

Abstract
PURPOSE
To examine changes of select reduction-oxidation (redox) sensitive proteins from human donor retinal pigment epithelium (RPE) at four stages of age-related macular degeneration (AMD).
DESIGN
Experimental study.
METHODS
Human donor eyes were obtained from the Minnesota Lions Eye Bank and graded using the Minnesota Grading System (MGS) into four stages that correspond to stages defined by the age-related eye disease study (AREDS). Protein content in RPE homogenates was measured using Western immunoblotting with protein-specific antibodies.
RESULTS
The content of several antioxidant enzymes and specific proteins that facilitate refolding or degradation of oxidatively damaged proteins increased significantly in MGS stage 3. These proteins are involved in the primary (copper-zinc superoxide dismutase [CuZnSOD], manganese superoxide dismutase [MnSOD], and catalase) and secondary (heat shock protein [HSP] 27, HSP 90, and proteasome) defense against oxidative damage. Additionally, the insulin pro-survival receptor exhibited disease-related upregulation.
CONCLUSIONS
The pattern of protein changes identified in human donor tissue graded using the MGS support the role of oxidative mechanisms in the pathogenesis and progression of AMD. The MGS uses nearly identical clinical definitions and grading criteria of AMD that are used in the AREDS, so our results apply to clinical and epidemiologic studies using similar definitions. Results from our protein analysis of human donor tissue helps to explain altered oxidative stress regulation and cell-survival pathways that occur in progressive stages of AMD.
Age-related macular degeneration (amd) is the leading cause of blindness in developed countries1–9. The number of affected individuals in the United States alone is expected to increase nearly twofold, to approximately three million by the year 2020.10 Fortunately, therapeutic options are improving. The use of antioxidant vitamins has been shown to delay disease progression at an intermediate stage,11 and rapid innovation in the use of antiangiogenic therapies has resulted in new clinical methods to treat the exudative phase of AMD.12–18 However, developing new treatment and prevention strategies targeting earlier stages of the disease requires a better understanding of the underlying disease mechanisms.
Findings from the Age-Related Eye Disease Study (AREDS) clearly support the hypothesis that oxidative mechanisms play a significant role in the progression of AMD. Although several studies have shown that the intake of antioxidant-rich foods lowers the risk of AMD,19–24 others have not supported this conclusion.25–27 Cigarette smoking, a pro-oxidant, significantly increases the risk of AMD, and this association is well supported in numerous, well-designed studies.28–35
The retinal pigment epithelium (RPE) is subject to a particularly high level of oxidative stress because of locally elevated oxygen tension, high polyunsaturated lipid content (phagocytosed photoreceptor outer segments), focused light exposure, direct interface with oxidative biochemicals (free radicals) generated by photoreceptor outer segment phagocytosis, and secondary photosensitizing agents (lipofuscin) that accumulate with aging.36 The RPE regulates oxidative stress using protective mechanisms for reactive oxygen species detoxification by using antioxidant enzymes such as superoxide dismutases (e.g., cytosolic copper-zinc superoxide dismutase [CuZnSOD] or mitochondrial manganese superoxide dismutase [MnSOD]). After damage to proteins caused by oxidative stress, molecular chaperones such as the heat shock proteins (HSPs) and the ubiquitin-proteasome pathway are involved in oxidative repair mechanisms by refolding or degrading damaged proteins.37
The etiology of AMD is multifactorial and involves genetic and environmental elements. Animal and cell culture models used to investigate the role of oxidative stress cannot replicate the true biochemical mechanisms of the human condition. Therefore, our approach has been to use nonpreserved, human eye bank tissue from donors with AMD and grade it according to well-accepted standard definitions of disease progression using the Minnesota Grading System (MGS).38 Briefly, the MGS uses high-resolution, digital, stereoscopic fundus images of freshly prepared eye bank eyes, carefully graded by examining the bare RPE and identifying key features of disease progression, using the same clinical phenotypic definitions described in the AREDS.39 As a correlate, the Alabama Grading System provides a well-characterized method for evaluating postmortem globes that is ideal for studying histopathologic features.40 Because of details of the Alabama Grading System methodology, our study design preferred the MGS to examine specific biochemical changes that are best seen using nonpreserved tissue. Herein, we investigate known pathways involved in oxidative stress response mechanisms and correlate these findings with the specific stage of AMD. These data provide valuable insights into the protein expression patterns of selective redox and quality control proteins from the RPE involved in early and late AMD.
METHODS
Donor eyes were obtained from the minnesota lions Eye Bank and maintained at 4 C in a moist chamber until dissection and photography. All tissue was acquired with consent for use in medical research from the donor or donor’s family according to the Declaration of Helsinki. An Internal Review Board exemption from the University of Minnesota was obtained for this study. The neurosensory retina from one globe was dissected to expose the RPE cells and high-resolution, digital, stereoscopic images were obtained and graded independently by two clinicians (X.F. and T.W.O.).
The globes were classified using the MGS into four progressive stages (MGS1-MGS4; Figure 1) according to AREDS criteria based on area of drusen, pigmentary abnormalities, geographic atrophy, choroidal neovascularization, fibrosis, and disciform scar formation. Exclusion criteria included diabetic retinopathy, advanced glaucoma, or atypical retinal disease not consistent with AMD. MGS1 served as a control group. MGS2 represents an early stage of AMD with numerous small drusen and pigmentary changes. MGS3 is an intermediate stage of the disease defined by numerous intermediate size drusen, a single large drusen, or noncentrally located geographic atrophy. MGS4 represents end-stage AMD with central geographic atrophy (atrophic or aAMD) or choroidal neovascularization (exudative or eAMD; MGS4). A minimum of five samples per stage was used for each analysis.
FIGURE 1.

Minnesota Grading System (MGS) stages 1–4. Representative, digital images (stereo not shown) of human eye bank eyes for each stage38 that corresponds to the clinical definitions used in the Age-Related Eye Disease Study (AREDS).39 Images demonstrate MGS1 (Top left), MGS2 (Top right; inset showing small hard drusen), MGS3 (Bottom left), and MGS4 (Bottom right).
Globes were frozen in liquid nitrogen and stored at −80 C. RPE cells were harvested and treated as previously described to obtain the RPE homogenates.41,42 Protein concentrations were determined with the bicinchoninic acid protein assay (Pierce, Rockford, Illinois, USA) using bovine serum albumin as a standard.
