

Abstract
Streptococcus agalactiae, a prokaryote that causes infections in neonates and immunocompromised adults, has a serine/threonine protein kinase (STK) signalling cascade. The structure of one of the targets, a family II inorganic pyrophosphatase, has been solved by molecular replacement and refined at 2.80 Å resolution to an R factor of 19.2% (Rfree = 26.7%). The two monomers in the asymmetric unit are related by a noncrystallographic twofold axis, but the biological dimer is formed by a crystallographic twofold. Each monomer contains the pyrophosphate analogue imidodiphosphate (PNP) and three metal ions per active site: two Mn2+ ions in sites M1 and M2 and an Mg2+ ion in site M3. The enzyme is in the closed conformation. Like other family II enzymes, the structure consists of two domains (residues 1–191 and 198–311), with the active site located between them. The conformation of Lys298 in the active site is different from those observed previously and it coordinates to the conserved DHH motif in a unique way. The structure suggests that Ser150, Ser194, Ser195 and Ser296 are the most likely targets for the Ser/Thr kinase and phosphatase because they are surface-accessible and either in the active site or in the hinge region between the two domains.
1. Introduction
Phosphorylation provides cells with the means to transduce signals in a manner that is analogous to an electrical switch. While kinases covalently attach phosphate to a protein, hydrolytic phosphatases reverse the process. Serine and threonine residues may undergo phosphorylation by a serine/threonine kinase (STK). This was previously thought to occur solely in eukaryotes (Alex & Simon, 1994), but in recent years serine/threonine signalling has been observed in many prokaryotes including Bacillus subtilis (Yang et al., 1996; Adler et al., 1997), Mycobacterium tuberculosis (Boitel et al., 2003) and Yersinia pseudotuberculosis (Hakansson et al., 1996). Serine/threonine phosphorylation has also been linked with virulence (Cozzone, 2005). Ser/Thr phosphorylation has also been observed in Streptococcus agalactiae, an organism responsible for infections among newborns and immuno-compromised adults (Rajagopal et al., 2003). Here, visible changes such as differences in chain length occur in mutant bacterial strains. Mutations in both the stp gene and in the stk gene make the bacteria less virulent, which is manifested in higher LD50 values in a rat sepsis model. M. tuberculosis (Walburger et al., 2004), S. pneumoniae (Echenique et al., 2004), Pseudomonas aeruginosa (Wang et al., 1998), Listeria monocytogenes (Herro et al., 2005) and Yersinia enterocolitica (Galyov et al., 1993) are other examples of bacteria in which these two genes are involved in controlling virulence.
From a structural perspective, the best-studied prokaryotic serine/threonine signalling system is that from M. tuberculosis (Greenstein et al., 2005). Structures are available of the intracellular subunit of one of the 11 M. tuberculosis STKs, PknB (PDB code 1mru; Young et al., 2003), and of the M. tuberculosis serine/threonine phosphatase (STP; PDB code 1txo; Pullen et al., 2004), which belongs to the PP2C (enzymes resembling human PP2C) subfamily. However, there are no structures available of bacterial Ser/Thr phosphorylation targets. Indeed, only a handful of prokaryotic STK and STP targets have been found to date. In S. agalactiae, the targets include adenylosuccinate synthetase (Rajagopal et al., 2005), the response regulator CovR (Rajagopal et al., 2006) and, surprisingly, a family II inorganic pyrophosphatase (Rajagopal et al., 2003).
Pyrophosphatases are essential enzymes (Chen et al., 1990) that provide the thermodynamic pull for many biochemical reactions that produce pyrophosphate from 5′-nucleoside triphosphates during anabolism (Lahti, 1983). There are two distinct families of soluble inorganic pyrophosphatases (EC 3.6.1.1): family I and family II (Shintani et al., 1998; Young et al., 1998). The two families show no structural similarity: family I PPases are single-domain OB-fold proteins (Heikinheimo et al., 1996), while family II PPases are two-domain proteins. In addition, the identities of the metal-coordinating ligands and the bound metal ions are different (Merckel et al., 2001; Ahn et al., 2001). Both family I and family II enzyme structures have been characterized in detail. Despite the convergence of the active-site structures in these two families (Merckel et al., 2001), the two families are not related by sequence.
