genetic link may be much more prevalent than currently believed if complex haplotypes (combination of polymorphisms) are involved.
11. Expert opinion
A number of antipsychotic and antidepressant drugs are known to increase the risk of ventricular arrhythmias and SCD by prolonging the QT interval and/or inducing TdP arrhythmias. These include: i) typical antipsychotics such as chlorpromazine, pimozide, thioridazine, perphenazine, trifl uoperazine, haloperidol, and droperidol; ii) atypical antipsychotics such clozapine, quetiapine, risperidone, sultopride, ziprasidone, and loxapine; iii) tricyclic anti-depressants such as amitryptyline, amoxapine, clomipramine, desipramine, citalopram, doxepin, imipramine, nortryptyline, and trimipramine; and iv) other antidepressants such as fl uoxetine, sertraline, and venlafaxine.
A number of antipsychotic and antidepressant drugs can increase the risk of ventricular arrhythmias and SCD by inducing a BS phenotype. These include: i) the anti-psychotics, trifl uorperazine and loxapine; ii) the tricyclic antidepressants amitryptyline, desipramine, and nortryptyline; and iii) other antidepressants such as maprotiline and lithium.
Antipsychotics can increase cardiac risk even at low doses, whereas antidepressants do it generally at high doses or in the setting of drug combination.
Not all antipsychotics display the same level of risk: the newer atypical antipsychotics olanzapine, risperidone, and quetiapine display a much lower level of risk than the older typical antipsychotics, especially those in the phenothiazine group.
Commonly used antipsychotic and antidepressant drugs should be used with great care in cases of LQTS or BS or when combined with agents known to prolong QT intervals or predispose to acquired forms of BS. The QT-prolonging drugs include a number of anti-arrhythmics, antihistaminics, sympathomimetics, antibiotics and antifungal agents. See [22] for a complete list of QT-prolonging agents.
Although antipsychotics and antidepressants display a overall low risk of long QT-mediated TdP and BS-mediated polymorphic VT, deaths have been attributed to anti-psychotics, especially the typical antipsychotics in the phenothiazine group. The degree of risk varies and depends on underlying pathology, genetic predisposition, the drug used, and concomitant use of other medications.
These observations call for great care in the administration of these agents in psychiatric medicine, particularly the need for a careful clinical history, as well as an ECG at baseline and after drug administration. Commonly used antipsychotic and antidepressant drugs should be used with great care in cases of LQTS or BS or when combined with agents known to prolong QT intervals or predispose to acquired forms of BS.
High-risk antipsychotics and antidepressants should be avoided in patients with known congenital LQTS or BS, as well as in the setting of an acute systemic disease, including acute cardiac infarction patients and those with renal dysfunction.
In order to minimize the cardiac risk, patients should be screened for relevant clinical risk factors. Major risk factors include structural heart disease, congenital LQTS, BS, family history of sudden death, and previous episode of drug-induced QT prolongation or TdP. Secondary risk factors include old age, electrolyte imbalance, renal or hepatic disease, or concomitant use of other QT-prolonging agents or drugs inducing the Brugada phenotype. In addition, the lowest possible dose of antidepressant and antipsychotic should be prescribed, combination of antipsychotics avoided, and the clinical symptoms closely monitored.
Serial ECGs are recommended at the start of antipsychotic drug treatment with high-risk agents (such as typical anti-psychotic drugs). Close monitoring is recommended to be continued if major risk factors or multiple risk factors exist, if high-risk antipsychotic/antidepressant drug combinations are used, as well as in cases of suspected drug overdose. Review of antipsychotic or antidepressant therapy, including cessation and change of medication, should be considered if the ECG shows major prolongation of the QT interval (QTc > 500 ms), QTc prolongation > 60 ms, T-wave abnormalities, marked bradycardia, or a BS phenotype.