Using one-dimensional gel electrophoresis, RPE proteins were electrophoretically separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis43 with a 13% and 4% polyacrylamide resolving (14 × 12.5 cm) and stacking gel, respectively, at constant amperage (25 mA per gel) for ~5.5 hours. For each antibody, protein loads between 5 and 25 μg produced a linear signal. Loads from preparations of human RPE cells were 15 to 20 μg per lane.
Next, we used Western immunoblot analysis of one-dimensional gels. RPE proteins were electrophoretically transferred to polyvinylidene diflouride membranes using a semi-dry transfer cell apparatus (Bio-Rad, Hercules, California, USA). Western immunoblotting using the BSP/NBIT (5-bromo-4-chlor-3′-iodolyl phosphate p-toluidine/nitro blue tetrazolium chloride) substrate (Bio-Rad) was performed as previously described.44 Primary antibodies and their dilutions are provided in Table 1. Densitometric analysis was performed on the immunoreaction of individual protein bands to determine the relative content of the different proteins in the RPE preparations (SigmaScan Pro, Systat Software, Inc, San Jose, California, USA).
TABLE 1.
Antibody Information Used for Western Immunoblot Analysis of Retinal Pigment Epithelial Proteins in Age-Related Macular Degeneration
| Primary antibody | Type* | Dilution | Company |
|---|---|---|---|
| Alpha 7 | M | 1:1,000 | Biomol International, Exeter, United Kingdom |
| 20S C2 | P | 1:1,000 | Affinity Bioreagents, Golden, Colorado, USA |
| CuZnSOD | P | 1:3,000 | Stressgen, Victoria, British Columbia, Canada |
| MnSOD | P | 1:3,000 | Stressgen, Victoria, British Columbia, Canada |
| Catalase | P | 1:2,000 | Abcam, Cambridge, United Kingdom |
| HSP27 | M | 1:500 | Affinity Bioreagents, Golden, Colorado, USA |
| HSP60 | M | 1:5,000 | BD Biosciences, San Jose, California, USA |
| HSP70 | P | 1:10,000 | Stressgen, Victoria, British Columbia, Canada |
| HSP90 | M | 1:1,000 | Santa Cruz Biotechnology, San Diego, California, USA |
| Enolase | P | 1:200 | Santa Cruz Biotechnology, San Diego, California, USA |
| InR | P | 1:500 | Upstate, Lake Placid, New York, USA |
M = monoclonal; P = polyclonal; LMP = low molecular weight protein; CuZnSOD = copper-zinc superoxide disumutase; HSP = heat shock protein; MnSOD = manganese superoxide dismutase; InR = insulin receptor.
Data were normalized to a standard reaction in all blots and to a chemiluminescent HSP70 reaction as a loading control (LC). Preliminary studies showed the housekeeping proteins glyceraldehyde phosphate dehydrogenase (GAPDH) and actin exhibited altered expression between stages and therefore could not be used as the loading control. However, HSP70 immunoreactivity did not change between MGS stages for an independent series of blots.
The results are expressed as mean ± SEM (standard error of the mean). Differences among experimental groups were evaluated by linear regression analysis using the statistic package in Origin 7.5. Significance was defined at P < .05.
RESULTS
There were 22 males and 18 females with relatively balanced distribution between groups (slightly greater male:female ratio in MGS2; 8:3). This trend is consistent with donor ratios of the Minnesota Lions Eye bank, rather than reflecting prevalence demographics of AMD. Time from death to tissue freezing was 16.6 ± 1.2 hours (n = 40). The estimated smoking history was not significantly different between the four groups. Comparison of the donor ages by MGS category showed that while MGS1, 2, and 3 were not different (67 ± 8, 67 ± 11, 70 ± 9, respectively), donors in MGS4 were significantly older than the other groups (83 ± 11, P = .0014); therefore, we cannot exclude an age effect in MGS4 data.
The cellular response to elevated oxidative stress is mediated by the activation of several redox-sensitive transcription factors (Table 2). We measured the gene products regulated by these transcription factors to assess the primary and secondary oxidative stress response in donor eyes with AMD. The primary defense response includes antioxidant enzymes that are involved in detoxifying or eliminating free radicals. When the primary defense is inadequate, reactive oxygen species can damage cellular proteins, cause them to unfold, lose function, and aggregate.37 Secondary defenses protect against cytotoxicity from reactive oxygen species damaged proteins and include chaperones from the HSP family and proteases, such as the proteasome, that refold or degrade damaged proteins, respectively.45 Finally, cellular responses to oxidative stress involve upregulation of pro-survival factors that prevent apoptosis until the primary and secondary defenses have counteracted the oxidative injury.
TABLE 2.
Transcription Factors and Gene Product, Measured in Response to Oxidative Stress in the Retinal Pigment Epithelium in Age-Related Macular Degeneration
| Redox Sensitive Transcription Factors | Target Gene Products | |||
|---|---|---|---|---|
| FOXO52,66–68 | MnSOD | Catalase | InR | |
| NF-kB52,66,69 | MnSOD | Catalase | CuZnSOD | |
| Nrf-270 | MnSOD | Alpha 6 | Alpha 7 | HSP90 |
| HSF-171,72 | HSP27 | HSP70 | ||
FOXO = forkhead box O 3A; Nrf-2 = nuclear factor-2; MnSOD = manganese superoxide dismutase; NF-kB = nuclear factor kappa-beta; CuZnSOD = copper-zinc superoxide disumutase; HSP = heat shock protein; HSF-1 = heat shock factor; InR = insulin receptor.
The relative content of three antioxidant enzymes (CuZnSOD, MnSOD, and catalase) was compared in donors at all MGS levels. The content of all three antioxidant enzymes showed a linear increase with disease progression, although the extent of increase was lowest for CuZnSOD. Protein content increased 140% for CuZn-SOD, whereas MnSOD and catalase content increased 200% and 320%, respectively, compared with MGS1 (Figure 2).
FIGURE 2.
