

Abstract
Malignant glioma, in particular glioblastoma multiforme (GBM), represents one of the most devastating cancers currently known and existing treatment regimens do little to change patient prognosis. Conditionally replicating adenoviral vectors (CRAds) represent attractive experimental anti-cancer agents with potential for clinical application. However, early protein products of the wild type adenovirus backbone—such as E1A—limit CRAds’ replicative specificity. In this study, we evaluated the oncolytic potency and specificity of CRAds in which p300/CPB and/or pRb binding capacities of E1A were ablated to reduce non-specific replicative cytolysis. In vitro cytopathic assays, quantitative PCR analysis, Western blot, and flow cytometry studies demonstrate the superior anti-glioma efficacy of a double-mutated CRAd, Ad2/24CMV, which harbors mutations that reduce E1A binding to p300/CPB and pRb. When compared to its single-mutated and wild type counterparts, Ad2/24CMV demonstrated attenuated replication and cytotoxicity in representative normal human brain while displaying enhanced replicative cytotoxicity in malignant glioma. These results have implications for the development of double-mutated CRAd vectors for enhanced GBM therapy.
Keywords: glioma, adenovirus, oncolytic virus, virotherapy, gene therapy, Ad2/24
INTRODUCTION
Glioblastoma multiforme (GBM) represent the most aggressive form of human brain cancer. Current therapeutic modalities, including surgery, radiotherapy, and chemotherapy have limited efficacy, with median survival measured in months rather than years [Kristiansen et al., 1981; Nakagawa et al., 1998; de Bouard et al., 2007]. The low therapeutic impact of treatments against GBM has prompted significant efforts to seek out novel treatment strategies in order to combat this fatal disease.
The use of conditionally replicating adenoviruses (CRAds) for the treatment of cancer has represented an active area of research investigation. With cytotoxic replication as the presumed goal for oncolytic virotherapy, selective replication has been obtained by engineering specific elements residing in the early region of the adenovirus genome, termed E1A and E1B [Kirn et al., 2001]. The E1A and E1B proteins are expressed early after adenoviral entry into a host cell and together they mediate stable production of adenoviral progeny [Babiss et al., 1985; Debbas and White, 1993]. Particular regions of these genes can be deleted to abolish non-tumor replicative cytotoxicity and achieve a more targeted anti-tumoral response. Vectors of this class that have gained recent attention are ONYX-015 and Ad5Δ24. ONYX-015 lacks the 55K E1B protein, which blocks p53-dependent transcription and apoptosis and thus aids in viral replication and progeny production [Yew and Berk, 1992]. E1B 55K ablation in ONYX-015 has demonstrated selective anti-cancer effects by localizing replication to cancer cells with dysfunctional p53-mediated signaling pathways [Heise et al., 1997].
The Ad5Δ24 vector, as described by Fueyo et al. [2000] contains a 24 base pair (BP) deletion in the conserved region 2 (CR2) of E1A which encodes an Rb-binding domain. The binding of early viral genome products to Rb and Rb-related family members is important for mediating E2F-dependent transcriptional activity and viral and cellular DNA replication [Nevins, 1992]. Theoretically, cells with mutations in the Rb/p16 pathway—which include many types of malignancies [Weinberg, 1992]—will be permissive to Ad5Δ24 infection and cell lysis. On the other hand, healthy cells—ones with functional Rb/p16 pathways—would remain non-permissive to Ad5Δ24 due to inhibited E1A-pRb binding.
The use of such vectors has proved promising in many cancer models, including glioma [Fueyo et al., 2000]. However, these modifications alone do not endow a vector with optimal tumor specificity [Cherubini et al., 2006]. Other modifications have been endeavored, including deletion of E1A regions responsible for binding p300 and other related proteins [Sauthoff et al., 2004]. The reasoning behind such maneuvers is that E1A-p300 binding has been reported to oppose p300-mediated cell cycle regulation by mechanisms that have yet to be clarified. Ablation of E1A-p300 binding has been shown to enhance tumor-specific viral replication in a manner similar to abrogation of Rb binding [Sauthoff et al., 2004; Baluchamy et al., 2007].
Herein, we evaluate the oncolytic potential of CRAds which contain a 3 bp mutation in the N-terminal of E1A, which is reportedly important for interactions with p300/CBP and associated proteins, and/or a 24 bp deletion in CR2, which mediates Rb-E1A binding (Fig. 1). Certain CRAds also harbor a CMV promoter/enhancer cassette upstream of E1A to allow for promoter-independent analysis of each E1A modification on adenoviral replication and cytotoxicity [Hitt and Graham, 1990]. We hypothesized that the use of a vector possessing these deletional modifications would allow for glioma tissue-specific viral oncolysis, with a greater therapeutic index than currently available oncolytic vectors.
Fig. 1.