Acknowledgments
Declaration of interest
Supported by grant HL47678 from NHLBI (CA) and NYS and Florida Grand Lodges F and AM.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Abdelmawla N, Mitchell AJ. Sudden cardiac death and antipsychotics. Part 1. Risk factors and mechanisms. Adv Psychiatr Treat. 2006;12:35–44. [Google Scholar]
- 2.Abdelmawla N, Mitchell AJ. Sudden cardiac death and antipsychotics. Part 2. Monitoring and prevention. Adv Psychiatr Treat. 2006;12:100–9. [Google Scholar]
- 3.Dessertenne F. La tachycardie ventriculaire a deux foyers opposes variables. Arch Mal Coeur Vaiss. 1966;59:263–72. [PubMed] [Google Scholar]
- 4.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–11. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 5.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 6.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511–9. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 7.Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 9.Splawski I, Tristani-Firouzi M, Lehmann MH, et al. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–40. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 10.Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel {beta}4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–42. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cronk LB, Ye B, Kaku T, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–6. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.••.Schwartz PJ, Priori SG, Napolitano C. The long QT syndrome. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: from Cell to Bedside. Philadelphia, PA: WB Saunders; 2000. pp. 597–615. Comprehensive review of clinical characteristics of LQTS. [Google Scholar]
- 13.Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–23. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–33. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 15.Bednar MM, Harrigan EP, Anziano RJ, et al. The QT interval. Prog Cardiovasc Dis. 2001;43:1–45. doi: 10.1053/pcad.2001.21469. [DOI] [PubMed] [Google Scholar]
- 16.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–83. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 17.Sipido KR, Volders PG, de Groot SH, et al. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–44. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 18.Volders PG, Sipido KR, Vos MA, et al. Downregulation of delayed rectifier K(+) currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999;100:2455–61. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- 19.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maltsev VA, Sabbah HN, Higgins RS, et al. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 21.Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2:S9–15. doi: 10.1016/j.hrthm.2004.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. [Last accessed February 26, 2008];QT Drug Lists by Risk Groups. Available from: URL: www.torsades.org/medical-pros/drug-lists/drug-lists.htm.
- 23.Vieweg WV, Wood MA. Tricyclic antidepressants, QT interval prolongation, and torsade de pointes. Psychosomatics. 2004;45:371–7. doi: 10.1176/appi.psy.45.5.371. [DOI] [PubMed] [Google Scholar]
- 24.Mehtonen OP, Aranko K, Malkonen L, Vapaatalo H. A survey of sudden death associated with the use of antipsychotic or antidepressant drugs: 49 cases in Finland. Acta Psychiatr Scand. 1991;84:58–64. doi: 10.1111/j.1600-0447.1991.tb01421.x. [DOI] [PubMed] [Google Scholar]
- 25.Ray WA, Meredith S, Thapa PB, et al. Antipsychotics and the risk of sudden cardiac death. Arch Gen Psychiatry. 2001;58:1161–7. doi: 10.1001/archpsyc.58.12.1161. [DOI] [PubMed] [Google Scholar]
- 26.Hennessy S, Bilker WB, Knauss JS, et al. Cardiac arrest and ventricular arrhythmia in patients taking antipsychotic drugs: cohort study using administrative data. BMJ. 2002;325:1070. doi: 10.1136/bmj.325.7372.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liperoti R, Gambassi G, Lapane KL, et al. Conventional and atypical antipsychotics and the risk of hospitalization for ventricular arrhythmias or cardiac arrest. Arch Intern Med. 2005;165:696–701. doi: 10.1001/archinte.165.6.696. [DOI] [PubMed] [Google Scholar]
- 28.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 29.•.Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus Report. Eur Heart J. 2002;23:1648–54. doi: 10.1053/euhj.2002.3382. First consensus conference report on Brugada syndrome. [DOI] [PubMed] [Google Scholar]
- 30.Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–9. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 31.Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome. Report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–70. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 32.•.Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2:429–40. doi: 10.1016/j.hrthm.2005.01.005. Second consensus conference report on Brugada syndrome. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada syndrome: from bench to bedside. Oxford: Blackwell Futura; 2005. [Google Scholar]
- 34.Fondazione Salvatore Maugeri. [last accessed February 26, 2008];Inherited arrhythmias database. Available from: URL: www.fsm.it/cardmoc.