Family II PPases are dimeric metalloenzymes which utilize divalent transition-metal ions, preferably Mn2+ or Co2+ (Zyryanov et al., 2004), which bind a bridging nucleophilic water molecule in the active site. This feature is widely used in hydrolytic metalloenzymes (Christianson & Cox, 1999). Early structural work revealed that the enzymes are capable of domain movement about a flexible hinge (Ahn et al., 2001; Merckel et al., 2001) and that the enzyme cycles between an open conformation into which substrate binds and a closed conformation during the hydrolysis step (Fabrichniy et al., 2004). Currently, the PDB contains family II PPase structures from S. mutans, B. subtilis and S. gordonii (Fabrichniy et al., 2004, 2007; Merckel et al., 2001; Ahn et al., 2001), all with high sequence homology to S. agalactiae PPase. The most recent B. subtilis structure was solved with imidodiphosphate (PNP), a substrate analogue, in the active site (Fabrichniy et al., 2007), thus mimicking the substrate-bound state of the enzyme. The S. agalactiae PPase structure that we present here is the first one of a known signalling-associated family II PPase.
2. Materials and methods
Expression, purification and crystallization of the family II inorganic pyrophosphatase in complex with the PNP ligand [(HO)2PONPO(OH)2] were performed as described by Rantanen et al. (2006). Crystals were grown from 0.4 Munbuffered monobasic ammonium phosphate (Rantanen et al., 2006). We collected the data using Cu Kα radiation generated by a Rigaku RU3000 rotating-anode generator operated at 50 kV and 20 mA and monochromated with confocal mirrors. An R-AXIS IV image-plate detector was used to collect the data at 100 K and the crystal diffraction limit was 2.8 Å. Using 0.5° frames with 12 min exposure times, a total of 100° of data were collected. The data were processed using XDS (Kabsch, 1993) and the data-collection statistics showed that the crystal belonged to space group R32, with unit-cell parameters a = 182.0, c = 132.6 Å, and that 99.2% completeness was reached with an overall I/σ(I) of 12.3. The R factor was 16.3% and Rmerged-F was 8.5% overall (Table 1) as described previously (Rantanen et al., 2006).
Table 1.
Data-collection and refinement statistics for the family II inorganic pyrophosphatase.
| Values in parentheses are for the highest resolution shell. | |
|---|---|
| Data collection | |
| Space group | R32 |
| Resolution range (Å) | 19.5–2.80 (2.9–2.8) |
| Wavelength (Å) | 1.5418 (Cu Kα) |
| Unit-cell parameters (Å) | a = b = 182.0, c = 132.6 |
| Reflections measured | 126779 (8798) |
| Unique reflections | 20708 (1035) |
| Completeness (%) | 99.2 (96.4) |
| Redundancy | 6.1 (4.4) |
| I/σ (I) | 12.3 (6.23) |
| R (%) | 16.3 (30.3) |
| Rmerged-F†(%) | 8.5 (14) |
| Refinement | |
| No. of reflections/No. in test set | 20708/1035 |
| Refined atoms (total/water/metal) | 4923/154/6 |
| Rwork‡(%) | 19.2 (30) |
| Rfree§(%) | 26.7 (35) |
| R.m.s.d. bond length (Å) | 0.012 |
| R.m.s.d. bond angle (°) | 1.49 |
| Overall B value (Å2) | 18.2 |
| Missing residues | A34 to A1, B34 to B1 |
| Ramachandran plot, residues in | |
| Most favoured region (%) | 86.5 |
| Additionally allowed region (%) | 13.5 |
Rmerged-F = Σ|AI(h,P) − AI(h,Q)|/0.5 Σ[AI(h,P) + AI(h,Q)], where AI = I1/2 if I ≥ 0 and AI = −I1/2 if I < 0. P and Q are two subsets of data (Diederichs & Karplus, 1997).
Rwork = Σ||Fobs| − |Fcalc|/Σ |Fobs|
Rfree is the same as Rwork but for a subset that represents 5% of the data.
The structure of the family II inorganic pyrophosphatase was solved by molecular replacement using MOLREP (Collaborative Computational Project, Number 4, 1994; Vagin & Teplyakov, 1997) and the CCP4i graphical interface (Potterton et al., 2003). We used the S. mutans enzyme structure (PDB code 1i74; Merckel et al., 2001) as a model. This enzyme has 77% sequence identity with the S. agalactiae enzyme. The initial R factor after molecular replacement was 43.2% and the correlation coefficient was 0.38. The structure was manually built into the electron density and refined using REFMAC (Murshudov et al., 1997). We used Coot for manual model building (Emsley & Cowtan, 2004). We used noncrystallographic symmetry restraints because the crystals only diffracted to 2.8 Å. After the R factor had fallen to 30%, we added metal ions, the PNP ligand and the water molecules. We refined the overall temperature factor in REFMAC5 and used an overall weighting term of 0.07; other geometric restraints were kept at the default settings. The scaling model that we used was simple scaling and the NCS restraints (between residues 2–311 of the A and B monomers) were set at a medium level. The refined structure (Table 1) has an R factor of 19.2% (for 5% test reflections, Rfree = 26.7%). The stereochemistry was checked using PROCHECK (Laskowski et al., 1993) and none of the refined residues were in the disallowed region of the Ramachandran plot. Figures were generated using PyMOL (DeLano, 2002), while three-dimensional superpositions were calculated using O (Jones et al., 1991).
