

Abstract
The sodium/myo-inositol cotransporter is a plasma membrane protein responsible for concentrative cellular accumulation of myo-inositol in a variety of tissues. When cells in kidney and brain are exposed to a hyperosmolar salt condition (hypertonicity) due to the operation of urinary concentration mechanism and pathological conditions, respectively, they survive the stress of hypertonicity by raising the cellular concentration of myo-inositol. Transcription of the sodium/myo-inositol cotransporter gene is markedly stimulated in response to hypertonicity, leading to an increase in the activity of the cotransporter, which in turn drives the osmoprotective accumulation of myo-inositol. To understand the molecular mechanisms by which hypertonicity stimulates transcription, we analyzed the 5′-flanking region of the cotransporter gene for cis-acting regulatory sequences. We identified five tonicity-responsive enhancers that are scattered over 50 kilobase pairs. All the enhancers are variations of the same type of enhancer interacting with the transcription factor named tonicity-responsive enhancer binding protein. In vivo methylation experiments demonstrated that exposure of cells to hypertonicity increases the binding of tonicity-responsive enhancer binding protein to the enhancer sites, indicating that all of these enhancers are involved in the transcriptional stimulation. We conclude that the sodium/myo-inositol cotransporter gene is regulated by a large region (∼50 kilobase pairs) upstream of the gene.
The sodium/myo-inositol cotransporter (SMIT)1 is a plasma membrane protein catalyzing concentrative uptake of myo-inositol (MI) using the electrochemical gradient of sodium across the cell membrane (1). Since two sodium ions are coupled per molecule of MI (2), SMIT can transport MI against a 1,000-fold concentration gradient, i.e. 50 mm in a cell versus 50 μm in the plasma. The level of SMIT activity determines the steady-state cellular concentration of MI at the point where uptake and leak balance out.
SMIT is most abundantly expressed in the kidney medulla (1, 3), which is hypertonic most of the time because of the operation of the urinary concentration mechanisms. The high level of SMIT expression in the renal medulla is secondary to the hypertonicity of this tissue in that SMIT mRNA abundance changes pari passu with the tonicity of the medulla (3, 4). The changes in mRNA abundance are primarily due to changes in transcription (5). When SMIT is inhibited under hypertonic conditions in cultured cells (6) or in kidneys in vivo (7), cells undergo necrosis demonstrating the importance of maintaining a high level of SMIT activity in a hypertonic environment.
How elevated SMIT activity protects the renal cells is explained by the theory of compatible osmolytes (8). It is useful to note that osmolarity inside a mammalian cell is always in equilibrium with interstitial osmolarity because blood facing plasma membranes are highly permeable to water and very compliant mechanically. Immediately after cells are exposed to hypertonicity, cellular ionic strength is elevated due to osmosis. When the cells are kept in hypertonicity for more than several hours, they accumulate small organic solutes called compatible osmolytes and, as a result, lower cellular ionic strength toward the isotonic level. If and when accumulation of compatible osmolytes is prevented, cells do not survive (6, 7, 9), probably because of the effects of elevated cellular ionic strength (8). The major compatible osmolytes in the hypertonic medulla are MI, betaine, sorbitol, taurine, and glycerophosphorylcholine (10). Like MI, accumulation of betaine and sorbitol is also regulated at the level of transcription; hypertonicity stimulates transcription of the genes for the sodium- and chloride-coupled betaine/γ-aminobutyric acid transporter (BGT1) and aldose reductase (AR: catalyzes synthesis of sorbitol), leading to an increase in their activity, which results in an increase in cellular concentration of betaine and sorbitol (11). The signal for stimulation of transcription is most likely the cellular ionic strength because induction of AR correlates highly with the sum of cellular sodium and potassium concentration (12). The abundance of BGT1 (13) and SMIT (14) mRNA also correlates positively with cellular ionic strength.
During hypernatremia, which results in systemic hypertonicity, brain (15), and eye (16) accumulate compatible osmolytes including MI. SMIT mRNA is expressed throughout brain in neurons and glial cells (17) and in eye (18). Hypernatremia increases the abundance of SMIT mRNA in brain (17, 19) and eye (18), presumably due to an increase in transcription. Thus, the SMIT gene in non-renal cells responds to hypertonicity in the same way as it does in renal cells.
Studies of regulatory cis-elements involved in the regulation of transcription uncovered two tonicity-responsive enhancers (TonEs) within 185 bp upstream of the BGT1 gene (20).2 The AR gene is regulated by three TonEs located about 1 kb upstream of the transcription start site (21). All the TonEs of the BGT1 and AR genes are functionally the same; they share a high level of sequence similarity, and all of them specifically bind to a nuclear protein named TonE-binding protein (TonEBP) (22). Cloning of TonEBP revealed that it is a transcription factor mediating the effect of TonE.3 When kidney-derived MDCK cells are exposed to hypertonicity, the activity of TonEBP is stimulated leading to the binding of TonEBP to TonE sites of the BGT1 gene and concurrent stimulation of transcription (22). Thus, stimulation of TonEBP activity is central to the hypertonicity-induced stimulation of transcription.
In this study, the 5′-flanking region of the SMIT gene is analyzed to identify regulatory cis-elements. Five TonEs spread over 50 kb are identified. All of them appear to contribute to the regulation of SMIT gene transcription, indicating that regulation of SMIT involves unusually but not unprecedentedly long range interactions between regulatory sequences and promoter.