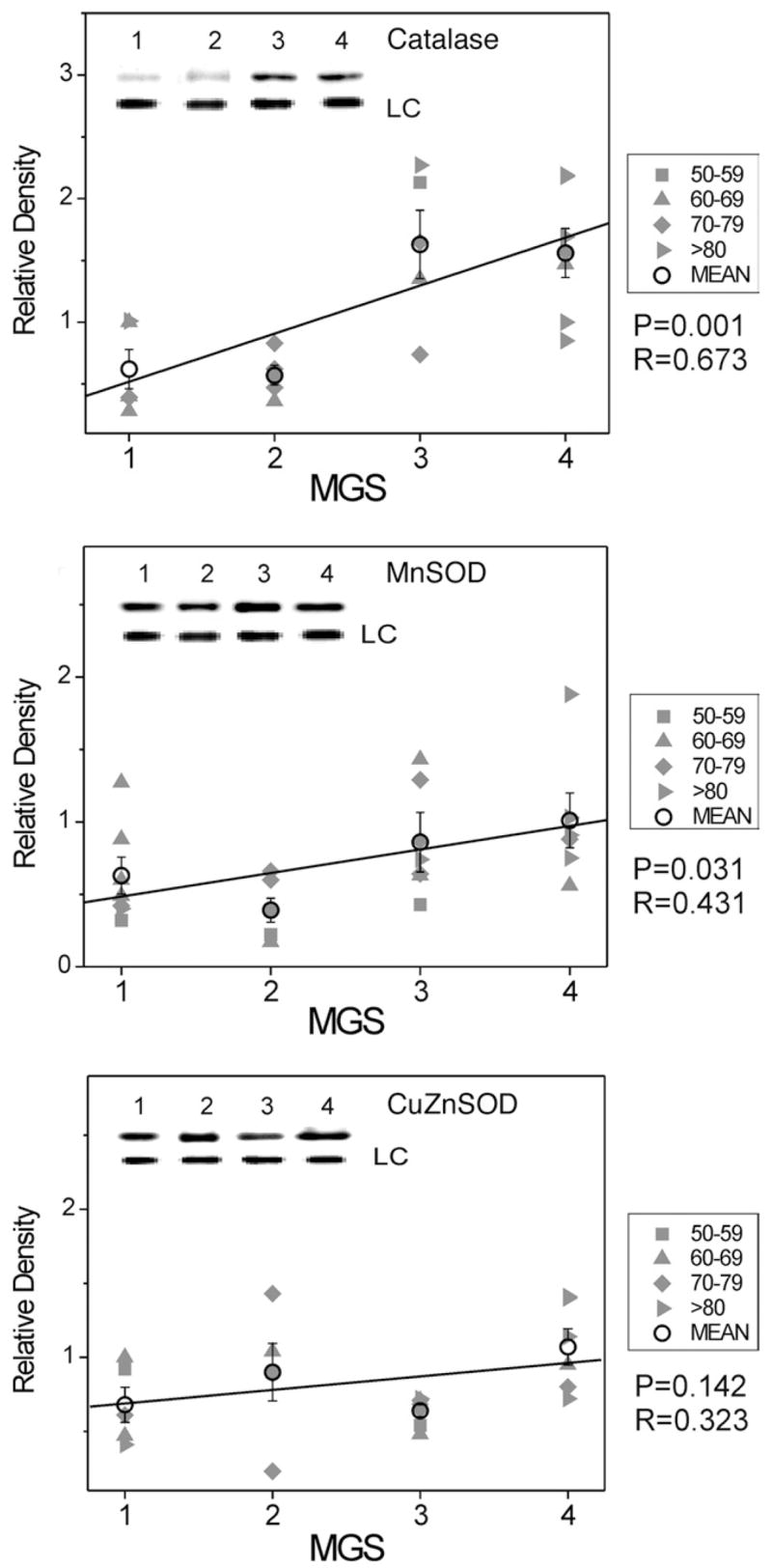
Select redox proteins in Minnesota Grading System (MGS) stages of age-related macular degeneration (AMD); antioxidant enzyme content. Retinal pigment epithelial (RPE) proteins (15 μg) were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis followed by Western immunoblotting. Data were normalized to a standard reaction in all blots and to heat shock protein 70 (HSP70) as a loading control (LC). Individual donor relative densities from four age groups are shown (gray symbols). Group mean ± standard error of the mean (SEM) and the results of linear regression analysis are shown. n = 5–7 donors/stage. Inset shows representative Western immunoblot of antioxidant enzymes catalase (Top), manganese superoxide dismutase (MnSOD) (Middle), and copper-zinc superoxide dismutase (CuZnSOD) (Bottom) at MGS 1–4 and the LC-HSP70.
HSPs, representing the second line of defense, consist of a large family of proteins that are categorized based on their molecular mass.42 In the current study, we determined the relative content of four different family members (HSP27, HSP60, HSP70, and HSP90). For HSP60 and HSP70, no change in content was observed throughout the disease. However, both HSP27 and HSP90 exhibited a 300% and 250% increase, respectively, from MGS1 to MGS4 (Figure 3).
FIGURE 3.
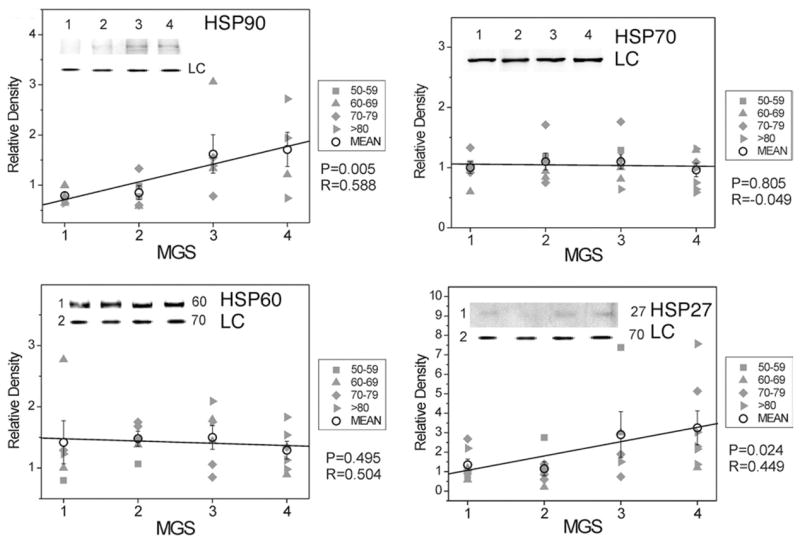
Select redox proteins in Minnesota Grading System (MGS) stages of age-related macular degeneration (AMD); heat shock protein (HSP) content. Retinal pigment epithelial (RPE) proteins (15 μg) were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis followed by Western immunoblotting. Data were normalized to a standard reaction in all blots and to HSP70 as a loading control (LC). Individual donor relative densities from four age groups are shown (gray symbols). Group mean ± standard error of the mean (SEM) and the results of linear regression analysis are shown. n = 5–7 donors/stage. Inset shows representative Western immunoblot of heat shock proteins HSP90 (Top left), HSP70 (Top right), HSP60 (Bottom left), and HSP27 (Bottom right) at MGS 1–4 and the LC.