To calculate the transformation matrices and the r.m.s.d per Cα atom values (root-mean-square deviation per Cα), we used the appropriate active-site residues (His9, Asp13, Asp15, Asp77, His99, His100, Asp151, Lys207, Arg297 and Lys298; S. agalactiae numbering) for active-site superpositions. To superpose all Cα atoms, we first superimposed every tenth residue using lsq_explicit and then improved the fits using lsq_improve (for 1i74 and 2haw). For the open conformation (1k23) structure we used a similar procedure, but only for the N-terminal domain (residues 2–191). The search for sequence motifs in S. agalactiae PPase was performed using the MOTIFSCAN server (http://scansite.mit.edu/motifscan_seq.phtml) using low-stringency settings (Obenauer et al., 2003).
3. Results and discussion
3.1. Structure solution
With two monomers per asymmetric unit in space group R32, the Matthews volume VM (Matthews, 1968) was 3.1 Å3 Da−1. This corresponds to a solvent content of 60.1% (Rantanen et al., 2006). Using MOLREP and a monomer of the S. mutans PPase (Merckel et al., 2001) as a model, good rotation and translation solutions were found (Rf/σ = 8.1 and 4.9; Tf/σ = 32.9 and 169) for two monomers per asymmetric unit. The structure was successfully refined at 2.80 Å resolution using 20 708 and 1035 reflections in the working and test sets, respectively. The final R factor was 19.2% (for 5% test reflections, Rfree = 26.7%).
The protein structure (covering residues 2–311) was refined with the PNP ligand in the active site. We also refined three metal ions per monomer (two transition-metal ions and one Mg2+) in the active site. We used an anomalous difference map (Fig. 1) to locate the two transition-metal ions, which are bound as expected at sites M1 and M2 (Merckel et al., 2001). Family II enzymes purified from Escherichia coli actually have a mixture of bound Fe3+ and Mn2+, giving the enzyme its distinct yellow colour (Merckel et al., 2001; Fabrichniy et al., 2007). The S. agalactiae enzyme was also yellow-coloured both after purification and crystallization steps, but our data collected at a home source cannot distinguish between these two metal ions. We therefore chose to use Mn2+ at 100% occupancy, as this is the physiological metal ion (Parfenyev et al., 2001). In addition, Fe3+ and Mn2+ are isoelectronic and so there should be no difference in the scattering power at low 2sinθ/λ. A 1.8Å resolution structure of B. subtilis PPase also showed a partial occupancy of a fourth metal ion in the active site in the presence of a substrate analogue (Fabrichniy et al., 2007), but this metal ion was not visible in our structure. Finally, there was electron density in the (Fo − Fc) maps at 4σ between the unique Trp31 residue and Tyr112 in both monomers (data not shown). The shape of the density did not correspond to any component of the crystallization or purification buffers, but could be fitted with a Trp amino acid (Trp1001), which stacks between Tyr112A and Trp31A. Within 4 Å are Phe23A, Tyr94A and Val97A. The Tyr112A hydroxyl is 3.9 Å from His161B Nε2. While it is clear that some organic, probably aromatic, molecule is bound in this pocket, its identity cannot be determined at this resolution.
Figure 1.
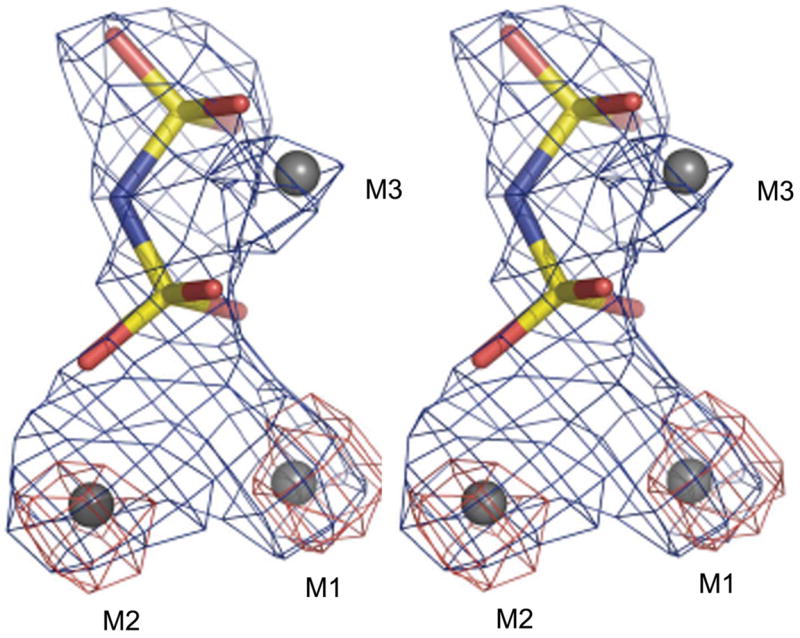
Stereoview of the fit of the S. agalactiae PPase metal ions M1–M3 (grey) and the PNP ligand to the electron density. The PNP P atoms are in yellow, O atoms in red and bridging N atom in blue. An (Fo − Fc) OMIT map calculated without the metal ions and the PNP after ten cycles of refinement is shown in blue and the anomalous difference map is shown in red. The maps were contoured at 5.5σ and 4.0σ, respectively.