Experimental Procedures
Cell Culture
MDCK cells were maintained in defined medium as described previously (5). HeLa cells were maintained in Eagle's spinner modification of minimum essential medium (Biofluids) supplemented with 5% horse serum. Medium was made hypertonic by adding 200 mm raffinose to defined medium or 100 mm NaCl to Eagle's spinner modification of minimum essential medium.
Restriction Mapping of P1 Clones and Localization of SacI Fragments
DNA from two P1 clones, 3283 and 3284 (23), was digested with NotI, ClaI, or MluI and then size-fractionated by pulse-field electrophoresis using the following settings: initial A time 1 s, final A time 10 s, A/B ratio of 1,200 V, run time 20 h in 0.5× TBE containing 45 mm Tris, 45 mm borate, and 1 mm EDTA. Southern blots were prepared and probed with T7 and SP6 oligonucleotides (specific for the P1 vector sequence flanking the cloning site) as well as various restriction fragments to delineate the restriction map shown in Fig. 1. To localize the S14 and S31 fragments (Fig. 1), P1 clones 3283 and 3284 were linearized with NotI digestion and then a series of SacI partial digestions with progressively less amount of enzyme were obtained. Southern blots of the partially digested DNA was hybridized to T7 oligonucleotide to determine the location of S14 and S31 fragments. The results were confirmed by hybridization of S14 and S31 fragments to Southern blots of P1 DNA.
Fig. 1. Map of the SMIT gene and TonE sequences.
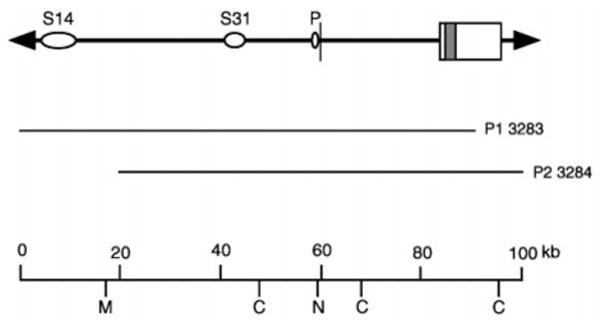
The restriction map shown at the bottom is constructed from two P1 clones 3283 and 3284 using pulse-field gel electrophoresis and Southern blot analysis: M, MluI; C, ClaI; N represents two NotI sites at −248 and −129 relative nucleotide positions relative to the SMIT gene. The first exon of the SMIT gene is indicated by a vertical bar and the second exon by a box within which the entire protein coding region (shaded box) is located. Ovals labeled S14 and S31 indicate SacI fragments in which TonE sequences are identified (see Figs. 2 and 3 and Table II). The narrow oval P denotes position of a TonE sequence (TonEp) at −331 relative nucleotide position (Table II).
Subcloning of 5′-Flanking Region of the Human SMIT Gene and Reporter Plasmid Construction
In order to obtain subclones of the 5′-flanking region of the human SMIT gene, DNA from the P1 3283 was digested with SacI and shot-gun cloned in pBluescriptII (Stratagen). Clones derived from the 5′-flanking region were identified by hybridization to Southern blots of NotI-digested P1 clone 3283. Nine non-overlapping SacI clones that cover 47 kb out of the 60-kb 5′ franking region of human SMIT gene (see Fig. 1) were obtained. To test tonicity-responsive regulatory activity of the SacI fragments, each SacI fragment was subcloned in front of the SMIT promoter ((−128/+134) fragment) and the luciferase reporter gene using pGL2-basic (Promega) (23). Some of the SacI fragments displaying enhancer activity were divided into smaller fragments (see Fig. 2), which were again tested for tonicity-responsive enhancer activity as above. Likewise, synthetic DNA fragments (Table I and Fig. 3) were also tested for tonicity-responsive enhancer activity.
Fig. 2. Localization of tonicity-responsive enhancer activity.
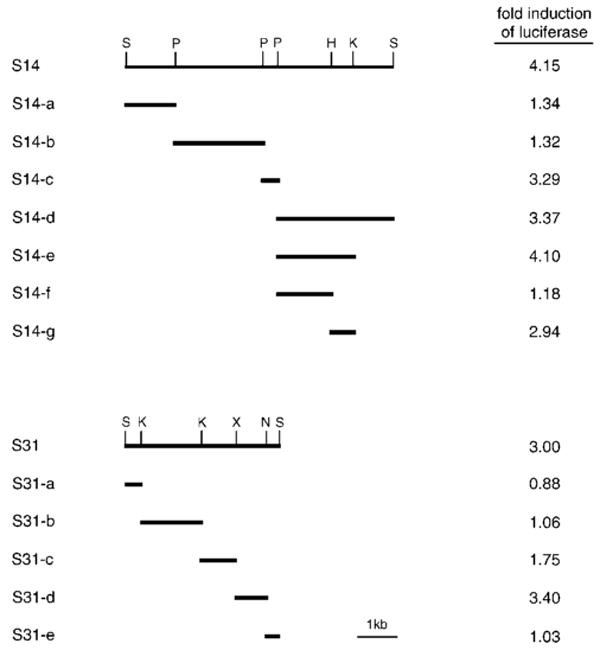
SacI restriction fragments S14 (top) and S31 (bottom) were subcloned from the P1 clone 3283 (Fig. 1). S14 and S31 and their smaller restriction derivatives shown were individually cloned in front of the SMIT promoter and the Photinus luciferase gene. Each construct was transfected into MDCK cells, and the transfected cells were cultured in isotonic or hypertonic medium. Activity of luciferase was measured from cell lysates, and -fold induction of luciferase by hypertonicity was calculated by dividing the activity in hypertonic condition by the activity in isotonic condition (mean values are shown; n = 2–5) as described under “Experimental Procedures.” Luciferase expression driven by the SMIT promoter alone was stimulated 11% (1.11-fold induction) by hypertonicity (n = 6). S, SacI; P, PstI; H, HindIII; K, KpnI; X, XbaI; N, NheI.