- 35.Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome. A novel gene locus on chromosome 3. Circulation. 2002;105:707–13. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 36.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brugada P, Brugada J, Brugada R. Arrhythmia induction by antiarrhythmic drugs. PACE. 2000;23:291–2. doi: 10.1111/j.1540-8159.2000.tb06751.x. [DOI] [PubMed] [Google Scholar]
- 38.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–5. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki T, Mitamura H, Miyoshi S, et al. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–70. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 40.Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol. 2002;25:395–8. doi: 10.1002/clc.4950250809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med. 2002;346:1591–2. doi: 10.1056/NEJM200205163462020. [DOI] [PubMed] [Google Scholar]
- 42.Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol. 2001;12:275. doi: 10.1046/j.1540-8167.2001.00275.x. [DOI] [PubMed] [Google Scholar]
- 43.Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol. 2001;12:1192–4. doi: 10.1046/j.1540-8167.2001.01192.x. [DOI] [PubMed] [Google Scholar]
- 44.Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol. 2001;34:345–9. doi: 10.1054/jelc.2001.26318. [DOI] [PubMed] [Google Scholar]
- 45.Nogami A, Nakao M, Kubota S, et al. Enhancement of J-ST-segment elevation by the glucose and insulin test in Brugada syndrome. PACE. 2003;26:332–7. doi: 10.1046/j.1460-9592.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 46.Araki T, Konno T, Itoh H, et al. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J. 2003;67:93–5. doi: 10.1253/circj.67.93. [DOI] [PubMed] [Google Scholar]
- 47.Akhtar M, Goldschlager NF. Brugada electrocardiographic pattern due to tricyclic antidepressant overdose. J Electrocardiol. 2006;39:336–9. doi: 10.1016/j.jelectrocard.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Darbar D, Yang T, Churchwell K, et al. Unmasking of Brugada syndrome by lithium. Circulation. 2005;112:1527–31. doi: 10.1161/CIRCULATIONAHA.105.548487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rouleau F, Asfar P, Boulet S, et al. Transient ST segment elevation in right precordial leads induced by psychotropic drugs: relationship to the Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:61–5. doi: 10.1046/j.1540-8167.2001.00061.x. [DOI] [PubMed] [Google Scholar]
- 50.Chow BJ, Gollob M, Birnie D. Brugada syndrome precipitated by a tricyclic antidepressant. Heart. 2005;91:651. doi: 10.1136/hrt.2004.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolognesi R, Tsialtas D, Vasini P, et al. Abnormal ventricular repolarization mimicking myocardial infarction after heterocyclic antidepressant overdose. Am J Cardiol. 1997;79:242–5. doi: 10.1016/s0002-9149(96)00727-8. [DOI] [PubMed] [Google Scholar]
- 52.Bigwood B, Galler D, Amir N, Smith W. Brugada syndrome following tricyclic antidepressant overdose. Anaesth Intensive Care. 2005;33:266–70. doi: 10.1177/0310057X0503300219. [DOI] [PubMed] [Google Scholar]
- 53.Zareba W, Lin DA. Antipsychotic drugs and QT interval prolongation. Psychiatr Q. 2003;74:291–306. doi: 10.1023/a:1024122706337. [DOI] [PubMed] [Google Scholar]
- 54.Glassman AH, Bigger JT., Jr Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry. 2001;158:1774–82. doi: 10.1176/appi.ajp.158.11.1774. [DOI] [PubMed] [Google Scholar]
- 55.Stroup TS, Lieberman JA, McEvoy JP, et al. Effectiveness of olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia after discontinuing perphenazine: a CATIE study. Am J Psychiatry. 2007;164:415–27. doi: 10.1176/ajp.2007.164.3.415. [DOI] [PubMed] [Google Scholar]
- 56.Kuehn BM. FDA warns antipsychotic drugs may be risky for elderly. JAMA. 2005;293:2462. doi: 10.1001/jama.293.20.2462. [DOI] [PubMed] [Google Scholar]