3.2. Structure and structural comparison
S. agalactiae PPase (Fig. 2) has a typical family II tertiary and quaternary structure. The N-terminal domain, constituted of a five-stranded parallel β-sheet and α-helices, is connected by a hinge region to the C-terminal domain, which consists of a mixed five-stranded β-sheet and α-helices. Residues 99–115 (Merckel et al., 2001) form the monomer–monomer interface in S. mutans PPase and (data not shown) in the S. agalactiae enzyme. This is not surprising, as the sequence is 100% conserved in this region.
Figure 2.
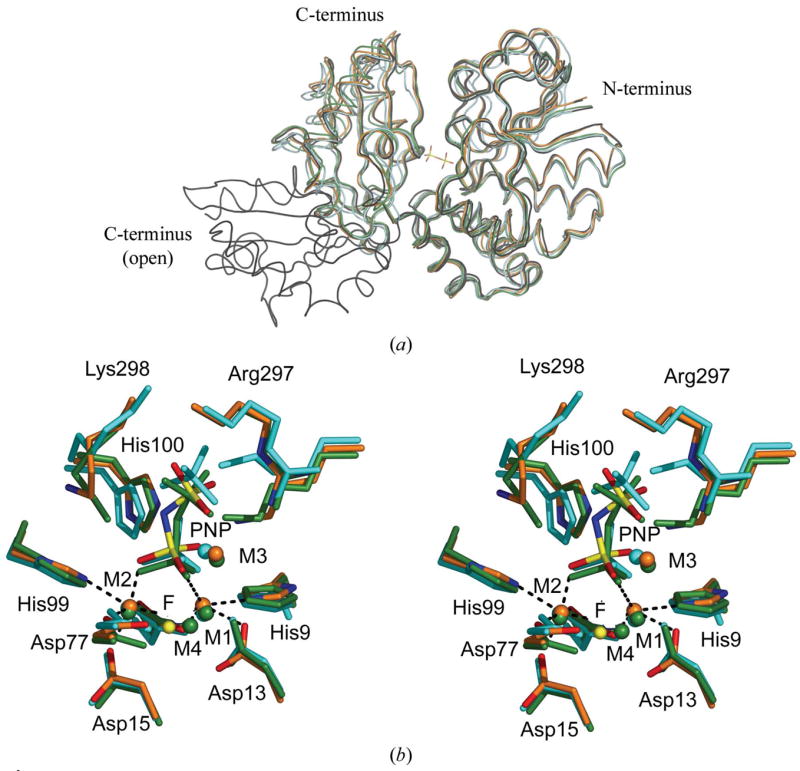
(a) Superposition of the S. agalactiae PPase monomer on four previously solved family II PPase structures. On the right-hand side is the larger N-terminal domain; on the left, the smaller C-terminal domain. All structures are presented as loop models. The PNP ligand of S. agalactiae PPase is included to mark the active site. The S. agalactiae PPase is in orange, the S. mutans PPase (PDB code 1i74) is in cyan, the S. gordonii PPase (PDB code 1k20) is in black, the open-conformation B. subtilis PPase (PDB code 1k23) is in black and the PNP-complexed B. subtilis PPase (PDB code 2haw) is in green. The superpositions were calculated using O (Jones et al., 1991). (b) Stereoview of the superposition of the active sites of three family II PPases. The active-site residues and ligands are shown as sticks and the metal ions as spheres. The B. subtilis PNP complex (PDB code 2haw) is shown in green. S. mutans (PDB code 1i74) in complex with sulfates is shown in cyan. The S. agalactiae PPase is shown in orange, with N atoms in blue, O atoms in red and P atoms in yellow. The metal ions, shown as spheres, were coloured with the same colours as the superposed molecules. The F− ion in 2haw is shown as a yellow sphere for comparison. The coordination geometries at M1 and M2 in S. agalactiae PPase are marked with black dashes. The superpositions were made using O (Jones et al., 1991).