The catalytic core of the proteasome consists of four stacked rings, each containing seven subunits.46 The α-subunits make-up the two outer rings of the complex. Because these subunits are part of every catalytic core, their content provides a valid estimate of the total amount of proteasome present in the cell. Therefore, we measured the content of Alpha 6- and Alpha 7-subunit to evaluate proteasome content changes through progressive MGS stages. We observed a 350% and 200% increase (respectively) in content of these Alpha-subunits through disease progression (Figure 4).
FIGURE 4.
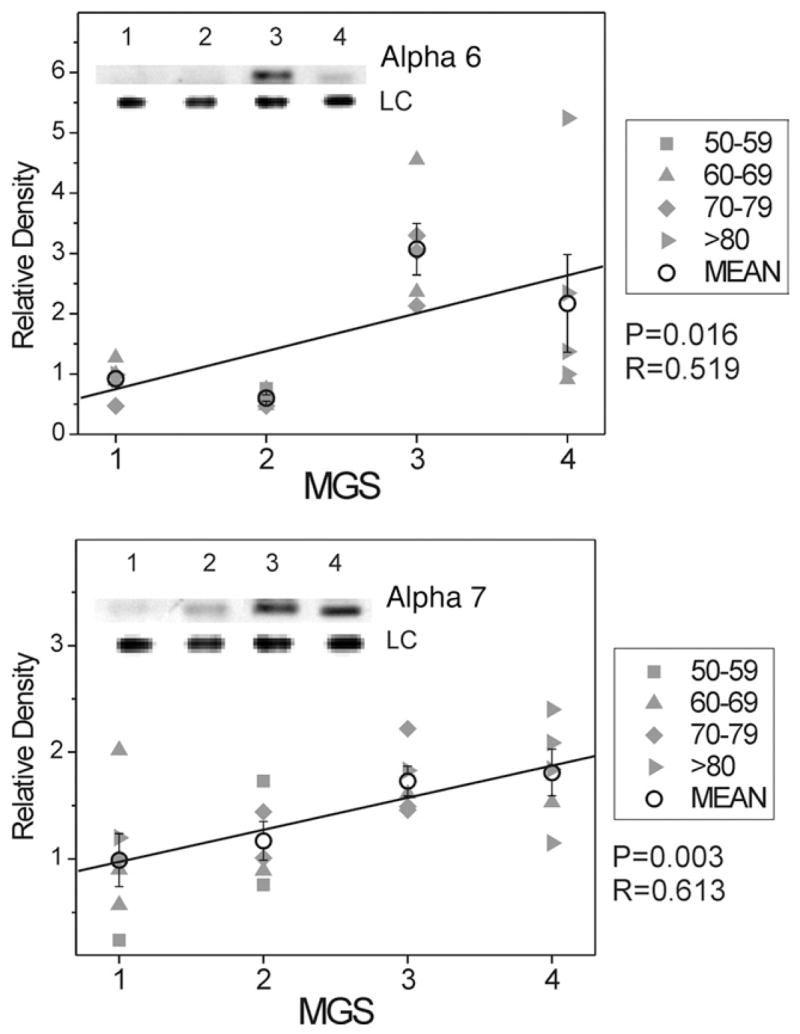
Select redox proteins in Minnesota Grading System (MGS) stages of age-related macular degeneration (AMD); total proteasome content. Retinal pigment epithelial (RPE) proteins (15 μg) were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis followed by Western immunoblotting. Data were normalized to a standard reaction in all blots and to heat shock protein 70 (HSP70) as a loading control (LC). Individual donor relative densities from four age groups are shown (gray symbols). Group mean ± standard error of the mean (SEM) and the results of linear regression analysis are shown. n = 5–7 donors/stage. Inset shows representative Western immunoblot for proteasome subunits Alpha 6 (Top) and Alpha 7 (Bottom) at MGS 1–4 and the LC.
Although there are numerous additional redox sensitive proteins, we focused on the insulin receptor because it is an important component of the survival pathway and crucial for the survival of post-mitotic tissues such as the RPE.47 The insulin receptor is a tyrosine kinase with an α- and β-chain that activates the pro-survival Akt/PKB pathway. The content of the β-chain of the insulin receptor was increased approximately 200% (Figure 5).
FIGURE 5.