Table I. Synthetic double-stranded TonE sequences used for luciferase reporter gene constructs and electrophoretic mobility shift assay.
Each DNA contains one, two, or three TonE sequences with 10 bp of flanking sequence (5 bp from each side) per each TonE. Sticky ends for cloning into HindIII and EcoRI sites are added (lowercase letters). TonE sequences are underlined.
| Name | Sequence |
|---|---|
| TonEA | agctGCAAGTGGAAAACTACCAAGA |
| CGTTCACCTTTTGATGGTTCTttaa | |
| TonEB1 | agctCAGAGATAGAATTCCACATTT |
| GTCTCTATCTTAAGGTGTAAAttaa | |
| TonEB2/3 | agctTTAGCTGGAAAATTCCAAACA |
| AATCGACCTTTTAAGGTTTGTttaa | |
| TonEC1 | agctAGAGGTGGAAAATTACAGGCA |
| TCTCCACCTTTTAATGTCCGTttaa | |
| TonEC2 | agctTGGCATGGAAAGTTACTCAAA |
| ACCGTACCTTTCAATGAGTTTttaa | |
| TonEC3 | agctTAAAAAGGGAGTTCCAGCATC |
| ATTTTTCCCTCAAGGTCGTAGttaa | |
| TonEp | agctAGGCATGGAAAGTTCCAGCCG |
| TCCGTACCTTTCAAGGTCGGCttaa | |
| TonEB1 (2/3) | agctCAGAGATAGAATTCCACATTTTTAGCTGGAAAATTCCAAACA |
| GTCTCTATCTTAAGGTGTAAAAATCGACCTTTTAAGGTTTGTttaa | |
| TonEC123 | agctAGAGGTGGAAAATTACAGGCATGGCATGGAAAGTTACTCAAATAAAAAGGGAGTTCCAGCATC |
| TCTCCACCTTTTAATGTCCGTACCGTACCTTTCAATGAGTTTATTTTTCCCTCAAGGTCGTAGttaa |
Fig. 3. Characterization of the SMIT TonEs.
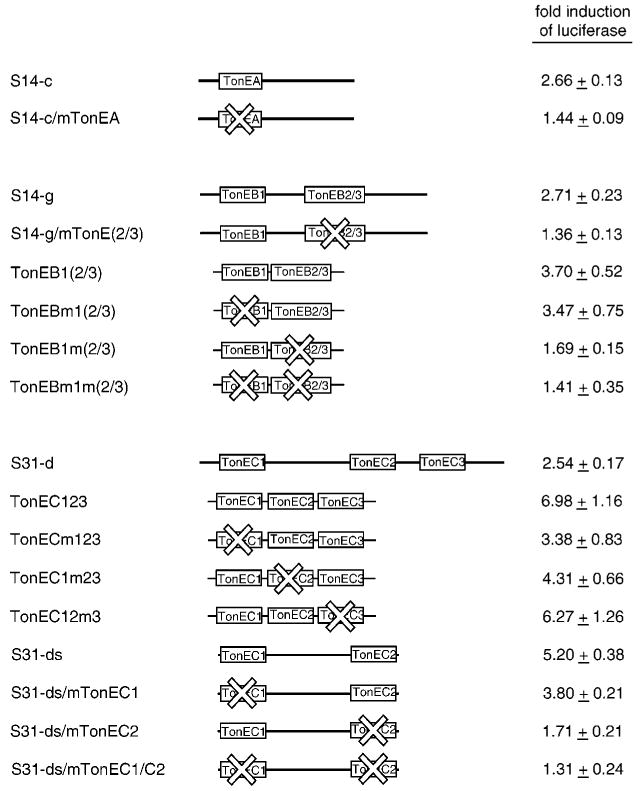
DNA fragments S14-c (top), S14-g (middle), and S31-d (bottom) are shown as thick lines with positions of TonE sequences indicated (not to scale) as boxes. S31-ds was derived from S31-d using PCR. TonEA in S14-c and TonEB(2/3) in S14-g were inactivated (as indicated by X marks) by mutating key nucleotide residues as described under “Experimental Procedures” to generate S14-c/mTonEA and S14-g/mTonE(2/3), respectively. Likewise, TonEC1 and TonEC2 in S31-ds were mutated to generate S31-ds/mTonEC1, S31-ds/mTonEC2, and S31-ds/mTonEC1/C2. TonEB1(2/3) (middle) and TonEC123 (bottom) are described in Table I. In other DNAs, TonE sequences marked by X were mutated by changing the 2nd G to T, the 5th A to T, and the 9th pyrimidine to G. Each of the wild-type and mutant DNA fragments was tested for its tonicity-responsive enhancer activity as described in Fig. 2. -Fold induction of luciferase in response to hypertonicity in cells transfected with each construct is shown: mean ± S.E., n = 4–9.