The overall r.m.s.d. per Cα atom (residues 2–311) is 1.2 Å between S. agalactiae and S. mutans (Merckel et al., 2001) PPases and 0.90 Å between the S. agalactiae enzyme and the substrate-bound B. subtilis enzyme (Fabrichniy et al., 2007). These two structures are closest to S. agalactiae PPase in terms of active-site content and it is the active-site content that determines whether the structure is ‘open’ or ‘closed’ (Fig. 2a). Not surprisingly, the active-site residues superimpose even more closely: for the ten active-site residues (His9, Arg13, Asp15, Asp77, His99, His100, Asp151, Lys207, Arg297 and Lys298; Fig. 2b), the r.m.s.d. per Cα atom is 0.81 Å between S. agalactiae and S. mutans (Merckel et al., 2001), 0.32 Å between S. agalactiae and S. gordonii (Ahn et al., 2001) and 0.32 Å between S. agalactiae and the substrate-bound B. subtilis enzyme (Fabrichniy et al., 2007). The overlap is best in the region surrounding the N-terminal domain side of the active-site cleft (Fig. 2). In the empty B. subtilis structure (Ahn et al., 2001) the overall structure is very open. This is a consequence of a 70° rotation about the hinge, which places the end of the C-terminal domain 37 Å away from the end of our C-terminal domain when the N-terminal domains are superposed.
The active-site geometry (Fig. 2b) is very similar to that of previous family II PPase structures; M1 and M2 are in the same position as M1 andM2 from S. mutans PPase, S. gordonii PPase and substrate-bound B. subtilis PPase (Merckel et al., 2001; Fabrichniy et al., 2007; Ahn et al., 2001). The low-affinity M1 and high-affinity M2 ions prefer different coordination geometries; M2 can shift between trigonal bipyramidal and octahedral geometry, explaining the preference in family II PPases for transition-metal ions such as Mn2+ and Co2+ (Fabrichniy et al., 2004). As in the S. mutans product-like structure and B. subtilis substrate-like structures (Merckel et al., 2001; Fabrichniy et al., 2007), M2 is coordinated by Asp15, Asp77, Asp151, His99 and a P1 oxygen. At M1, the coordination is from His9, Asp13, Asp77 and a P1 oxygen, as in the Mn2+-containing B. subtilis PPase core structure (Fabrichniy et al., 2004). We did not place Wn, the nucleophilic water, in the density between M1 and M2 because it did not refine properly, presumably owing to the low resolution of the data. Similar to our previous observations (Fabrichniy et al., 2004), the geometry at M1 is more distorted than at M2. A magnesium ion is bound at the M3 site between the P1 and P2 oxygen ligands of the PNP substrate as in the substrate-bound B. subtilis PPase 2haw, but M4 (Fabrichniy et al., 2007) does not seem to be present. The distance from the probable Wn site to the electrophilic P atom of P1 would still be 3.1 Å as the substrate is also in a slightly different position. This movement may reflect differences in the active-site contents in the structures, as the catalytic hydroxide ion can move by up to 1 Å upon substrate/product binding (Fabrichniy et al., 2004, 2007).
The conformation of Lys298 (Fig. 3) is probably the largest difference between the S. agalactiae PPase active-site structure and that of B. subtilis (Fabrichniy et al., 2007). In the latter, the — Lys296 headgroup binds to terminal O atoms on both P1 and P2 (Fabrichniy et al., 2007), but in our structure it points away from the substrate and makes a hydrogen bond to the carbonyl O atom of His99. His99 is part of the important DHH motif (Fabrichniy et al., 2007) that is involved in positioning the active site correctly for catalysis. In this respect, our structure resembles the S. gordonii and S. mutans structures more than the B. subtilis structure (Merckel et al., 2001; Ahn et al., 2001), but these structures contain sulfate ions, mimicking the enzyme state with product, rather than substrate, bound. The interactions are also somewhat different. In S. gordonii PPase (PDB code 1k20; Ahn et al., 2001) Lys298 is hydrogen bonded to both the Val118 and the His99 carbonyl O atoms, while in S. mutans PPase (PDB code 1i74; Merckel et al., 2001) it is hydrogen bonded only to water molecules.
Figure 3.