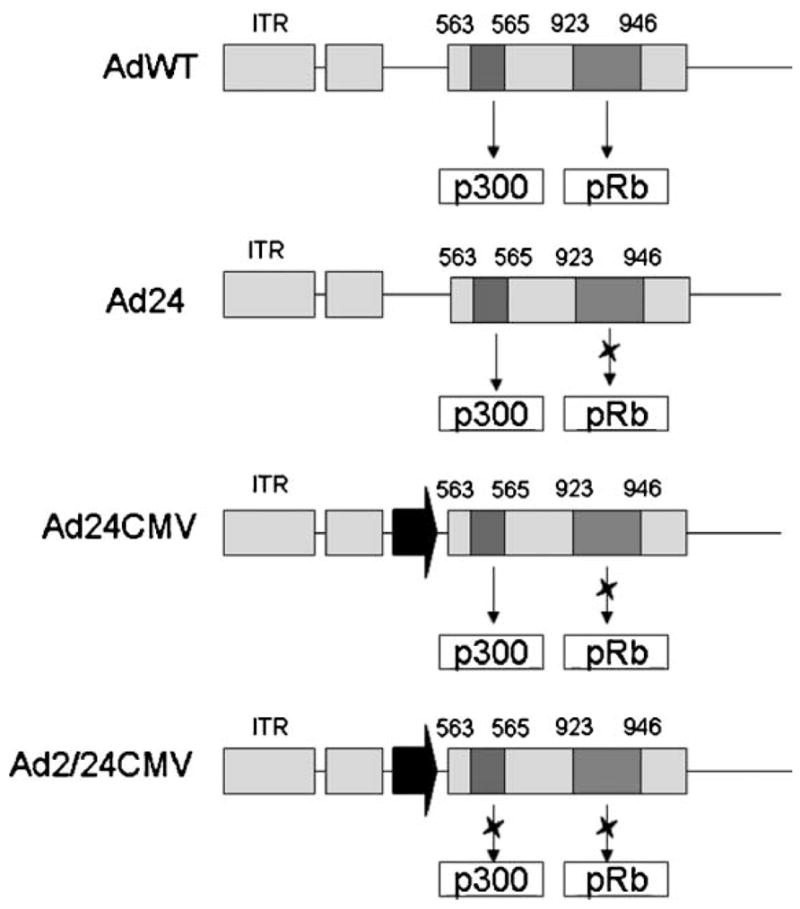
Schematic representation of deletional mutations applied to CRAds. Blue and red boxes indicate regions of E1A gene which encode high-affinity binding domains for the host proteins p300 and pRB, respectively. An X placed over the arrow pointing towards p300 or pRb boxes illustrates the ablation of these regions from a particular construct. ITR; inverted terminal repeat at left of adenovirus genome. Black arrow in Ad24CMV and Ad2/24CMV diagram represents the presence of CMV promoter.
MATERIALS AND METHODS
Cell Lines and Tissues
HEK293- human embryonic kidney cells, human U87MG, U373MG, A172, and mouse glioma GL261 cells were purchased from ATCC (Manassas, VA). Mouse glioma cell line GL261 and human glioma cells No.10, U87MG, A172, U373MG, and Kings were maintained in a humidified 37°C atmosphere containing 5% CO2 and cultured with DMEM supplemented with 10% FBS (CellGrow). Normal human astrocytes (NHA) were purchased and maintained according to vendor recommendations (Cambrex Bioscience, Hopkinton, MA). In order to perform infections, the cells were infected in medium with 2% FBS.
Human primary tissue samples consisting of GBMs (T25, T26) or normal brain (NB) were obtained from surgically resected specimens according to a protocol approved by the Institutional Review Board of the University of Chicago. Representative tissue samples, as confirmed by neuropathologist as grade IV astrocytoma, were converted into tissue slices using a previously described technique [Kirby et al., 2004].
Viruses
AdWT and Ad24CMV have been previously described [Banerjee et al., 2004]. Ad2/24CMV is CR1, CR2-mutated CMV-controlled E1A competent vector [Nettelbeck et al., 2002]. Briefly, to rescue Ad24CMV and Ad2/24CMV, several shuttle plasmids were generated. All of these plasmids contain a fragment of Ad genome from 522 to 3,924 bp (Genebank sequence BK000408). To generate a shuttle plasmid for Ad2/24CMV, the CMV promoter/enhances sequence was incorporated upstream of two deletions: 923–946 and 563–565 bp on the backbone of plasmid pSc. This plasmid was then linearized by PmeI digestion and used for homological recombination with Ptg3602 adenoviral backbone (Transgene, Strasbourg, France). To obtain viral stock, the resultant adenoviral backbone was linearized by PacI and transfected into MeWo cells by using Lipofectamine (Invitrogen, Carlsbad, CA). Ad24 is competent vector having CR1, 24 bp mutation, required for Rb protein binding [Fueyo et al., 2003; Gomez-Manzano et al., 2006]. All viruses were purified on cesium chloride gradients. The vp concentration was determined at 260 nm, and standard plaque assay on HEK293 cells was performed to determine infectious particles [Mittereder et al., 1996]. The pfu titers and vp/pfu ratio were as follows: AdWT (6 ×1010 pfu/ml, ratio 30:1), Ad24 (2 ×1010 pfu/ml, ratio 20:1), Ad24CMV (1.1 ×1011 pfu/ml, ratio 10:1), Ad2/24 CMV (2.2 ×1010 pfu/ml, ratio 30:1).
Cell Killing Assays
Cells were infected with AdWT, Ad24, Ad24CMV or Ad2/24CMV as previously described [Rivera et al., 2004]. For crystal violet staining, 5 ×104 cells were plated on 24-well plates and infected with each virus in different doses: 1,000, 100, 10, and 1 vp per cell. Oncolysis was evaluated at day 10-post-infection [Preuss et al., 2004].
Evaluation of Virus Replication and Brain Slices’ Toxicity
Primary brain slices were cultured for 1 hr at 37°C and then infected with either media (mock) or 500 vp of AdWT, Ad24, Ad24CMV, or Ad2/24CMV vectors according to a previously developed protocol [Kirby et al., 2004]. Four hours later, slices were rinsed with PBS several times and a fresh portion of growth media was added. Portions of infected slices were harvested on days 1 and 2 (T25 and T26) or on days 3, 6 or 9 after infection (NB samples).