Site-directed Mutagenesis
TonE sequences in the S14-c and S14-g fragments (Fig. 3) were mutated to inactivate their enhancer activity using PCR. To mutate TonEA, fragment S14-c was separately amplified with two pairs of primers. The first pair was gggctgcatTGGGTGTTTTTATGGGA (primer A1; the sequence at one end of S14-c is in uppercase letters, and the PstI site added is shown in lowercase letters) and GCTCTTGGTcGTTaTCaACTTGC (TonEA portion is underlined (antisense strand); lowercase letters represent mutations) while the second pair was GCAAGTtGAtAACgACCAAGAGC (TonEA portion is underlined (sense strand); lowercase letters represent mutations) and gggctgacgGCGGAACAGCAGAT (primer A2; the sequence at one end of S14-c is in uppercase letters, and the PstI site added is shown in lowercase letters). To generate S14-c with mutations in TonEA, aliquots (1 μl each) of the two PCR products were mixed and subjected to PCR amplification using primer A1 and A2. The resulting mutations were confirmed by sequencing. TonEB2 in the S14-g was mutated in the same way using the following two sets of primers: gggctgcagGAATTCCACATTTCGTT (primer B1) and GATGTTTGGcATTaTCaAGCTAA (TonEB2/3 is underlined; antisense strand); TTAGCTtGAtAATgCCAAACATC (TonEB2/3 is underlined; sense strand) and gggctgcagAAGCTTCTTTCCTAGTC (primer B2). These mutations in TonEB2 are also expected (22) to inactivate TonEB3, which overlaps with TonEB2 in the antisense direction (see Table II). All the mutant fragments were sequenced completely to confirm the mutations and also to verify that the sequence outside the TonE regions remain unchanged.
Table II. Sequence of the SMIT TonEs.
Sequencing of the small DNA fragments S14-c, S14-g, and S31-d (Fig. 2) revealed seven TonE sequences that conform to the consensus shown near the bottom. TonEB2 and TonEB3 overlap in reverse directions sharing 10 bp as follows: TGGAAAATTCCA, where TonEB2 is in bold letters and the complementary strand of TonEB3 is underlined. TonEp (p for proximal) is at the −331/−321 position. Taking active SMIT TonEs into consideration, an updated TonE consensus is derived (shown at the bottom). Y represents C or T; N represents A, G, C, or T.
| Name | Location | Sequences | Remarks |
|---|---|---|---|
| TonEA | S14-c | TGGAAAACTAC | |
| TonEB1 | S14-g | TGGAATTCTAT | Not active |
| TonEB2 | S14-g | TGGAAAATTCC | Overlap with TonEB3 |
| TonEB3 | S14-g | TGGAATTTTCC | Probably not active |
| TonEC1 | S31-d | TGGAAAATTAC | Same as “hTonE” |
| TonEC2 | S31-d | TGGAAAGTTAC | |
| TonEC3 | S31-d | TGGAACTCCCT | Not active |
| TonEp | −331/−321 | TGGAAAGTTCC | |
| Consensus | YGGAANNNYNY | Ref. 22 | |
| Updated consensus | TGGAAANNYNY |
To prepare S31-ds (Fig. 3), S31-d was PCR-amplified using primer C1 (gggctcgagAGAGGTGGAAAATTACAGGCA; the sequence of TonEC1 is underlined, and the XhoI site added is shown in lowercase letters) and primer C2 (cccctgcagTGAGTAACTTTCCATGCCACC; the antisense sequence of TonEC2 is underlined, and the PstI site added is shown in lowercase letters). Primer mC1 (gggctcgagAGAGGTtGAtAATgACAGGCA) was used in place of primer C1 to mutate TonEC1 in S31-ds. Likewise, primer mC2 (cccctgcagTGAGTcACTaTCaATGCCACC) was used to mutate TonEC2. All these fragments were sequenced completely for verification.
Cell Transfection
The reporter plasmid constructs were transfected into MDCK cells using the DEAE-dextran method as described previously (22). Briefly, 2 μg of each reporter construct or 10 ng of β-actin construct (the luciferase gene under the strong promoter of the β-actin gene) was transfected with 50 ng of pRL/CMV, a plasmid containing the Renilla luciferase gene under control of CMV promoter. Transfected cells were cultured in isotonic defined medium for 20 h and then for an additional 20 h in isotonic or hypertonic medium. Cell extracts were prepared, and the activity of Photinus and Renilla luciferase was determined using dual-luciferase reporter assay system (Promega). For each extract, the activity of the Photinus luciferase was divided by the activity of the Renilla luciferase to correct for transfection efficiency. Under each tonicity condition, i.e. isotonic or hypertonic, the corrected activity from cells transfected with a test construct was again divided by that from cells transfected with the β-actin construct. The resulting luciferase activity standardized for the β-actin promoter was used to calculate -fold induction of luciferase by hypertonicity: activity of luciferase in hypertonic medium divided by activity of luciferase in isotonic medium. Each experiment (n = 1) was performed in duplicate dishes.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
MDCK cells or HeLa cells cultured in isotonic or hypertonic medium were chilled to 4 °C, and nuclear extracts were prepared as described previously (22). To prepare probes for EMSA shown in Table I, single-stranded oligonucleotides were synthesized and purified. 200 pmol each of complementary oligonucleotides were annealed in 100 μl containing 100 mm NaCl to obtain a double-stranded probe. Five μg of nuclear extract was incubated initially for 10 min at room temperature in 20 μl containing 20 mm HEPES (pH 7.9), 100 mm KCl, 0.1 mm EDTA, 10% glycerol, 1 mm dithiothreitol, and 1.5 μg of poly(dA-dT), 5 mm MgCl2. The mixture was then incubated for an additional 20 min after adding 32P-labeled probe with or without an unlabeled competitor. The reaction was electrophoresed on a 4% polyacrylamide gel (79:1, acrylamide:bis) in 0.5× TBE buffer. The gel was dried and exposed to a PhosphorImage screen. The radioactivity was visualized and quantified using PhosphorImager and ImageQuant software (Molecular Dynamics).