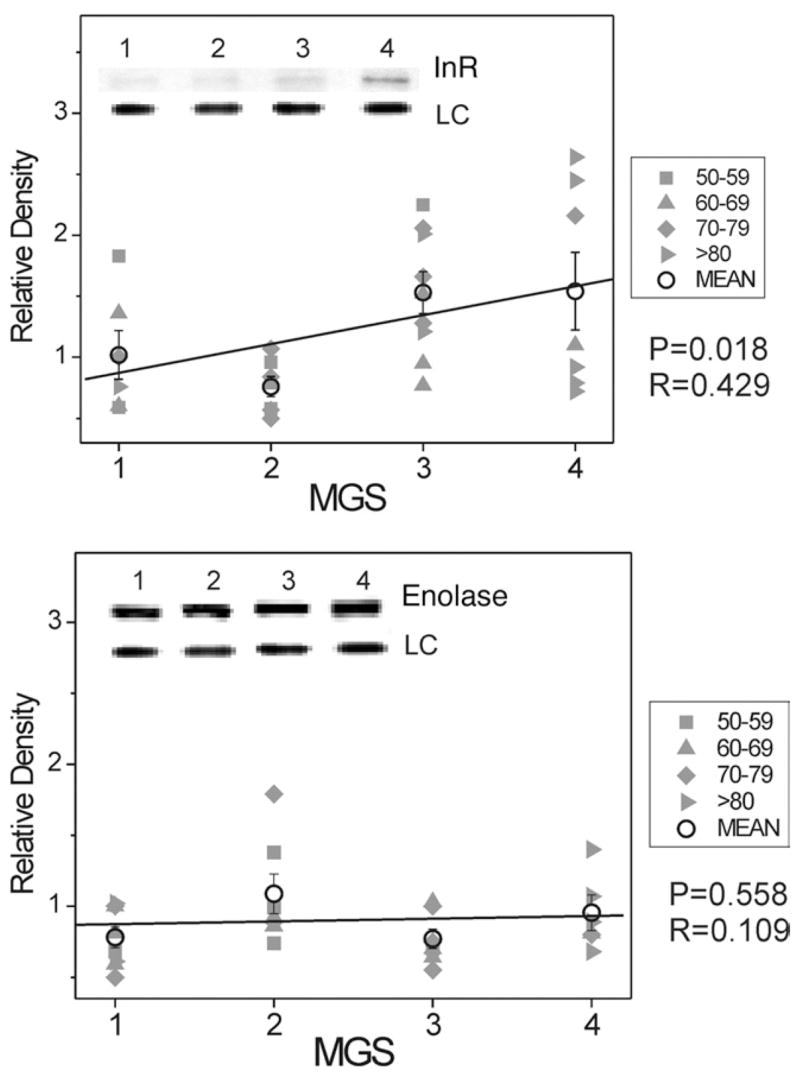
Select redox proteins in Minnesota Grading System (MGS) stages of age-related macular degeneration (AMD); insulin receptor and enolase content. Retinal pigment epithelial (RPE) proteins (15 μg) were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis followed by Western immunoblotting. Data were normalized to a standard reaction in all blots and to heat shock protein 70 (HSP70) as a loading control (LC). Individual donor relative densities from four age groups are shown (gray symbols). Group mean ± standard error of the mean (SEM) and the results of linear regression analysis are shown. n = 5–9 donors/stage. Inset shows representative Western immunoblot for insulin receptor β subunit (Top) and enolase (Bottom) at MGS 1–4 and the LC.
As a negative control, we also compared the relative content of enolase, a glycolytic enzyme that is not redox-sensitive. As expected from the hypothesis that oxidative stress specifically plays a role in AMD pathogenesis, enolase content did not change during AMD progression (Figure 5).
DISCUSSION
The oxidative stress hypothesis of amd proposes that cumulative oxidative damage to proteins, lipids, and DNA leads to disease progression.48,49 The primary advantage of our approach is in studying responses to oxidative stress using human donor eyes that have been carefully examined for phenotypic features of AMD, thereby avoiding some limitations of cell culture, dichotomous grading systems (± AMD), or animal model systems. A direct measure of reactive oxygen species is technically not feasible because of their extremely short half-lives; therefore, we chose to examine the content of several proteins that are regulated by redox-sensitive transcription factors as an indicator of increased oxidative stress. Herein, we focused on specific proteins that mediate primary and secondary defenses against oxidative stress and cell survival.
Our data suggest that intracellular antioxidant proteins and pathways are directly upregulated with progressive clinical stages of disease, represented by the MGS, of eye bank eyes with AMD. We propose that the primary RPE response to oxidative stress is the upregulation of antioxidant enzymes that detoxify or eliminate free radicals generated by rod outer segment phagocytosis or other pro-oxidant events. MnSOD and CuZnSOD convert superoxide anions to hydrogen peroxide, which is then converted to water by peroxidases such as catalase.52,53 Superoxide dismutase (SOD) and catalase upregulation may thus reflect increased reactive oxygen species production since oxidative stress induces their expression through redox-sensitive transcription factors (Table 2).
Previous studies by Frank and associates50 examined RPE from eyes with advanced AMD using electron microscopic immunocytochemistry and found marginally increased CuZn-SOD; however, they did not find changes in catalase content. There may be several reasons for the discrepancy. First, we separated specific clinical stages of AMD in our study using the MGS, compared with early vs late AMD comparison. Next, some of the tissue used in previous studies was processed at 48 hours postmortem as compared with fresh tissue from our data (15–18 ± 3 hours; Table 3). Finally, in some of their tissue specimens, macular drusen were not observed. Liles and associates51 reported no change in SOD activity and a decrease in catalase activity in the RPE from human donor eyes with AMD. We did not measure enzymatic activity, and it is possible that the observed increases in SOD and catalase protein content were not accompanied by a corresponding increase in enzymatic activity.
TABLE 3.
Donor Demographics
| MGS Grade | Sample Size | Gender
|
Age (yrs)* |
TAD (hrs) | Smoking history1 (%) | Cause of Death ( # ) | ||
|---|---|---|---|---|---|---|---|---|
| M | F | Mean | Range | |||||
| 1 | 10 | 6 | 4 | 67 ± 8 | 53–80 | 15.6 ± 3 | 7 (70) | Cancer (5), COPD (2), Multiorgan failure (2), HTN (1) |
| 2 | 11 | 8 | 3 | 67 ± 11 | 64–75 | 16.2 ± 4 | 4 (40) | Cancer (6), Respiratory (4), CVA (1) |
| 3 | 11 | 5 | 6 | 70 ± 9 | 54–91 | 18.2 ± 3 | 5 (50) | Cancer (7), COPD (2), Alzheimer (1), Surgical complication (1) |
| 4 | 8 | 3 | 5 | 83 ± 11 | 61–98 | 15.9 ± 3 | 5 (62) | Cancer (2), CHF (1), COPD (1), CVA (2), Respiratory (1), Alzheimer (1) |
M = male; F = female; TAD = Time after death to tissue freezing (mean ± SD); COPD = chronic obstructive pulmonary disease; HTN = hypertension; CVA = cerebro-vascular accident; CHF = chronic heart failure.
Mean ± SD.
Number of donors with a positive smoking history.