Quantitative Real-Time PCR and RT-PCR
Cells were cultured and infected as described above. At the indicated time points, cell culture media was removed, and the adherent cells were collected by trypsinization. The solution was then centrifuged and supernatant was removed. RNAs obtained from the pelleted cells were isolated using the standard protocol provided by Qiagen for RNAeasy isolation. Contaminating DNA was removed by DNase digestion (Qiagen). Reverse transcription to yield cDNA was carried out using GeneAmp RNA core kit (Applied Biosystems, Foster City, CA) using 1 mg total RNA in a volume of 10 ml according to the manufacturer’s instruction. cDNA (4 μl; 1:50 dilution) or isolated DNA in a total volume of 8 μl SYBR samples were incubated at 30 min at 48°C; 10 min at 95°C; and 40 cycles of 15 sec at 95°C and 1 min at 60°C with primers recognizing E1A area, as described earlier [Sonabend et al., 2008]. Data was analyzed with LightCycler software version 3. Results were analyzed using the comparative CT (ΔCT) method as described by the manufacturer. Data are presented as fold increase of each RNA or DNA compared with AdWT-infected cell controls after normalization with the GAPDH content of each sample.
Western Blot Analysis
Human U373MG glioma and NHA cells were infected with 1,000 vp/cell of AdWT, Ad24, Ad24CMV, Ad2/24CMV or with media (mock). Total cell lysates were prepared from cells 24 hr post-infection. Thirty micrograms from each sample was subjected to 10% SDS–Tris-glycine gel electrophoresis and transferred to a PVDF membrane. The membrane was blocked with 5% non-fat milk and incubated with adenovirus type 5 E1A ab-1 (M58, Lab vision, CA) or actin-loading control (AC-15, Abcam Co., Cambridge, MA) followed by incubation with rabbit anti-mouse secondary (ab 6728, Abcam Co.) antibodies conjugated with horseradish peroxidase for 1 hr. Signals were detected with West Pico Peroxidase kit (Pierce, IL). The image was developed with a Kodak 440 image station (Kodak Molecular Imaging, CT). Western blotting results were repeated twice independently.
Statistical Analysis
Statistical significance was determined by standard t-testing with P values less than 0.05 considered statistically significant.
RESULTS
In Vitro Assessment of CRAd Cytotoxicity
To evaluate the cytotoxicity of CRAds with modification at E1A region (Fig. 1), we conducted infections in the human glioma cell lines U87MG, U373MG, A172, No10, Kings and a mouse glioma cell line GL261 with AdWT, Ad24, Ad24CMV, and Ad2/24CMV at different doses (Fig. 2). Ten days after infection, the cytopathic effect was observed by crystal violet staining. Ad24CMV and Ad2/24CMV demonstrated the broadest cytopathic index, with cell killing observed amongst all cell lines. Ad2/24CMV also showed the most efficient cell killing in this experiment, with cytotoxicity observed at 1 vp/cell in some subsets (U87MG, GL261, and U373MG).
Fig. 2.
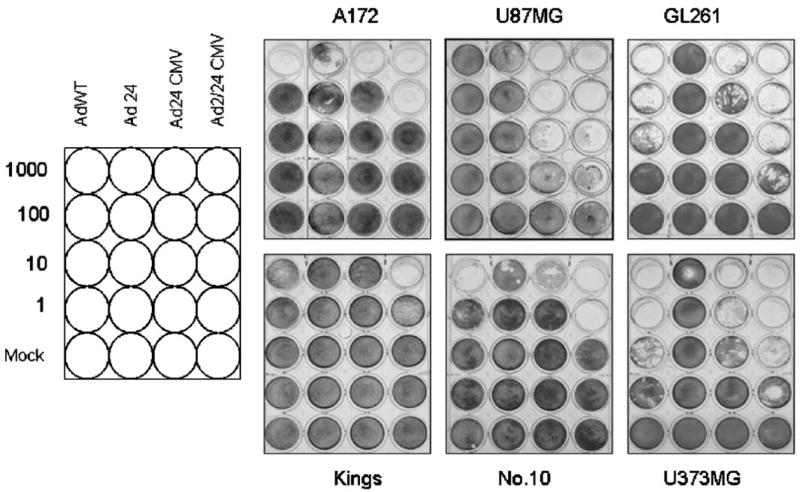
Analysis of induced cytopathic effect by conditionally replicating adenoviruses. Oncolytic potential of mutant vectors was evaluated by crystal violet staining in U87MG, U373MG, GL261, A172, Kings, and N.10 cells. Cells were infected with the panel of viruses at the indicated doses (vp/cell). After 10 days of culture, adherent cells were stained with crystal violet.
CRAd E1A Transcription in Glioma Cells
To assess the effects of the different deletion mutations on E1A transcription, we infected the same panel of glioma cell lines as in Figure 2 at 10 vp/cell and 24 hr later, the number of E1A transcripts were quantified using real-time PCR techniques in the same cell lines (Fig. 3A). E1A expressions were first normalized to GAPDH and then we analyzed the ratio between averages exhibited by each vector to the average present by AdWT. Both Ad24CMV and Ad2/24CMV had E1A transcript levels greater than those observed for Ad24 and WT, possibly due to the presence of the CMV promoter. Ad2/24CMV transcript number was greater than Ad24CMV in the majority of the cell lines tested, including A172 (Ad2/24CMV: 271.5; Ad24CMV: 40.3), U87 (Ad2/24CMV: 793.2; Ad24CMV: 295), Kings (Ad2/24CMV: 2759; Ad24CMV: 52.7) U373MG (Ad2/24CMV: 19.5; Ad24CMV: 5.78) Ad2/24CMV yielded lower transcripts than Ad24CMV in GL261 (Ad2/24CMV: 37.6; Ad24CMV: 61.8) and No.10 (Ad2/24CMV: 128; Ad24CMV: 271).
Fig. 3.