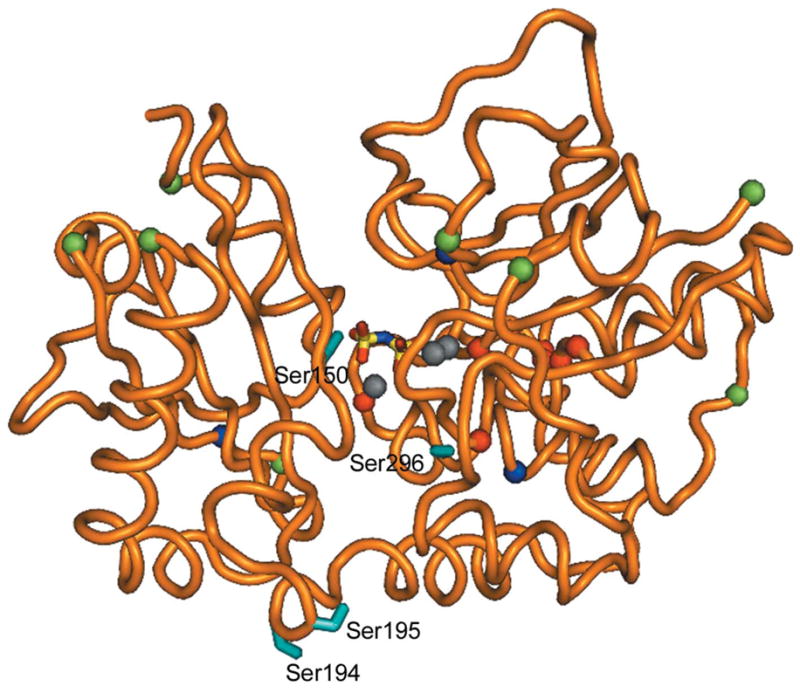
The position of the serines in S. agalactiae PPase. The protein (monomer) is shown as a loop representation in orange. The side chains of the potentially important serines (Ser150, Ser194, Ser195 and Ser296) where phosphorylation may have an effect on the enzyme activity (see text) are shown as cyan sticks. The serines excluded by MS are shown as red spheres at the Cα atoms, surface-exposed serines are shown as green spheres and totally buried serines as blue spheres. Grey spheres indicate the metal ion and the PNP ligand is shown in sticks and coloured by atom to mark the position of the active site.
In the wild-type B. subtilis PPase structure the occupancy of PNP was 85%, with an additional 15% occupancy of phosphate at the leaving-group P2 site (Fabrichniy et al., 2007). In our structure, an (Fo − Fc) OMIT map without the PNP ligand (data not shown) indicated that the P2 site may have a higher occupancy; the P2 peak was 11σ, while the peak at P1 was only 8σ. PNP may have undergone partial hydrolysis as in B. subtilis PPase (Fabrichniy et al., 2007), but the resolution was too low to allow the use of partial occupancies. Modelling the activesite ligands as two phosphate ions led to an (Fo − Fc) map with negative density at P1. In addition, the continuous density we observe is not consistent with two phosphates, which would be separated by around 4 Å as in yeast PPase (Heikinheimo et al., 2001). PNP is thus still present in our structure, albeit probably not at full occupancy.
3.3. Possible phosphorylation sites
Phosphoaminoacid analysis demonstrates that the S. agalactiae PPase is phosphorylated at one of its 23 serines, not on a threonine (Rajagopal et al., 2003), but the exact site of phosphorylation is not known. We showed by mass-spectrometric analysis of phosphorylated trypsin-digested PPase that certain serine-containing peptide fragments did not contain the phosphate group (Rajagopal et al., 2003), although we were then and are still unable to identify the exact site (L. Rajagopal, unpublished work; M. Rantanen, unpublished work). There are eight unphosphorylated Ser residues: Ser14, Ser19, Ser20, Ser120, Ser122, Ser123, Ser157 and Ser163 (Fig. 3).
Other locations can be eliminated based on this structure and the expected mechanism of the STK and STP. First, the site of phosphorylation needs to be accessible to the STK and STP (Fig. 2). Serine residues that are not accessible to solvent in any of the currently known conformations include Ser14, Ser19, Ser20, Ser120, Ser122 and Ser123 (eliminated above) and in addition Ser84, Ser146 and Ser264 (Fig. 3).
Secondly, any physiological effect on the enzyme should be inhibitory, as family II PPases are diffusion-controlled enzymes with a kcat ten times that of yeast PPase (Shintani et al., 1998). Increasing the catalytic rate of the enzyme would not have any effect, but decreasing its rate might lead to an increase in the mutation rate (Lahti, 1983; Kukko-Kalske & Heinonen, 1985) and so could help the bacterium escape host defences. Consequently, the serine should be located at a site where phosphorylation would inhibit the enzyme. There are three obvious places: near the monomer–monomer interface, near the hinge region which allows active-site closure and near the active site. On this basis Ser2, Ser64, Ser86, Ser135, Ser250, Ser262, Ser271 and Ser308 are unlikely candidates because they are not close to any of these three sites. This leaves just four likely candidates: Ser150 and Ser296 near the active site, and Ser194 and Ser195 near the hinge region. Conversely, scans using MOTIFSCAN (Obenauer et al., 2003) did not identify any possible STK sites consistent with the biological data.