In Vivo Footprinting
In order to methylate G residues of the genomic DNA in vivo, HeLa cells in isotonic or hypertonic medium were incubated in the same medium containing 0.1% dimethyl sulfate for 2 min at room temperature. Cells were washed with phosphate-buffered saline twice to remove dimethyl sulfate, and DNA was isolated. The methylated DNA was converted to a single-stranded form and cleaved at sites immediately 3′ to the methylated G residues by treatment with piperidine (Sigma) at 90 °C for 30 min. The cleaved G residues were detected using ligation-mediated PCR as described (22). The cleaved DNA was annealed to primer AP1 (CTCACTGTTCAACAAAAGCCC), BP1 (GTGACCTCATGGGTGGTGGT), CP1 (GATAGAATGAGGTGGGAGGA), or pP1 (GAATGTTCCAGAACCCCTG) to synthesize the first strand DNA covering the TonEA, TonEB2, TonEC2, or TonEp region, respectively. A staggered double-stranded linker (22) was ligated, and 18 cycles of PCR were performed at the specific annealing temperature using a nested primer: AP2 (CTCCCATGCAGTGAAGAGCTGGCCC), BP2 (TGGGGAAGACAGCAGCAGAAGCAAG), CP2 (GAGGCAGGCAGCTTGGAACCAAGAA), or pP2 (TTCCAGAACCCCTGCGAGCAGCCGTT). Two additional rounds of PCR were performed using a 32P-labeled nested primer. The reaction was electrophoresed on a sequencing gel with sequencing ladder. Radioactivity was detected and quantified as described above.
Results
Localization of Tonicity-responsive Enhancer Activity in the 5′-Flanking Region of the SMIT Gene
Previously, we cloned the canine SMIT gene and a total of 15 kb of flanking sequence (11 kb from the 5′ side and 4 kb from the 3′ side) from overlapping λ clones (24). When we examined the entire cloned region including the flanking regions for tonicity-responsive regulatory activity, we detected only a small tonicity-responsive enhancer activity, less than 2-fold stimulation of the SMIT promoter in response to hypertonicity (see below), which is localized within (−2,900/−119) relative nucleotide region. Other investigators (16) studying the bovine SMIT gene reported a TonE sequence similar to those of the BGT1 and AR genes (22) at (−346/−336) relative nucleotide position. This enhancer stimulates the SMIT promoter about 2-fold in response to hypertonicity (16). Because transcription of the SMIT gene is stimulated over 10-fold under the same hypertonic conditions (5, 16), we anticipated more regulatory sequences outside (further upstream and/or downstream) the cloned region. To explore this possibility, we decided to search further upstream of the gene for more tonicity-responsive regulatory sequences. We turned to the two P1 clones (23), which contain the human SMIT gene and about 60 kb of 5′-flanking sequence (Fig. 1). The structure and sequence of the human SMIT gene is very similar to that of the canine gene (23). In addition, like the canine SMIT gene, the immediate 5′-flanking region of the human SMIT gene, the (−2,444/−130) relative nucleotide region, also displayed small tonicity-responsive enhancer activity (23).
About 80% of the 60-kb 5′-flanking region was subcloned in nine non-overlapping, SacI restriction fragments. In order to test tonicity-responsive regulatory activity of these fragments, each DNA fragment was cloned in front of the human SMIT promoter and the Photinus luciferase gene to generate an expression construct as described under “Experimental Procedures.” Two of the SacI fragments, S14 (7 kb) and S31 (4 kb), displayed significant, albeit small, tonicity-responsive enhancer activity for the SMIT gene promoter (Fig. 2). Partial digestion and Southern blot analyses of the P1 clones localized S14 and S31 at 50 and 15 kb upstream of the SMIT gene, respectively (Fig. 1). None of the SacI fragments displayed negative regulatory (or suppressor) activity, indicating that, like the BGT1 and AR genes, the SMIT gene is regulated only by enhancers.
Characterization of SMIT Tonicity-responsive Enhancers
The DNA fragments S14 and S31 were divided into smaller restriction fragments and tested for tonicity-responsive enhancer activity (Fig. 2). The enhancer activity of S14 was narrowed to two small fragments: S14-c (357 bp) and S14-g (588 bp). Likewise, the enhancer activity of S31 was confined to an 877-bp fragment named S31-d. Sequencing of these fragments revealed that each of them contains one or more sequences (Table II) that fit the consensus of TonEs for the BGT1 and AR genes (22): YGGAANNNYNY (Y is C or T; N is any nucleotide). We investigated whether these TonEs are indeed responsible for the enhancer activity.
S14-c has only one TonE named TonEA. When three of the key residues (22) of TonEA were mutated to inactivate it (see “Experimental Procedures”), the enhancer activity of S14-c was lost; -fold induction of luciferase decreased from 2.66 (S14-c) to 1.44 (S14-c/mTonEA, Fig. 3). It was concluded that TonEA was responsible for the enhancer activity of S14-c.