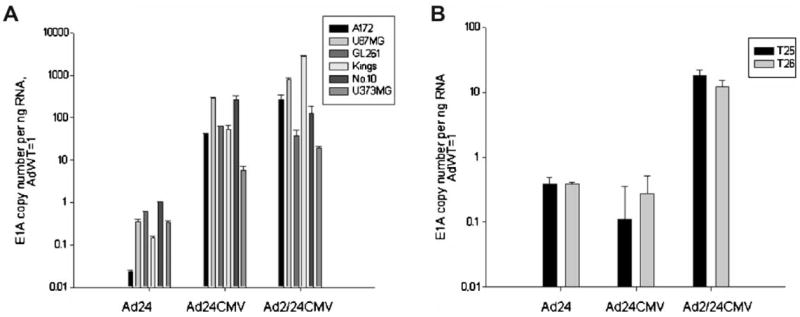
Effect of E1A mutations on adenoviral transcription in glioma. A: The mouse glioma cell line GL261 and human glioma cell lines were infected with AdWT, Ad24, Ad24CMV, and Ad2/24CMV vectors at 10 vp/cell. Twenty-four hours after infection, transcripts were quantified by real-time PCR using primers recognizing adenovirus E1A. E1A transcript copy number for each mutant virus is represented as fold E1A copy/ng RNA over AdWT. B: The same analysis as for (A) was performed in two GBM tissue samples, T25 and T26.
We also tested the E1A expression of our CRAds in two primary glioblastoma samples resected from patients, designated T25 and T26 (Fig. 3B). Primary samples were infected with AdWT, Ad24, Ad24CMV, and Ad2/24CMV at 500 vp/cell and then E1A transcripts were quantified and analyzed in the same manner as in Figure 3A. Whereas CR2-mutated vectors did not show significant improvement of E1A expression, Ad2/24CMV, which harbors modifications in both N-terminus and CR2, displayed significant increase in E1A mRNA expression in both T25 (17.82 ± 4.1-fold over AdWT) and T26 (12.13 ± 3.22-fold over AdWT).
In Vitro Replication in Glioma Cells
Next, we wished to investigate the effect of the N-term/CR2 mutations on adenoviral replication in glioma cells. We performed in vitro infection assays using the same panel of glioma cell lines as well as the two primary glioblastoma samples, T25 and T26. As a quantitative measure of replication efficiency, E1A DNA copy numbers were quantified by real-time PCR analysis and normalized to AdWT E1A DNA expression (Fig. 4).
Fig. 4.

Analysis of viral replication in glioma. A: Human glioma cells U87MG, U373MG, A172, N.10, Kings, mouse glioma cells GL261, and (B) primary GBM tissue T25 and T26 were infected with AdWT, Ad24, Ad24CMV, and Ad2/24CMV viruses at a dose of 1,000 (A) and 500 (B) vp/cell. After 4 hr adsorption, cells were rinsed with PBS and allowed to continue incubation with growth media. Twenty-four and 48 hr after infection with each virus, replication activity was quantified by measuring the amount of E1A DNA copies/ng DNA by qPCR. Bar graphs represent fold change of E1A DNA copies/ng DNA over AdWT.
At both time points (days 1 and 2), Ad2/24CMV E1A DNA expression significantly exceeded that of Ad24 and Ad24CMV in all cell lines and both primary samples (P <0.05). The only exception was the mouse glioma cell line, GL261, where Ad24CMV E1A copy numbers were significantly greater than Ad2/24CMV at day 1 (2.5 × 104 ± 1.05 × 102 and 710.04 ± 86.1 copies) and non-significantly less than Ad2/24CMV at day 2 (1.3 × 105 ± 3.07 × 104 and 1.04 × 106 ±4. 105 copies). Ad24CMV E1A copies were roughly equal to or less than those of Ad24 at both time points in U87MG, Kings, No. 10 (only day 1). In case of T25, they were 0.7 ± 0.4 and 1.5 ± 0.34-fold increased over AdWT at days 1 and 2 and in T26 they were 0.3 ± 0.12 and 0.34 ± 0.23-fold increased at days 1 and 2, respectively. In comparison to all other glioma cell lines, replication of Ad24, Ad24CMV, and Ad2/24CMV was not as robust in the cell line U373. However, in primary samples, T25 and T26, E1A DNA copies were 2–10-fold increased relative to AdWT (P <0.05).
Specificity of E1A Expression
The suitability of a viral vector for oncolytic therapy is determined by its specificity of E1A protein expression and cytolysis in targeted tissue. Thus, we wished to examine the therapeutic potential of N-terminal/CR2-modified CRAds by assessing how the N-term/CR2 mutations might affect E1A protein expression, viral replication, and cytotoxicity in cultured cells representing NB cell types.
To analyze the effects of N-term/CR2 mutations on E1A protein expression in non-transformed cells, we infected U373MG and NHA cells with 1,000 vp of AdWT, Ad24, Ad24CMV, and Ad2/24CMV. Twenty-four hours post-infection, cell lysates were analyzed for the amount of E1A protein by Western blot (Fig. 5A). AdWT was able to direct E1A protein expression in both types of cells; however, more E1A protein was present in lysates recovered from NHA compared to U373MG (lane 2). Similar results were observed for Ad24 and Ad24CMV (lanes 3 and 4, respectively). In contrast, Ad2/24CMV infection resulted in decreased amounts of E1A protein in NHA cells relative to U373MG infection (lane 5).
Fig. 5.