3.4. Structural implications
As mentioned above, the S. agalactiae PPase is in the closed conformation in our crystal form, consistent with it binding substrate. The most striking difference from the PNP-bound B. subtilis substrate complex 2haw (Fabrichniy et al., 2007) is the conformation of Lys298 and the absence of the substrate-associated M4 that we found previously (Fabrichniy et al., 2007). In addition, Tyr112 near the dimer interface has a new conformation compared with that in 1i74 (Merckel et al., 2001), extending the dimer interface somewhat. These changes are probably because the crystals were grown from 0.4 M unbuffered monobasic ammonium phosphate (Rantanen et al., 2006), which has a pH of about 4.2 (Fluka Biochemica product No. 77104; http://www.sigmaaldrich.com). Consequently, the active site in general and PNP in particular are more protonated than when crystallized from buffered ammonium sulfate solution as in 2haw (Fabrichniy et al., 2007). This would cause Lys298 to swing away from the substrate. In addition, M4 binding would be weaker, especially as the P1 phosphate is one of the M4 ligands (Fig. 2b).
The similarity of family II PPases to each other suggests that family II PPases from other pathogenic bacterial species may also undergo reversible phosphorylation. A search for conservation of serines and possibly for the conservation of surface residues could reveal how the family II PPases are recognized by their STKs and STPs. We have also solved the structure of the STP from S. agalactiae (M. Rantanen, unpublished work), but a simple docking experiment (data not shown) did not reveal how these two enzymes recognize each other, suggesting that a conformational change may occur in one or both proteins prior to complex formation. Having identified the four most likely candidates for phosphorylation (Ser150, Ser194, Ser195 and Ser296), we now intend to identify the exact site and attempt to produce cocrystals of S. agalactiae family II PPase with STP or with STK.
Acknowledgments
We would like to thank Professor Arto Annila for funding. This work was supported by grants from the Sigrid Juselius Foundation and from the Academy of Finland (grant Nos. 1105157 and 114752) to AG, who is a member of the Biocentrum Helsinki research organization, and by the National Institutes of Health grant No. RO1 AI056073 to CER and a CHRMC Basic Science Steering Committee Award to LR.
Footnotes
PDB Reference: family II inorganic pyrophosphatase, 2enx, r2enxsf.
References
- Adler E, Donella-Deana A, Arigoni F, Pinna LA, Stragler P. Mol Microbiol. 1997;23:57–62. doi: 10.1046/j.1365-2958.1997.1801552.x. [DOI] [PubMed] [Google Scholar]
- Ahn S, Milner AJ, Fütterer K, Konopka M, Ilias M, Young TW, White SA. J Mol Biol. 2001;313:797–811. doi: 10.1006/jmbi.2001.5070. [DOI] [PubMed] [Google Scholar]
- Alex LA, Simon MI. Trends Genet. 1994;10:133–138. doi: 10.1016/0168-9525(94)90215-1. [DOI] [PubMed] [Google Scholar]
- Boitel B, Ortiz-Lombardia M, Duran R, Pompeo F, Cole ST, Cervenansky C, Alzari PM. Mol Microbiol. 2003;49:1493– 1508. doi: 10.1046/j.1365-2958.2003.03657.x. [DOI] [PubMed] [Google Scholar]
- Chen J, Brevet A, Fromant M, Leveque F, Schmitter JM, Blanquet S, Plateau P. J Bacteriol. 1990;172:5686–5689. doi: 10.1128/jb.172.10.5686-5689.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson DW, Cox JD. Annu Rev Biochem. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Cryst. 1994;D50:760–763. [Google Scholar]
- Cozzone AJ. J Mol Microbiol Biotechnol. 2005;9:198–213. doi: 10.1159/000089648. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- Diederichs K, Karplus PA. Nature Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- Echenique J, Kadioglu A, Romao S, Andrew PW, Trombe MC. Infect Immun. 2004;72:2434–2437. doi: 10.1128/IAI.72.4.2434-2437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley K, Cowtan K. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fabrichniy IP, Lehtiö L, Salminen A, Zyryanov AB, Baykov AA, Lahti R, Goldman A. Biochemistry. 2004;43:14403– 14411. doi: 10.1021/bi0484973. [DOI] [PubMed] [Google Scholar]