Increased antioxidant enzyme expression suggests a compensatory response to high oxidative stress. However, this response may be inadequate to completely detoxify reactive oxygen species because markers of oxidative damage, such as advanced glycation end products, Ne-(carboxymethyl) lysine, and carboxyethylpyrrole are all increased in human donor eyes with advanced AMD.55–58 Therefore, upregulation of secondary defenses, including HSPs and the proteasome, are necessary to protect the RPE from damage caused by protein unfolding and aggregation. After exposure to an oxidative stress, HSP27 in cultured RPE cells has been shown to be upregulated.59,60 These findings are in agreement with our results that demonstrate increased HSP27 content. HSP90 content also increases, but not mitochondrial HSP60 and cytosolic HSP70. The specific upregulation of HSP27 and HSP90 may reflect differences in the transcription factors regulating their expression (Table 2). However, we previously found changes in a subset of HSP60 and HSP70 proteins using two-dimensional gel electrophoresis that may not be evident using Western immunoblotting, which measures total protein content.41
The total proteasome content also increases with clinically relevant disease progression. Other investigators have reported an increase in proteasome content after exposure of cells to reactive oxygen species45 that confers protection against oxidative damage.61 Taken together, these data are consistent with the idea that proteasome content can be regulated by the cellular redox status and is an important component in the cellular defense against oxidative damage.
The insulin receptor as well as MnSOD and catalase are regulated by the redox-sensitive transcription factor Fork-head box O 3A (FOXO).62 Previous studies have shown that insulin receptor activation increases survival rates in different tissues, including the RPE.47,63–65 Increased insulin receptor content suggests that the RPE, in critical stages of AMD, may upregulate pro-survival signaling, in response to increased oxidative stress.
Certainly, age is the primary risk factor for AMD. Not surprisingly, when comparing the ages of all donors used in this study, MGS4 donors were significantly older than the other groups. However, significant biochemical changes were observed at MGS3, in which donor age did not differ from MGS1. Therefore, the changes we observed most likely reflect AMD progression.41,54 Age-matched controls would be ideal; however, we are limited by tissue availability for this direct comparison.
Our results provide evidence for increased content of redox-sensitive proteins in human donor eyes with AMD, and identify specific proteins that may mediate the upregulation of defense mechanisms that protect the RPE from oxidative stress.
In summary, we present biochemical evidence of altered oxidative stress regulation and cell-survival pathways that occur during clinically relevant stages of AMD progression that may serve to help identify potential targets of future therapy and intervention.
Acknowledgments
This study was supported by the national institute on aging grant no. AG025392 (T.W.O.), NATIONAL EYE Institute, Grant EY014176, Bethesda, Maryland, (D.A.F.), Minnesota Lions Macular Degeneration Center, Minneapolis, Minnesota and Minnesota Lions Eye Bank, and an unrestricted grant to from Research to Prevent Blindness Foundation, Inc, New York, New York. The authors indicate no financial conflict of interest. Involved in conduct and design of study (A.D., D.A.F., T.W.O.); collection, management, analysis, and interpretation of the data (A.D., C.L.N., X.F., D.A.F., T.W.O.); and preparation, review, or approval of the manuscript (A.D., C.L.N., X.F., D.A.F., T.W.O.).
Biographies
Biosketch
Alejandra Decanini, MD, graduated from medical school in 2005 from Anahuac University, Mexico City, Mexico. Dr Decanini completed her transitional internship at the ABC Medical Center, Santa Fe, Mexico, followed by a year of social service providing free medical consultation in a rural community. Dr Decanini has participated in several clinical and basic science research protocols presented and published in various Mexican medical societies. She is currently a research fellow at the University of Minnesota, Minneapolis, Minnesota, studying macular degeneration using proteomics.

Biosketch
Deborah A. Ferrington, PhD, received her degree in Biochemistry from the University of Kansas, Lawrence, Kansas, in 1997. She is an NIH-funded Associate Professor of Ophthalmology at the University of Minnesota, Minneapolis, Minnesota, with joint appointments in Biochemistry, Biophysics, and Molecular Biology and Gerontology. Dr Ferrington was awarded the Fesler-Lampert Chair in Aging Studies and regularly publishes her work on normal and pathological aging of the retina in leading eye research journals.

References
- 1.Leibowitz HM, Krueger DE, Maunder LR, et al. The Framingham Eye Study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610. [PubMed] [Google Scholar]
- 2.Klein BE, Klein R. Cataracts and macular degeneration in older Americans. Arch Ophthalmol. 1982;100:571–573. doi: 10.1001/archopht.1982.01030030573002. [DOI] [PubMed] [Google Scholar]
- 3.Hyman LG, Lilienfeld AM, Ferris FL, Fine SL. Senile macular degeneration: a case-control study. Am J Epidemiol. 1983;118:213–227. doi: 10.1093/oxfordjournals.aje.a113629. [DOI] [PubMed] [Google Scholar]
- 4.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1992;99:933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- 5.Klein R, Wang Q, Klein BE, et al. The relationship of age-related maculopathy, cataract, and glaucoma to visual acuity. Invest Ophthalmol Vis Sci. 1995;36:182–191. [PubMed] [Google Scholar]
- 6.Mitchell P, Smith W, Attebo K, Wang JJ. Prevalence of age-related maculopathy in Australia the Blue Mountains Eye Study. Ophthalmology. 1995;102:1450–1460. doi: 10.1016/s0161-6420(95)30846-9. [DOI] [PubMed] [Google Scholar]
- 7.Klaver CC, Wolfs RC, Vingerling JR, et al. Age-specific prevalence and causes of blindness and visual impairment in an older population: the Rotterdam Study. Arch Ophthalmol. 1998;116:653–658. doi: 10.1001/archopht.116.5.653. [DOI] [PubMed] [Google Scholar]