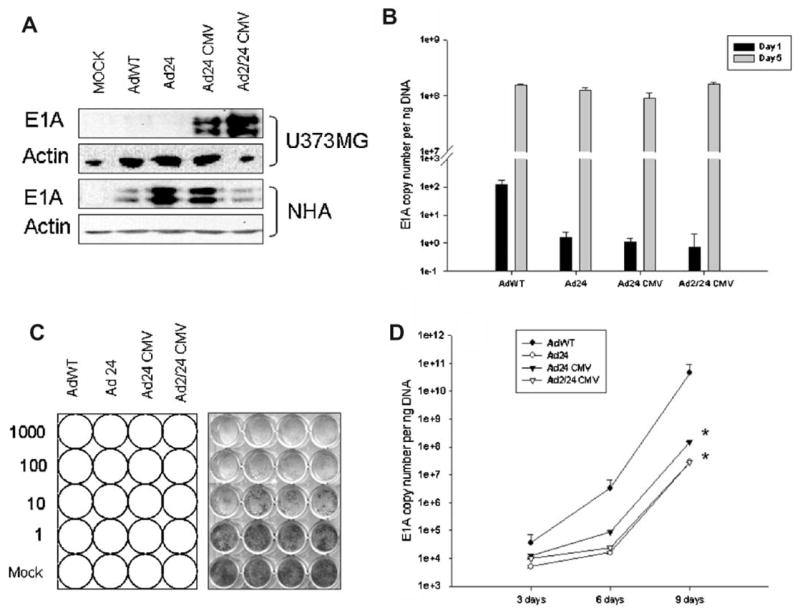
Specificity of protein production and replication mediated by E1A-mutant CRAds. A: Human glioma U373MG or normal human astrocytes (NHA) cells were infected with AdWT, Ad24, AdCMV24, or Ad2/24CMV vectors at 1,000 vp/cell. After 4 hr adsorption, virus containing media was aspirated and replaced with fresh media supplemented with 10% FBS. Cell lysates were examined 24 hr after viral infection for E1A protein expression by Western blot. Replications assays were also conducted for NHA (B) and normal human brain (D). CRAd-induced toxicity in NHA was analyzed by crystal violet staining (C). E1A detection was achieved using a polyclonal antibody against Ad5 E1A proteins. Mock- and AdWT-infected cells were used as comparative controls and actin antibodies were used for quality control. Data represent results from two independent experiments. E1A DNA copies were quantified and bar graphs represent copies of E1A DNA/ng DNA. “*” shows P-value <0.05.
To determine the replication activity of each vector in normal cells, we conducted quantitative analysis of E1A DNA copy numbers in both cultured NHA and NB tissue resected from patient using real-time PCR (Fig. 5B,D, respectively). E1A DNA content was quantified at days 1 and 5 in NHA and at days 3, 6, and 9 in NB. In NHA, Ad24, Ad24CMV, and Ad2/24CMV expressed significantly less E1A DNA than wild type at day 1 (1.59 ± 1.11; 1.11 ± 0.84; 0.722 ± 0.34 vs. 1.27 × 102 ± 47.2 copies; P <0.05) while analysis at day 5 yielded similar expression patterns among all virus types. In NB, E1A DNA expression appeared to increase in a time-dependent manner. Thus, AdWT E1A DNA content was significantly greater than all modified viruses at all time points. For instance, Ad24, Ad24CMV, and Ad2/24CMV E1A DNA values were similar at day 3 (5.1 × 103 ± 512; 1.2 × 104 ± 1.1 × 103; 9.6 × 103 ± 650 copies per ng DNA); however, however, Ad24 and Ad2/24CMV E1A expression was attenuated at days 6 and 9, relative to Ad24CMV. Thus we observed significant inhibition of E1A for Ad24 and Ad2/24CMV expressions in comparison with Ad24CMV (1.6 × 104 ± 195.1 and 2.4 × 104 ± 301.6 vs. 8.6 × 104 ± 624.25) at day 6 and (2.8 × 108 ± 4.3 × 105 and 2.7 × 107 ± 2.52 × 105copies vs. 1.5 ×108 ± 1.6 ×106; P <0.05) at day 9.
We also determined the cytotoxicity of each vector in NHA by crystal violet staining (Fig. 5C). Cytotoxicity was demonstrated for all viruses at all doses. Complete cell killing was observed for all constructs at 1,000 vp/cell infections (row 1) and nearly complete cell killing at 100 vp/cell infections (row 2). At doses of 10 and 1 vp/cell (rows 3 and 4, respectively) AdWT showed a higher cytotoxic effect than the other attenuated vectors.
DISCUSSION
Current therapies for the treatment of malignant glioma do little to change patient prognosis, necessitating the development of novel strategies for the treatment of the aggressive disease. Conditionally replicating adenoviral (CRAd) therapy has arisen as a potential therapy that may be included in treatment regimens for different malignancies, including glioma [Freytag et al., 2007; Sonabend et al., 2007]. CRAds that harbor deletion mutations in the early regions of the E1A gene that encode high-affinity binding regions for the proteins p300/CBP and pRb have been suggested as an effective mechanism for achieving oncolytic specificity in many different tumor models, including malignant melanoma [Nettelbeck et al., 2002], ovarian cancer [Lockley et al., 2006], and disseminated gastric cancer [Kangasniemi et al., 2006], to name a few.
The proteins p300/CPB, pRB, and their related family members are crucial regulators of cellular transcriptional activity and cell cycle; they are also targets of adenoviral early region protein products [Ghosh and Harter, 2003]. p300 and cyclic AMP-response element-binding protein (CBP) are transcriptional co-activating enzymes which mediate the activity of a diverse set of genes throughout the human genome. CBP and p300 control gene activity via acetylation of histones and by association with other proteins that complex with DNA. Adenoviral E1A binding to p300, CBP, and other related proteins is mediated by regions in the N-terminus and conserved region 1 (CR1) of E1A [Gallimore and Turnell, 2001; Frisch and Mymryk, 2002]. These diverse associations regulate transcriptional activity/inactivity schemas of viral and cellular DNA which can ultimately promote S phase induction, viral replication, and eventually cell lysis.