- 57.Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1:210–7. doi: 10.1016/j.hrthm.2004.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antzelevitch C, Fish JM. Therapy for the Brugada syndrome. In: Kass R, Clancy CE, editors. Handbook of Experimental Pharmacology. New York: Springer-Verlag; 2006. pp. 305–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sala M, Coppa F, Cappucciati C, et al. Antidepressants: their effects on cardiac channels, QT prolongation and Torsade de pointes. Curr Opin Investig Drugs. 2006;7:256–63. [PubMed] [Google Scholar]
- 60.••.Redfern WS, Carlsson L, Davis AS, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. Excellent review of acquired LQTS. [DOI] [PubMed] [Google Scholar]
- 61.Belardinelli L, Aantzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–25. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Antzelevitch C, Shimuzu W. Cellular mechanisms underlying the Long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Shimuzu W, Antzelevitch C. Effects of a K(+) channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation. 2000;102:706–12. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- 64.Antzelevitch C. Heterogeneity of cellular repolarization in LQTS: the role of M cells. Eur Heart J. 2001;(Suppl 3):K2–16. [Google Scholar]
- 65.Tsuboi M, Antzelevitch C. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7) Heart Rhythm. 2006;3:328–35. doi: 10.1016/j.hrthm.2005.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sicouri S, Timothy KW, Zygmunt AC, et al. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: effects of ranolazine. Heart Rhythm. 2007;4:638–47. doi: 10.1016/j.hrthm.2006.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimuzu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–22. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 68.Shimuzu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–47. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 69.Shimuzu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–86. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 70.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell Circ Res. 1991;68:1729–41. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 71.Antzelevitch C, Shimuzu W, Yan GX, et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–52. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 72.Anyukhovsky EP, Sosunov EA, Gainullin RZ, Rosen MR. The controversial M cell. J Cardiovasc Electrophysiol. 1999;10:244–60. doi: 10.1111/j.1540-8167.1999.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 73.Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–8. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]
- 74.Zygmunt AC, Eddlestone GT, Thomas GP, et al. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–97. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 75.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. Circ Res. 1995;76:351–65. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 76.Li GR, Feng J, Yue L, Carrier M. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol. 1998;275:H369–77. doi: 10.1152/ajpheart.1998.275.2.H369. [DOI] [PubMed] [Google Scholar]
- 77.Krishnan SC, Antzelevitch C. Flecainide-induced arrhythmia in canine ventricular epicardium. Phase 2 reentry? Circulation. 1993;87:562–72. doi: 10.1161/01.cir.87.2.562. [DOI] [PubMed] [Google Scholar]
- 78.•.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–9. doi: 10.1161/01.cir.93.2.372. First study dealing with the development of a model of BS. [DOI] [PubMed] [Google Scholar]
- 79.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–6. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 80.Fish JM, Antzelevitch C. Cellular and ionic basis for the sex-related difference in the manifestation of the Brugada syndrome and progressive conduction disease phenotypes. J Electrocardiol. 2003;36:173–9. doi: 10.1016/j.jelectrocard.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 81.Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol. 2001;96:517–27. doi: 10.1007/s003950170002. [DOI] [PubMed] [Google Scholar]