- 8.Buch H, Vinding T, Nielsen NV. Prevalence and causes of visual impairment according to World Health Organization and United States criteria in an aged, urban Scandinavian population: the Copenhagen City Eye Study. Ophthalmology. 2001;108:2347–2357. doi: 10.1016/s0161-6420(01)00823-5. [DOI] [PubMed] [Google Scholar]
- 9.Buch H, Vinding T, La Cour M, et al. Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: the Copenhagen City Eye Study. Ophthalmology. 2004;111:53–61. doi: 10.1016/j.ophtha.2003.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 11.The Age-Related Eye Disease Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS Report No. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spaide RF, Sorenson J, Maranan L. Combined photodynamic therapy with verteporfin and intravitreal triamcinolone acetonide for choroidal neovascularization. Ophthalmology. 2003;110:1517–1525. doi: 10.1016/S0161-6420(03)00544-X. [DOI] [PubMed] [Google Scholar]
- 13.Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. New Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 14.Spaide RF, Sorenson J, Maranan L. Combined photodynamic therapy and intravitreal triamcinolone for nonsubfoveal choroidal neovascularization. Retina. 2005;25:685–690. doi: 10.1097/00006982-200509000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Avery RL, Pieramici DJ, Rabena MD, et al. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology. 2006;113:363–372. doi: 10.1016/j.ophtha.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Chan WM, Lai TY, Wong AL, et al. Combined photodynamic therapy and intravitreal triamcinolone injection for the treatment of subfoveal choroidal neovascularization in age-related macular degeneration: a Comparative Study. Br J Ophthalmol. 2006;90:337–341. doi: 10.1136/bjo.2005.081299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenfeld PJ, Heier JS, Hantsbarger G, Shams N. Tolerability and efficacy of multiple escalating doses of ranibizumab (Lucentis) for neovascular age-related macular degeneration. Ophthalmology. 2006;113:623–632. doi: 10.1016/j.ophtha.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 18.Spaide RF, Fisher YL. Intravitreal bevacizumab (Avastin) treatment of proliferative diabetic retinopathy complicated by vitreous hemorrhage. Retina. 2006;26:275–278. doi: 10.1097/00006982-200603000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Seddon JM, Ajani UA, Sperduto RD, et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420. [PubMed] [Google Scholar]
- 20.Mares-Perlman JA, Klein R, Klein BE, et al. Association of zinc and antioxidant nutrients with age-related maculopathy. Arch Ophthalmol. 1996;114:991–997. doi: 10.1001/archopht.1996.01100140199014. [DOI] [PubMed] [Google Scholar]
- 21.Cho E, Stampfer MJ, Seddon JM, et al. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann Epidemiol. 2001;11:328–336. doi: 10.1016/s1047-2797(01)00217-4. [DOI] [PubMed] [Google Scholar]
- 22.Seddon JM, Rosner B, Sperduto RD, et al. Dietary fat and risk for advanced age-related macular degeneration. Arch Ophthalmol. 2001;119:1191–1199. doi: 10.1001/archopht.119.8.1191. [DOI] [PubMed] [Google Scholar]
- 23.Cho E, Seddon JM, Rosner B, et al. Prospective study of intake of fruits, vegetables, vitamins, and carotenoids and risk of age-related maculopathy. Arch Ophthalmol. 2004;122:883–892. doi: 10.1001/archopht.122.6.883. [DOI] [PubMed] [Google Scholar]
- 24.van Leeuwen R, Boekhoorn S, Vingerling JR, et al. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA. 2005;294:3101–3107. doi: 10.1001/jama.294.24.3101. [DOI] [PubMed] [Google Scholar]
- 25.VandenLangenberg GM, Mares-Perlman JA, Klein R, et al. Associations between antioxidant and zinc intake and the 5-year incidence of early age-related maculopathy in the Beaver Dam Eye Study. Am J Epidemiol. 1998;148:204–214. doi: 10.1093/oxfordjournals.aje.a009625. [DOI] [PubMed] [Google Scholar]
- 26.Smith W, Mitchell P, Webb K, Leeder SR. Dietary antioxidants and age-related maculopathy: the Blue Mountains Eye Study. Ophthalmology. 1999;106:761–767. doi: 10.1016/S0161-6420(99)90164-1. [DOI] [PubMed] [Google Scholar]
- 27.Flood V, Smith W, Wang JJ, et al. Dietary antioxidant intake and incidence of early age-related maculopathy: the Blue Mountains Eye Study. Ophthalmology. 2002;109:2272–2278. doi: 10.1016/s0161-6420(02)01263-0. [DOI] [PubMed] [Google Scholar]
- 28.Khan JC, Thurlby DA, Shahid H, et al. Smoking and age-related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularization. Br J Ophthalmol. 2006;90:75–80. doi: 10.1136/bjo.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fraser-Bell S, Wu J, Klein R, et al. Smoking, alcohol intake, estrogen use, and age-related macular degeneration in Latinos: the Los Angeles Latino Eye Study. Am J Ophthalmol. 2006;141:79–87. doi: 10.1016/j.ajo.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Thornton J, Edwards R, Mitchell P, et al. Smoking and age-related macular degeneration: a review of association. Eye. 2005;19:935–944. doi: 10.1038/sj.eye.6701978. [DOI] [PubMed] [Google Scholar]
- 31.Tamakoshi A, Yuzawa M, Matsui M, et al. Smoking and neovascular form of age-related macular degeneration in late middle aged males: findings from a case-control study in Japan. Research Committee on Chorioretinal Degenerations. Br J Ophthalmol. 1997;81:901–904. doi: 10.1136/bjo.81.10.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith W, Mitchell P, Leeder SR. Smoking and age-related maculopathy: the Blue Mountains Eye Study. Arch Ophthalmol. 1996;114:1518–1523. doi: 10.1001/archopht.1996.01100140716016. [DOI] [PubMed] [Google Scholar]
- 33.Klein R, Klein BE, Linton KL, DeMets DL. The Beaver Dam Eye Study: the relation of age-related maculopathy to smoking. Am J Epidemiol. 1993;137:190–200. doi: 10.1093/oxfordjournals.aje.a116659. [DOI] [PubMed] [Google Scholar]
- 34.Vinding T, Appleyard M, Nyboe J, Jensen G. Risk factor analysis for atrophic and exudative age-related macular degeneration. An epidemiological study of 1000 aged individuals. Acta Ophthalmol (Copenh) 1992;70:66–72. doi: 10.1111/j.1755-3768.1992.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 35.The Eye Disease Case-Control Study Group. Risk factors for neovascular age-related macular degeneration. Arch Ophthalmol. 1992;110:1701–1708. doi: 10.1001/archopht.1992.01080240041025. [DOI] [PubMed] [Google Scholar]