Rb, and its family members p130 and p107, are well-studied proteins that regulate the cell cycle. In quiescent cells, Rb is non- or hypophosphorylated and bound to members of the E2F family of transcription factors [Chellappan et al., 1991]. S phase of the cell cycle is induced, in part, when cyclin-dependent kinases (Cdks) phosphorylate Rb, relieving its inhibition of E2F-mediated transcription of cell cycle genes [Knudsen and Wang, 1996]. S phase can also be induced when adenoviral E1A protein binds Rb, relieving its inhibition of E2F [Bandara and La Thangue, 1991; Zamanian and La Thangue, 1992]. E1A binding to Rb is achieved via regions in both CR1 and CR2 of E1A that associate with the pocket domains of Rb and is sufficient for inducting DNA synthesis and adenoviral replication in quiescent cells [Svensson et al., 1991; Chow et al., 1996].
The potential of CRAd-based tumor therapy is predicated upon a vector’s oncolytic potency and specificity. Inherent properties of E1A, as listed above, limit the replicative specificity of CRAds and diminish their potential for achieving high therapeutic indices in cancer models [Kirn, 2001]. The proliferative nature of malignant cells provides a pro-growth expression profile suitable for adenoviral replication. Additionally, the Rb-pathway of cell cycle regulation is commonly associated with deleterious mutations in many cancers, including glioma [Sehgal et al., 1998]. Thus, it is reasoned that E1A binding to p300, Rb, and related members is unnecessary for viral replication in cancer cells and thus dispensable in the context of oncolytic virotherapy. Moreover, abrogation of such binding capabilities would enhance the replicative selectivity of an oncolytic vector. In this study, we evaluated the oncolytic potency and specificity of CRAds containing mutations in two key regions of E1A responsible for non-specific cell transformation and lysis. One of the two mutations included a 3 bp deletion mutation in the N-terminal region of E1A. Mutations in this reason strongly reduce adenoviral transformation of quiescent cells [Sauthoff et al., 2004; Rasti et al., 2005]. The second mutation was a 24 bp deletion in the CR2 region of E1A, which encodes amino acids that are required for sequestration of Rb and induction of S phase in senescent cells [Svensson et al., 1991].
Our results indicate that N-term/CR2 mutations of E1A do not diminish the oncolytic potency of the CRAds in glioma cells. In fact, these deletions appear to endow the virus with an enhanced anti-tumor effect. This was shown in a broad panel of glioma cell lines as well as resected glioma tissue from two patients. Crystal violet cytotoxic assays, E1A transcript analysis, protein expression, and DNA expression confirm that N-terminal p300/CBP—and CR2 Rb-binding site mutations do not affect processes of glioma cell killing, E1A transcription, or replication, respectively. Furthermore, we have identified a double-mutant CRAd, Ad2/24CMV, as a superior vector that exhibits increased replication and glioma cell killing, when compared to wild type adenovirus, and its other mutant counterparts (Ad24, Ad24CMV). These results are in accordance with previously published reports which demonstrate that doubly mutated vectors have enhanced, or similar, oncolytic capacities compared to wild type vectors. [Nettelbeck et al., 2002].
It is debatable whether or not the amplified replication and cell killing observed was the result of CMV promoter activity [Kasparov, 2007]. However, our results are more aligned with earlier reports suggesting that promoter activity does not serve a predictive function in regards to adenoviral replication and cell killing [Hitt and Graham, 1990; Nettelbeck et al., 2002]. We found that replication assays (E1A DNA quantification) were much more suitable for predicting the relative cytotoxicity amongst the tested viruses (compare Figs. 2 vs. 4). Thus, the CMV promoter application proved to be a useful system for an evaluation of the effects of E1A deletion mutations on CRAd replication and cytotoxicity within the context of malignant glioma.
The Ad2/24CMV vector demonstrated the greatest potential for selective replication among the viruses tested (Fig. 4). This was confirmed by down-regulated E1A protein expression and decreased DNA replication in NHA 1 day after infection with Ad2/24CMV (Fig. 5A,B, respectively). A more convincing, and clinically relevant, exhibition of Ad2/24CMV selective replication was revealed in the DNA assays conducted in NB tissue. The use of normal human brain is superior to NHA as the latter represents a commercially available cell line derived from fetal tissue still undergoing cell division. In these studies, Ad2/24CMV displayed a significant attenuation of genome amplification when evaluated relative to AdWT and Ad24CMV (Fig. 5D).
Our immunoblot results support similar findings revealed by Hitt et al. which suggest that little E1A protein expression is required for induction of viral replication. For instance, Figure 5A (Western blot) shows that infection of NHAs with the different viruses yielded differential E1A expression; yet, by day 5 post-infection, DNA copy numbers were similar amongst all viruses (Fig. 5B; ~1Eþ8). We suggest the existence of an E1A threshold, which is necessary for viral replication to ensue in a particular host cell. It remains to be determined if the threshold is cell-type specific. Thus, our observations in NHA could be the result of strong CMV promoter activity in NHA and/or the expression of CAR on the surface of NHA, two important factors that would enable stable E1A protein expression and subsequent viral replication in these cells [Pulliam et al., 1988; Fueyo et al., 2003]. In this regard, the application of combinatorial targeting methodologies that include tumor-specific promoters, E1A deletions, and fiber-knob modifications would be a feasible approach for attenuating E1A expression in normal tissues below “threshold levels.” We are currently conducting studies to identify such a vector for malignant glioma.