S14-g has three TonE sequences named TonEB1, TonEB2, and TonEB3. TonEB2 and TonEB3 overlap over a 10-bp span in antisense directions (Table II). When S14-g was mutated to inactivate both TonEB2 and TonEB3, the enhancer activity of S14-g was completely lost (2.71-fold induction to 1.36-fold induction, Fig. 3). These results indicate that (i) TonEB(2/3) is essential for the enhancer activity of S14-g and (ii) TonEB1 alone is not active. To further explore the roles of TonEB1 and TonEB2/3, a synthetic DNA named TonEB1(2/3) was prepared by combining sequences of TonEB1 and TonEB(2/3) and their flanking sequences (10 bp from each; Table I). TonEB1(2/3) displayed an enhancer activity higher than that of S14-g (3.70-versus 2.71-fold induction; Fig. 3) probably because TonE sequences are closer to the promoter (see also below). Mutation of TonEB(2/3) inactivated the enhancer function of TonEB1(2/3) to 1.69-fold induction, as expected. On the other hand, mutation of TonEB1 did not affect the enhancer activity of TonEB1(2/3) (3.47-fold induction), indicating that TonEB1 is not a functional TonE. Inspection of inactive TonE sequences TonEB1 and TonEC3 (see below) suggests that having pyrimidines in both the 6th and 7th position may disable TonE activity. Having only one pyrimidine in the 6th or 7th positions does not disable TonE (22). Since TonEB3 has T residues in the 6th and 7th position, we believe that TonEB3 is not active and TonEB2 is the only functional TonE in S14-g.
S31-d holds three TonEs named TonEC1, TonEC2, and TonEC3 (Table II). To study their function, a synthetic DNA named TonEC123 was prepared by combining the three S31-d TonE sequences with their flanking sequences (10 bp from each TonE; Table I). Interestingly, enhancer activity of TonEC123 (6.98-fold induction of luciferase) is more than twice that of S31-d (2.71-fold) (Fig. 3). Bringing TonEC1 and TonEC2 closer to each other (from 494 bp apart to 20 bp apart) and to the promoter (TonEC2 is 339 bp away from the promoter in S31-d compared with 26 bp away in TonE123) may have enhanced their ability to activate transcription. At any rate, mutations in either TonEC1 or TonEC2 reduced enhancer activity of TonEC123 to 3.38-fold or 4.31-fold, respectively (Fig. 3). On the other hand, mutations in TonEC3 did not affect enhancer activity of TonEC123. Bases on these and other (see below and Fig. 4) studies, we conclude that TonEC1 and TonEC2, but not TonEC3, contribute to the enhancer activity of S31-d.
Fig. 4. Binding of TonE sequences to nuclear extracts from MDCK and HeLa cells.
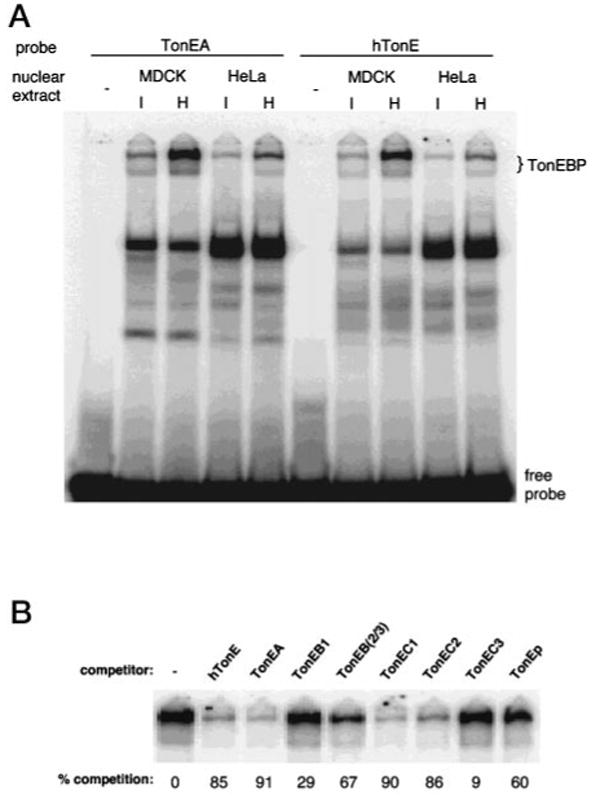
A, autoradiogram of an EMSA gel using 32P-labeled hTonE (right) or TonEA (left). Each lane was loaded with a binding reaction containing 5 μg of nuclear extract prepared from MDCK or HeLa cells cultured in isotonic (I) or hypertonic (H) medium for 24 h. Slowly migrating bands representing TonEBP and free probes are marked on the right. B, autoradiogram of an EMSA gel showing only the TonEBP bands. Each lane was loaded with a binding reaction containing 0.5 nm 32P-labeled hTonE, 5 μg of nuclear extract prepared from MDCK cells cultured in hypertonic medium, and 10 nm of a competitor TonE sequence indicated on the top. Percent competition (or reduction) of 32P-labeled hTonE binding to TonEBP by a TonE sequence was calculated from the radioactivity of the slow bands in the lane without (first lane) and in the lane with that particular TonE competitor. Means of triplicate determinations are shown at the bottom.
In order to explore the function of TonEC1 and TonEC2 further, S31-ds was prepared from S31-d by eliminating 116 bp upstream of TonEC1 and 336 bp downstream of TonEC2 including TonEC3 as described under “Experimental Procedures.” S31-ds displayed enhancer activity greater than S31-d (5.20-fold versus 2.54-fold induction of luciferase, Fig. 3) probably because TonEC2 is closer to the promoter. As expected, mutating TonEC1 or TonEC2 significantly decreased the enhancer activity of S31-ds. These data establish that TonEC1 and TonEC2 are functional when they are spaced apart at the natural distance.