- 82.Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90:889–96. doi: 10.1161/01.res.0000016960.61087.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kurita T, Shimuzu W, Inagaki M, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–4. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 84.Yan GX, Lankipalli RS, Burke JF, et al. Ventricular repolarization components on the electrocardiogram: cellular basis and clinical significance. J Am Coll Cardiol. 2003;42:401–9. doi: 10.1016/s0735-1097(03)00713-7. [DOI] [PubMed] [Google Scholar]
- 85.Antzelevitch C. Modulation of transmural repolarization. Ann NY Acad Sci. 2005;1047:314–23. doi: 10.1196/annals.1341.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fish JM, Antzelevitch C. Link between hypothermia and the Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:942–4. doi: 10.1046/j.1540-8167.2004.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fish JM, Welchons DR, Kim YS, et al. Dimethyl lithospermate B, an extract of danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation. 2006;113:1393–400. doi: 10.1161/CIRCULATIONAHA.105.601690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tukki R, Sogaard P, Vleugels J, et al. Delay in right ventricular activation contributes to Brugada syndrome. Circulation. 2004;109:1272–7. doi: 10.1161/01.CIR.0000118467.53182.D1. [DOI] [PubMed] [Google Scholar]
- 89.••.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–22. doi: 10.1056/NEJMra032426. Excellent review of acquired LQTS. [DOI] [PubMed] [Google Scholar]
- 90.Donger C, Denjoy I, Berthet M, et al. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96:2778–81. doi: 10.1161/01.cir.96.9.2778. [DOI] [PubMed] [Google Scholar]
- 91.Napolitano C, Schwartz PJ, Brown AM, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–6. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 92.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–8. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 93.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 94.Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 95.Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–6. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 96.Pollevick GD, Oliva A, Viskin S, et al. Genetic predisposition to post-myocardial infarction long QT intervals and Torsade de pointes [abstract] Heart Rhythm. 2007;4:S121. [Google Scholar]
- 97.Crotti L, Lundquist AL, Insolia R, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 98.Anson BD, Ackerman MJ, Tester DJ, et al. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–41. doi: 10.1152/ajpheart.00891.2003. [DOI] [PubMed] [Google Scholar]
- 99.Paavonen KJ, Chapman H, Laitinen PJ, et al. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG) Cardiovasc Res. 2003;59:603–11. doi: 10.1016/s0008-6363(03)00458-9. [DOI] [PubMed] [Google Scholar]
- 100.Bezzina CR, Verkerk AO, Busjahn A, et al. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36. doi: 10.1016/s0008-6363(03)00342-0. [DOI] [PubMed] [Google Scholar]
- 101.Bezzina CR, Shimizu W, Yang P, et al. Common sodium channel promoter haplotype in asian subjects underlies variability in cardiac conduction. Circulation. 2006;113:338–44. doi: 10.1161/CIRCULATIONAHA.105.580811. [DOI] [PubMed] [Google Scholar]
- 102.Ford GA, Wood SM, Daly AK. CYP2D6 and CYP2C19 genotypes of patients with terodiline cardiotoxicity identified through the yellow card system. Br J Clin Pharmacol. 2000;50:77–80. doi: 10.1046/j.1365-2125.2000.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roden DM. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc Res. 2001;50:224–31. doi: 10.1016/s0008-6363(00)00302-3. [DOI] [PubMed] [Google Scholar]
- 104.Table of CYP substrates, inhibitors and inducers. [last accessed February 26, 2008]; Available from: URL: medicine.iupui.edu/flockhart/clinlist.htm.