Our findings also illustrate the complexity of E1A protein function in the adenoviral life cycle. Recent data suggests that the N-terminal region of E1A possesses distinct amino acids that mediate binding of a milieu of different proteins involved in E1A-mediated S-phase induction; it was found that these proteins bound to these sites independent of other E1A-N-terminal binding proteins (i.e., no complex formation) [Rasti et al., 2005]. It is possible, though not proven experimentally, that E1A N-terminus regions have evolved to induce S-phase and subsequent viral replication in a number of different cell types via mediating interactions with differentially expressed proteins. This would be an interesting topic for further investigation; and, it might be able to explain non-specific replication of the Ad2/24CMV CRAds. It would be useful to further elucidate the molecular mechanisms responsible for endowing E1A mutant CRAds with replication and cell killing in malignant cells, yet decreased replication and cytopathic effect in normal cells. Also, identifying the relationship between the amount of E1A expressed and viral genome amplification/cytotoxicity observed would be helpful when evaluating relative CRAd cytotoxicities. Clarification in these areas would allow for the engineering of CRAds that more effectively target desired tissues.
In conclusion, we have demonstrated in a glioma model, for the first time, the superior oncolytic potency and specificity of a double-mutant CRAd which harbors deletion mutations in the N-terminal p300/CPB binding region in addition to the CR2 Rb-binding region. Our results demonstrate that these deletions alone (regardless of promoter) enhance CRAd oncolysis while reducing normal tissue cytotoxicity. These qualities make this vector suitable for advanced experimentation in designing CRAd-based therapies for malignant glioma.
Acknowledgments
National Cancer Institute; Grant number: R01CA122930; Grant sponsor: National Institute of Neurological Disorders and Stroke; Grant number: K08-NS046430; Grant sponsor: Alliance for Cancer Gene Therapy Young Investigator Award; Grant sponsor: American Cancer Society; Grant number: RSG-07-276-01-MGO.
References
- Babiss LE, Ginsberg HS, Darnell JE., Jr Adenovirus E1B proteins are required for accumulation of late viral mRNA and for effects on cellular mRNA translation and transport. Mol Cell Biol. 1985;5:2552–2558. doi: 10.1128/mcb.5.10.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluchamy S, Sankar N, Navaraj A, Moran E, Thimmapaya B. Relationship between E1A binding to cellular proteins, c-myc activation and S-phase induction. Oncogene. 2007;26:781–787. doi: 10.1038/sj.onc.1209825. [DOI] [PubMed] [Google Scholar]
- Bandara LR, La Thangue NB. Adenovirus E1a prevents the retinoblastoma gene product from complexing with a cellular transcription factor. Nature. 1991;351:494–497. doi: 10.1038/351494a0. [DOI] [PubMed] [Google Scholar]
- Banerjee NS, Rivera AA, Wang M, Chow LT, Broker TR, Curiel DT, Nettelbeck DM. Analyses of melanoma-targeted oncolytic adenoviruses with tyrosinase enhancer/promoter-driven E1A, E4, or both in submerged cells and organotypic cultures. Mol Cancer Ther. 2004;3:437–449. [PubMed] [Google Scholar]
- Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- Cherubini G, Petouchoff T, Grossi M, Piersanti S, Cundari E, Saggio I. E1B55K-deleted adenovirus (ONYX-015) overrides G1/S and G2/M checkpoints and causes mitotic catastrophe and endoreduplication in p53-proficient normal cells. Cell Cycle. 2006;5:2244–2252. doi: 10.4161/cc.5.19.3263. [DOI] [PubMed] [Google Scholar]
- Chow KN, Starostik P, Dean DC. The Rb family contains a conserved cyclin-dependent-kinase-regulated transcriptional repressor motif. Mol Cell Biol. 1996;16:7173–7181. doi: 10.1128/mcb.16.12.7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bouard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, Guillamo JS. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007 doi: 10.1215/15228517-2007-024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J, Zhang Y, Barton KN, Brown SL, Lu M, Savera A, Kim JH. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther. 2007;15:1016–1023. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Mymryk JS. Adenovirus-5 E1A: Paradox and paradigm. Nat Rev Mol Cell Biol. 2002;3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, Liu TJ, Jiang H, Lemoine MG, Suzuki K, Sawaya R, Curiel DT, Yung WK, Lang FF. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003;95:652–660. doi: 10.1093/jnci/95.9.652. [DOI] [PubMed] [Google Scholar]
- Gallimore PH, Turnell AS. Adenovirus E1A: Remodelling the host cell, a life or death experience. Oncogene. 2001;20:7824–7835. doi: 10.1038/sj.onc.1204913. [DOI] [PubMed] [Google Scholar]
- Ghosh MK, Harter ML. A viral mechanism for remodeling chromatin structure in G0 cells. Mol Cell. 2003;12:255–260. doi: 10.1016/s1097-2765(03)00225-9. [DOI] [PubMed] [Google Scholar]
- Gomez-Manzano C, Alonso MM, Yung WK, McCormick F, Curiel DT, Lang FF, Jiang H, Bekele BN, Zhou X, Alemany R, Fueyo J. Delta-24 increases the expression and activity of topoisomerase I and enhances the antiglioma effect of irinotecan. Clin Cancer Res. 2006;12:556–562. doi: 10.1158/1078-0432.CCR-05-1892. [DOI] [PubMed] [Google Scholar]
- Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- Hitt MM, Graham FL. Adenovirus E1A under the control of heterologous promoters: Wide variation in E1A expression levels has little effect on virus replication. Virology. 1990;179:667–678. doi: 10.1016/0042-6822(90)90134-d. [DOI] [PubMed] [Google Scholar]
- Kangasniemi L, Kiviluoto T, Kanerva A, Raki M, Ranki T, Sarkioja M, Wu H, Marini F, Hockerstedt K, Isoniemi H, Alfthan H, Stenman UH, Curiel DT, Hemminki A. Infectivity-enhanced adenoviruses deliver efficacy in clinical samples and orthotopic models of disseminated gastric cancer. Clin Cancer Res. 2006;12:3137–3144. doi: 10.1158/1078-0432.CCR-05-2576. [DOI] [PubMed] [Google Scholar]
- Kasparov S. Suitability of hCMV for viral gene expression in the brain. Nat Methods. 2007;4:379. doi: 10.1038/nmeth0507-379a. author reply 379. [DOI] [PubMed] [Google Scholar]
- Kirby TO, Rivera A, Rein D, Wang M, Ulasov I, Breidenbach M, Kataram M, Contreras JL, Krumdieck C, Yamamoto M, Rots MG, Haisma HJ, Alvarez RD, Mahasreshti PJ, Curiel DT. A novel ex vivo model system for evaluation of conditionally replicative adenoviruses therapeutic efficacy and toxicity. Clin Cancer Res. 2004;10:8697–8703. doi: 10.1158/1078-0432.CCR-04-1166. [DOI] [PubMed] [Google Scholar]
- Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001;8:89–98. doi: 10.1038/sj.gt.3301377. [DOI] [PubMed] [Google Scholar]
- Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- Kristiansen K, Hagen S, Kollevold T, Torvik A, Holme I, Nesbakken R, Hatlevoll R, Lindgren M, Brun A, Lindgren S, Notter G, Andersen AP, Elgen K. Combined modality therapy of operated astrocytomas grade III and IV. Confirmation of the value of postoperative irradiation and lack of potentiation of bleomycin on survival time: A prospective multicenter trial of the Scandinavian Glioblastoma Study Group. Cancer. 1981;47:649–652. doi: 10.1002/1097-0142(19810215)47:4<649::aid-cncr2820470405>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Lockley M, Fernandez M, Wang Y, Li NF, Conroy S, Lemoine N, McNeish I. Activity of the adenoviral E1A deletion mutant dl922–947in ovarian cancer: Comparison with E1A wild-type viruses, bioluminescence monitoring, and intraperitoneal delivery in icodextrin. Cancer Res. 2006;66:989–998. doi: 10.1158/0008-5472.CAN-05-2691. [DOI] [PubMed] [Google Scholar]
- Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K, Aoki Y, Fujimaki T, Tago M, Terahara A, Karasawa K, Sakata K, Sasaki Y, Matsutani M, Akanuma A. High-dose conformal radiotherapy influenced the pattern of failure but did not improve survival in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1998;40:1141–1149. doi: 10.1016/s0360-3016(97)00911-5. [DOI] [PubMed] [Google Scholar]
- Nettelbeck DM, Rivera AA, Balague C, Alemany R, Curiel DT. Novel oncolytic adenoviruses targeted to melanoma: Specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res. 2002;62:4663–4670. [PubMed] [Google Scholar]
- Nevins JR. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- Preuss MA, Lam JT, Wang M, Leath CA, III, Kataram M, Mahasreshti PJ, Alvarez RD, Curiel DT. Transcriptional blocks limit adenoviral replication in primary ovarian tumor. Clin Cancer Res. 2004;10:3189–3194. doi: 10.1158/1078-0432.ccr-03-0802. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Berens ME, Rosenblum ML. A normal human brain cell aggregate model for neurobiological studies. J Neurosci Res. 1988;21:521–530. doi: 10.1002/jnr.490210243. [DOI] [PubMed] [Google Scholar]
- Rasti M, Grand RJ, Mymryk JS, Gallimore PH, Turnell AS. Recruitment of CBP/p300, TATA-binding protein, and S8 to distinct regions at the N terminus of adenovirus E1A. J Virol. 2005;79:5594–5605. doi: 10.1128/JVI.79.9.5594-5605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera AA, Davydova J, Schierer S, Wang M, Krasnykh V, Yamamoto M, Curiel DT, Nettelbeck DM. Combining high selectivity of replication with fiber chimerism for effective adenoviral oncolysis of CAR-negative melanoma cells. Gene Ther. 2004;11:1694–1702. doi: 10.1038/sj.gt.3302346. [DOI] [PubMed] [Google Scholar]
- Sauthoff H, Pipiya T, Heitner S, Chen S, Bleck B, Reibman J, Chang W, Norman RG, Rom WN, Hay JG. Impact of E1a modifications on tumor-selective adenoviral replication and toxicity. Mol Ther. 2004;10:749–757. doi: 10.1016/j.ymthe.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Sehgal A, Ricks S, Boynton AL, Warrick J, Murphy GP. Molecular characterization of CXCR-4: A potential brain tumor-associated gene. J Surg Oncol. 1998;69:239–248. doi: 10.1002/(sici)1096-9098(199812)69:4<239::aid-jso9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Sonabend AM, Ulasov IV, Lesniak MS. Gene therapy trials for the treatment of high-grade gliomas. Gene Ther Mol Biol. 2007;11:79–92. [PMC free article] [PubMed] [Google Scholar]
- Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008 doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]
- Svensson C, Bondesson M, Nyberg E, Linder S, Jones N, Akusjarvi G. Independent transformation activity by adenovirus-5 E1A-conserved regions 1 or 2 mutants. Virology. 1991;182:553–561. doi: 10.1016/0042-6822(91)90596-4. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma gene and gene product. Cancer Surv. 1992;12:43–57. [PubMed] [Google Scholar]
- Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- Zamanian M, La Thangue NB. Adenovirus E1a prevents the retinoblastoma gene product from repressing the activity of a cellular transcription factor. EMBO J. 1992;11:2603–2610. doi: 10.1002/j.1460-2075.1992.tb05325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]