- 36.Choudhary S, Xiao T, Srivastava S, et al. Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol Appl Pharmacol. 2005;204:122–134. doi: 10.1016/j.taap.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 37.Marques C, Guo W, Pereira P, et al. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006;20:741–743. doi: 10.1096/fj.05-5080fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olsen TW, Feng X. The Minnesota Grading System of eye bank eyes for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:4484–4490. doi: 10.1167/iovs.04-0342. [DOI] [PubMed] [Google Scholar]
- 39.Age-Related Eye Disease Study Research G. The Age-Related Eye Disease Study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: the Age-Related Eye Disease Study Report No. 6. Am J Ophthalmol. 2001;132:668–681. doi: 10.1016/s0002-9394(01)01218-1. [DOI] [PubMed] [Google Scholar]
- 40.Curcio CA, Medeiros NE, Millican CL. The Alabama Age-Related Macular Degeneration Grading System for donor eyes. Invest Ophthalmol Vis Sci. 1998;39:1085–1096. [PubMed] [Google Scholar]
- 41.Nordgaard CL, Berg KM, Kapphahn RJ, et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- 42.Hipkiss AR. Accumulation of altered proteins and ageing: causes and effects. Exp Gerontol. 2006;41:464–473. doi: 10.1016/j.exger.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 44.Kapphahn RJ, Ethen CM, Peters EA, et al. Modified -A crystallin in the retina: altered expression and truncation with aging. Biochemistry. 2003;42:15310–15325. doi: 10.1021/bi034774e. [DOI] [PubMed] [Google Scholar]
- 45.Ding Q, Reinacker K, Dimayuga E, et al. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 2003;546:228–232. doi: 10.1016/s0014-5793(03)00582-9. [DOI] [PubMed] [Google Scholar]
- 46.Eleuteri AM, Cuccioloni M, Bellesi J, et al. Interaction of HSP90 with 20S proteasome: thermodynamic and kinetic characterization. Proteins. 2002;48:169–177. doi: 10.1002/prot.10101. [DOI] [PubMed] [Google Scholar]
- 47.Barber AJ, Nakamura M, Wolpert EB, et al. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276:32814–32821. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- 48.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 49.Cai J, Nelson KC, Wu M, et al. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 50.Frank RN, Amin RH, Puklin JE. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am J Ophthalmol. 1999;127:694–709. doi: 10.1016/s0002-9394(99)00032-x. [DOI] [PubMed] [Google Scholar]
- 51.Liles MR, Newsome DA, Oliver PD. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch Ophthalmol. 1991;109:1285–1288. doi: 10.1001/archopht.1991.01080090111033. [DOI] [PubMed] [Google Scholar]
- 52.Zhou LZ, Johnson AP, Rando TA. NF kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic Biol Med. 2001;31:1405–1416. doi: 10.1016/s0891-5849(01)00719-5. [DOI] [PubMed] [Google Scholar]
- 53.Valko M, Rhodes CJ, Moncol J, et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Ethen CM, Reilly C, Feng X, et al. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:2280–2290. doi: 10.1167/iovs.05-1395. [DOI] [PubMed] [Google Scholar]
- 55.Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu X, Meer SG, Miyagi M, et al. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–42035. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 57.Hammes HP, Hoerauf H, Alt A, et al. N(epsilon)(carboxymethyl)lysin and the AGE receptor RAGE colocalize in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:1855–1859. [PubMed] [Google Scholar]
- 58.Ishibashi T, Murata T, Hangai M, et al. Advanced glycation end products in age-related macular degeneration. Arch Ophthalmol. 1998;116:1629–1632. doi: 10.1001/archopht.116.12.1629. [DOI] [PubMed] [Google Scholar]
- 59.Arrigo AP, Virot S, Chaufour S, et al. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Signal. 2005;7:414–422. doi: 10.1089/ars.2005.7.414. [DOI] [PubMed] [Google Scholar]
- 60.Strunnikova N, Baffi J, Gonzalez A, et al. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:2130–2138. [PubMed] [Google Scholar]
- 61.Chondrogianni N, Tzavelas C, Pemberton AJ, et al. Over-expression of proteasome β-5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem. 2005;280:11840–11850. doi: 10.1074/jbc.M413007200. [DOI] [PubMed] [Google Scholar]
- 62.Puig O, Marr MT, Ruhf ML, Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003;17:2006–2020. doi: 10.1101/gad.1098703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miceli MV, Newsome DA. Insulin stimulation of retinal outer segment uptake by cultured human retinal pigment epithelial cells determined by a flow cytometric method. Exp Eye Res. 1994;59:271–280. doi: 10.1006/exer.1994.1108. [DOI] [PubMed] [Google Scholar]
- 64.Salceda R. Insulin-stimulated taurine uptake in rat retina and retinal pigment epithelium. Neurochem Int. 1999;35:301–306. doi: 10.1016/s0197-0186(99)00072-8. [DOI] [PubMed] [Google Scholar]
- 65.Jomary C, Cullen J, Jones SE. Inactivation of the Akt survival pathway during photoreceptor apoptosis in the retinal degeneration mouse. Invest Ophthalmol Vis Sci. 2006;47:1620–1629. doi: 10.1167/iovs.05-1176. [DOI] [PubMed] [Google Scholar]
- 66.Essers MA, Weijzen S, de Vries-Smits AM, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 68.Murphy CT, McCarroll SA, Bargmann CI, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 69.Rojo AI, Salinas M, Martin D, Perona R, Cuadrado A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappa B. J Neurosci. 2004;24:7324–7334. doi: 10.1523/JNEUROSCI.2111-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santos SD, Saraiva MJ. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience. 2004;126:657–663. doi: 10.1016/j.neuroscience.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 71.St Clair DK, Porntadavity S, Xu Y, Kiningham K. Transcription regulation of human manganese superoxide dismutase gene. Methods Enzymol. 2002;349:306–312. doi: 10.1016/s0076-6879(02)49345-7. [DOI] [PubMed] [Google Scholar]
- 72.Fiorenza MT, Bevilacqua A, Canterini S, Torcia S, Pontecorvi M, Mangia F. Early transcriptional activation of the hsp70.1 gene by osmotic stress in one-cell embryos of the mouse. Biol Reprod. 2004;70:1606–1613. doi: 10.1095/biolreprod.103.024877. [DOI] [PubMed] [Google Scholar]