- 105.Dorsey ST, Biblo LA. Prolonged QT interval and torsades de pointes caused by the combination of fl uconazole and amitriptyline. Am J Emerg Med. 2000;18:227–9. doi: 10.1016/s0735-6757(00)90027-5. [DOI] [PubMed] [Google Scholar]
- 106.Davison ET. Amitriptyline-induced Torsade de pointes: successful therapy with atrial pacing. J Electrocardiol. 1985;18:299–301. doi: 10.1016/s0022-0736(85)80055-8. [DOI] [PubMed] [Google Scholar]
- 107.Jerjes Sanchez DC, Garcia HN, Gonzalez CV, Rosado BA. Helicoidal ventricular tachycardia caused by amitriptyline. Presentation of a case. Arch Inst Cardiol Mex. 1985;55:353–6. [PubMed] [Google Scholar]
- 108.Flugelman MY, Pollack S, Hammerman H, et al. Congenital prolongation of Q-T interval: a family study of three generations. Cardiology. 1982;69:170–4. doi: 10.1159/000173500. [DOI] [PubMed] [Google Scholar]
- 109.Strasberg B, Coelho A, Welch W, et al. Doxepin induced torsade de pointes. PACE. 1982;5:873–7. doi: 10.1111/j.1540-8159.1982.tb06570.x. [DOI] [PubMed] [Google Scholar]
- 110.Alter P, Tontsch D, Grimm W. Doxepin-induced torsade de pointes tachycardia. Ann Intern Med. 2001;135:384–5. doi: 10.7326/0003-4819-135-5-200109040-00026. [DOI] [PubMed] [Google Scholar]
- 111.Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363–72. doi: 10.1136/heart.89.11.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Teschemacher AG, Seward EP, Hancox JC, Witchel HJ. Inhibition of the current of heterologously expressed HERG potassium channels by imipramine and amitriptyline. Br J Pharmacol. 1999;128:479–85. doi: 10.1038/sj.bjp.0702800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jo SH, Youm JB, Lee CO, et al. Blockade of the HERG human cardiac K(+) channel by the antidepressant drug amitriptyline. Br J Pharmacol. 2000;129:1474–80. doi: 10.1038/sj.bjp.0703222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park KS, Kong ID, Park KC, Lee JW. Fluoxetine inhibits L-type Ca2+ and transient outward K+ currents in rat ventricular myocytes. Yonsei Med J. 1999;40:144–51. doi: 10.3349/ymj.1999.40.2.144. [DOI] [PubMed] [Google Scholar]
- 115.Barber MJ, Starmer CF, Grant AO. Blockade of cardiac sodium channels by amitriptyline and diphenylhydantoin. Evidence for two use-dependent binding sites. Circ Res. 1991;69:677–96. doi: 10.1161/01.res.69.3.677. [DOI] [PubMed] [Google Scholar]
- 116.Curtis LH, Ostbye T, Sendersky V, et al. Prescription of QT-prolonging drugs in a cohort of about 5 million outpatients. Am J Med. 2003;114:135–41. doi: 10.1016/s0002-9343(02)01455-9. [DOI] [PubMed] [Google Scholar]
- 117.Witchel HJ, Hancox JC, Nutt DJ. Psychotropic drugs, cardiac arrhythmia, and sudden death. J Clin Psychopharmacol. 2003;23:58–77. doi: 10.1097/00004714-200302000-00010. [DOI] [PubMed] [Google Scholar]
- 118.Thomas D, Gut B, Wendt-Nordahl G, Kiehn J. The antidepressant drug fl uoxetine is an inhibitor of human ether-a-go-go-related gene (HERG) potassium channels. J Pharmacol Exp Ther. 2002;300:543–8. doi: 10.1124/jpet.300.2.543. [DOI] [PubMed] [Google Scholar]
- 119.Tie H, Walker BD, Valenzuela SM, et al. The heart of psychotropic drug therapy. Lancet. 2000;355:1825. doi: 10.1016/s0140-6736(05)73083-x. [DOI] [PubMed] [Google Scholar]
- 120.Tie H, Walker BD, Singleton CB, et al. Clozapine and sudden death. J Clin Psychopharmacol. 2001;21:630–2. doi: 10.1097/00004714-200112000-00023. [DOI] [PubMed] [Google Scholar]
- 121.Tamargo J. Drug-induced torsade de pointes: from molecular biology to bedside. Jpn J Pharmacol. 2000;83:1–19. doi: 10.1254/jjp.83.1. [DOI] [PubMed] [Google Scholar]
- 122.Antzelevitch C. The Brugada syndrome: diagnostic criteria and cellular mechanisms. Eur Heart J. 2001;22:356–63. doi: 10.1053/euhj.2000.2461. [DOI] [PubMed] [Google Scholar]