Sequencing of the (−2,444/−130) relative nucleotide region, which displayed a moderate tonicity-responsive enhancer activity (23), revealed a TonE sequence named TonEp at (−331/−321) position (Table II). TonEp is also found at (−346/−336) position of the bovine SMIT gene and has been shown to display small TonE activity: 1.5–2-fold induction (16). We conclude that TonEp is most likely responsible for the enhancer activity of the (−2,444/−130) region (see more below).
EMSA of SMIT TonEs
Previously, we identified a 200-kDa nuclear protein of MDCK cells named TonEBP, which displays very slow mobility in non-denaturing gel electrophoresis (22). Recent cloning of TonEBP3 demonstrate that it is a transcription factor mediating the enhancer activity of TonEs. Since the SMIT TonEs described above are of human origin, we searched for human cells to study the regulation of SMIT transcription. We found that HeLa cells expressed SMIT and AR mRNA, the abundance of which was stimulated ∼10-fold by hypertonicity as in MDCK and other kidney-derived cells (data not shown). Nuclear extracts of HeLa cells displayed TonEBP bands in EMSA gels when “hTonE,” a prototypical TonE (22), was used as a probe (Fig. 4A, right); TonEBP of HeLa cells were indistinguishable (data not shown) from TonEBP of MDCK cells in their binding specificity described previously (22). On the other hand, all the other bands including the dominant band in the HeLa lanes about a third of the way down do not display correct binding specificity, indicating that they are not TonEBP: they are either not competed by active TonE sequences or competed by inactive TonE sequences (data not shown). We conclude that HeLa cells possess functional TonEBP.
When TonEA was used as a probe, TonEBP are also detected from MDCK and HeLa cells as expected (Fig. 4A, left). To quantify binding of the SMIT TonEs to TonEBP, we measured the ability of each SMIT TonE to compete hTonE binding to TonEBP as described in the legend for Fig. 4B. Under these conditions, functional (or active) TonEs compete better than 50% (22). As shown in Fig. 4B, all the active SMIT TonEs (TonEA, TonEB(2/3), TonEC1, TonEC2, and TonEp) competed away more than 60% of hTonE binding, demonstrating that these TonEs are functional, consistent with the data presented in Fig. 4. On the other hand, TonEB1 and TonEC3 competed poorly, in keeping with their lack of enhancer activity (Fig. 4B). Collectively, the data in Figs. 3 and 4 prove that the SMIT TonEs reported here are functionally identical to the TonEs of the BGT1 and AR genes (22).
Increase in Site Occupancy of the SMIT TonEs in Response to Hypertonicity
When MDCK cells are exposed to hypertonicity, activity of TonEBP is markedly stimulated, leading to an increase in TonEBP binding to the TonE sites upstream of the BGT1 gene and stimulation of BGT1 transcription (22).2 The increase in TonEBP activity in HeLa cells in response to hypertonicity (Fig. 4) predicts that there should be a parallel increase in binding of TonEBP to the SMIT TonEs. To address this issue directly, we performed in vivo footprinting assays where methylation of G residues on HeLa genomic DNA by treatment with dimethyl sulfate was measured using quantitative PCR as described under “Experimental Procedures.” Nested PCR primers were designed to examine regions covering TonEA, TonEB2, TonEC2, and TonEp (Fig. 5). We did not detect any consistent change in methylation outside the TonE sites described below. In TonEA, methylation of the 2nd and 3rd G residues is decreased by 46% in hypertonicity. Likewise, methylation of the 2nd G in TonEB2 and TonEC2 was inhibited by 36 and 33%, respectively. In TonEB2 and TonEC2, the 3rd G residues were not methylated even in control DNA (data not shown), indicating that these nucleotides are inaccessible to dimethyl sulfate. It was not possible to examine the TonEp region in the sense direction because (−300/−50) region is GC-rich (83%) and repetitive. We managed to come up with a pair of PCR primers covering TonEp in the antisense direction. G residues complementary to the last two C residues were protected from methylation by 30% in hypertonicity. Collectively, the data in Fig. 5 demonstrate that activated TonEBP gets access to and binds to all the SMIT TonEs spread over 50 kb, providing in vivo functional support to the idea that all the TonEs contribute to the stimulation of transcription by hypertonicity.
Fig. 5. Changes in the occupancy of SMIT TonE sites in response to hypertonicity.