- Fabrichniy IP, Lehtiö L, Tammenkoski M, Zyryanov AB, Oksanen E, Baykov AA, Lahti R, Goldman A. J Biol Chem. 2007;282:1422–1431. doi: 10.1074/jbc.M513161200. [DOI] [PubMed] [Google Scholar]
- Galyov EE, Hakansson S, Forsberg A, Wolf-Watz H. Nature (London) 1993;361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- Greenstein AE, Grundner C, Echols N, Gay LM, Lombana TN, Miecskowski CA, Pullen KE, Sung PY, Alber T. J Mol Microbiol Biotechnol. 2005;9:167–181. doi: 10.1159/000089645. [DOI] [PubMed] [Google Scholar]
- Hakansson S, Galyov EE, Rosqvist R, Wolf-Watz H. Mol Microbiol. 1996;20:593–603. doi: 10.1046/j.1365-2958.1996.5251051.x. [DOI] [PubMed] [Google Scholar]
- Heikinheimo P, Lehtonen J, Baykov A, Lahti R, Cooperman BS, Goldman A. Structure. 1996;4:1491–1508. doi: 10.1016/s0969-2126(96)00155-4. [DOI] [PubMed] [Google Scholar]
- Heikinheimo P, Tuominen V, Ahonen AK, Teplyakov A, Cooperman BS, Baykov AA, Lahti R, Goldman A. Proc Natl Acad Sci USA. 2001;98:3121–3126. doi: 10.1073/pnas.061612498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herro R, Poncet S, Cossart P, Buchrieser C, Gouin E, Glaser P, Deutscher J. J Mol Microbiol Biotechnol. 2005;9:224–234. doi: 10.1159/000089650. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- Kukko-Kalske E, Heinonen J. Int J Biochem. 1985;17:575–580. doi: 10.1016/0020-711x(85)90288-5. [DOI] [PubMed] [Google Scholar]
- Lahti R. Microbiol Rev. 1983;47:169–178. doi: 10.1128/mr.47.2.169-178.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Matthews BW. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Merckel MC, Fabrichniy IP, Salminen A, Kalkkinen N, Baykov AA, Lahti R, Goldman A. Structure. 2001;9:289–297. doi: 10.1016/s0969-2126(01)00587-1. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantle LC, Yaffe MB. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfenyev AN, Salminen A, Halonen P, Hachimori A, Baykov AA, Lahti R. J Biol Chem. 2001;276:24511–24518. doi: 10.1074/jbc.M101829200. [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, Dodson EJ. Acta Cryst. 2003;D59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- Pullen KE, Ng HL, Sung PY, Good MC, Smith SM, Alber T. Structure. 2004;12:1947–1954. doi: 10.1016/j.str.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Rajagopal L, Clancy A, Rubens CE. J Biol Chem. 2003;278:14429–14441. doi: 10.1074/jbc.M212747200. [DOI] [PubMed] [Google Scholar]
- Rajagopal L, Vo A, Silvestroni A, Rubens CE. Mol Microbiol. 2005;56:1329–1346. doi: 10.1111/j.1365-2958.2005.04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal L, Vo A, Silvestroni A, Rubens CE. Mol Microbiol. 2006;62:941–957. doi: 10.1111/j.1365-2958.2006.05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantanen MK, Lehtiö L, Rajagopal L, Rubens CE, Goldman A. Acta Cryst. 2006;F62:891–894. doi: 10.1107/S174430910602954X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Uchiumi T, Yonezawa T, Salminen A, Baykov AA, Lahti R, Hachimori A. FEBS Lett. 1998;439:263–266. doi: 10.1016/s0014-5793(98)01381-7. [DOI] [PubMed] [Google Scholar]
- Vagin AA, Teplyakov A. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto- Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- Wang J, Li C, Yang H, Mushegian A, Jin S. J Bacteriol. 1998;180:6764–6768. doi: 10.1128/jb.180.24.6764-6768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kang CM, Brody MS, Price CW. Genes Dev. 1996;10:2265–2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- Young TA, Delagoutte B, Endrizzi JA, Falick AM, Alber T. Nature Struct Biol. 2003;10:168–174. doi: 10.1038/nsb897. [DOI] [PubMed] [Google Scholar]
- Young TW, Kuhn NJ, Wadeson A, Ward S, Burges D, Cooke GD. Microbiology. 1998;144:2563–2571. doi: 10.1099/00221287-144-9-2563. [DOI] [PubMed] [Google Scholar]
- Zyryanov AB, Vener AV, Salminen A, Goldman A, Lahti R, Baykov AA. Biochemistry. 2004;43:1065–1074. doi: 10.1021/bi0357513. [DOI] [PubMed] [Google Scholar]