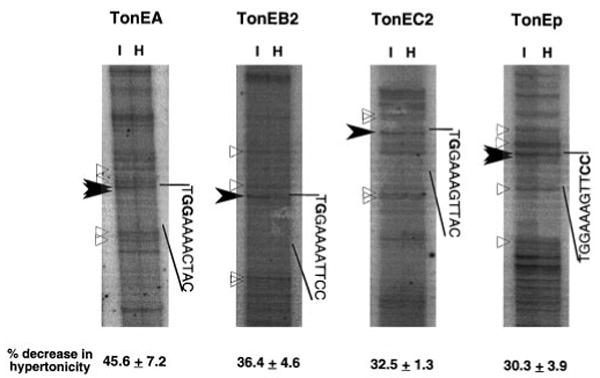
Site occupancy was determined by in vivo footprinting assay in which the efficiency of methylation of G residues by dimethyl sulfate in vivo was measured. HeLa cells cultured in isotonic (I) or hypertonic medium (H) for 24 h were treated with dimethyl sulfate before isolation of DNA. The DNA was cleaved with piperidine, and quantitative PCR was performed to detect methylated G residues. 32P-Labeled primers were used in the last two rounds of PCR to visualize amplified products in sequencing gels as shown. Each panel shows PCR products of a primer, and the region corresponding to its TonE site is demarcated along with the sequence. Bands representing the 2nd and 3rd G residues in TonEA, the 2nd G residue in TonEB2 and TonEC2, and the G residues complementary to the C residues in the 10th and 11th position of TonEp are indicated by filled arrowheads and bold letters. The radioactivity of these bands in each lane was divided by the radioactivity of the reference bands marked by open triangles to calculate the corrected intensity. Percent decrease in the corrected intensity of these bands in hypertonic cells relative to that in isotonic cells are shown at the bottom (mean and S.E., n = 4).
Discussion
When kidney-derived cells are challenged with hypertonicity, transcription of the SMIT (5), BGT1 (25), and AR (26) genes increases over 10-fold. Expression of the reporter gene controlled by two TonEs2 and the promoter within 185 bp of the BGT1 gene (20) or three TonEs ∼1 kb upstream of the AR gene and its promoter (21) is also stimulated over 10-fold by hypertonicity. On the other hand, ∼3 kb immediate upstream of the SMIT gene containing a TonE (TonEp) induces reporter gene expression at most 2-fold in response to hypertonicity (16, 23, 24), indicating that the major cis-regulatory sequences are outside this region. Our previous efforts (24) established that the putative major regulatory sequences are outside the 52 kb of the cloned canine genomic region containing the entire SMIT gene, 11 kb of 5′-flanking, and 4 kb of 3′-flanking sequence. The data presented in this paper demonstrate that the additional regulatory sequences are spread in the region 15–55 kb upstream of the human gene as a form of four TonEs: TonEA, TonEB2, TonEC1, and TonEC2. The five TonEs (including TonEp) would provide enough additive enhancer activity to account for greater than 10-fold stimulation of transcription in response to hypertonicity. The most convincing support for this idea is the in vivo footprinting data (Fig. 5), which show that at least four out of the five TonE sites are active in that binding to these sites is increased under hypertonicity as in the TonE sites of the BGT1 gene (22).2
The osmo-protective accumulation of compatible osmolytes is conserved throughout evolution (8). Interestingly, the role of transcription is also widely conserved. In bacteria, transcription of the Pro U operon is markedly stimulated by hypertonicity, resulting in an increase in the activity of the ATP-consuming betaine transporter and accumulation of betaine (27). In yeast, transcription of GPD1 is stimulated by hypertonicity leading to an increased synthesis and accumulation of glycerol (28), which is the major yeast compatible osmolyte. Exposure of plants including sugar beet, spinach, and barley to high salt environments results in transcriptional stimulation of the betaine aldehyde dehydrogenase gene and consequent synthesis and accumulation of betaine (29). The molecular basis for the foregoing transcriptional regulation is not clear. Transcriptional regulation is far better understood for accumulation of MI, betaine, and sorbitol in mammalian cells. This work and previous studies (22) collectively demonstrate that a cis-element TonE and its cognate transcription factor TonEBP play the central role in the hypertonicity-induced stimulation of the SMIT, BGT1, and AR genes.
The consensus sequence of TonE, YGGAAnnnYnY (Table II), was based on (a) seven known TonE sequences at the time and (b) analysis of nine TonE mutants where each mutant has a single nucleotide mutated in one of nine different positions (22). Inspection of five additional TonEs identified in this paper provides further insight into the consensus. Most TonEs, 10 out of 11 known TonEs so far (TonEC1 is the same as hTonE in Ref. 22), start with T rather than C. The sixth nucleotide (“n” in the consensus) is A in all the TonEs, even though mutating A to T affects its activity only moderately (22). Therefore, a more definitive consensus sequence is TGGAAAnnYnY (Table II). In DNA with random sequence, this sequence should be present on average 1 in every 16 kb. This should be an underestimate because certain nucleotides are flexible, i.e. the first and sixth nucleotides. In this regard, the 5′-flanking region of the SMIT gene is not particularly rich in TonEs: Five TonEs in ∼50 kb recovered from 60 kb of the 5′-flanking region or an average of one in every ∼10 kb, a value close to the expected frequency. This contrasts with the BGT1 and AR genes, where TonEs are more concentrated near the promoter; two TonEs within 185 bp upstream of the BGT1 gene and three TonEs within 1.2 kb upstream of the AR gene. It is possible that the five SMIT TonEs spread over 50 kb are physically close to the promoter, perhaps by way of multiple long range looping, and act additively to stimulate the SMIT promoter. This is based on the observation that bringing TonEC1 and TonEC2 closer (from 494 bp apart in S31-d to 20 bp apart in TonEC123; Fig. 3) increases the enhancer activity. The binding of TonEBP to the TonE sites in hypertonicity (Fig. 5) may facilitate the looping or reorganization of local chromatin structure to enable additive action of the bound TonEBP to the promoter.
Regulation of transcription by enhancers located far from the gene is well established. In the locus control region for the β-globin gene cluster, five enhancers spread over 15 kb are believed to function as a single unit or holocomplex where all the enhancers are brought close to each other (30). In adult erythrocyte precursors, the locus control region stimulates transcription of the β-globin gene located some 40 kb downstream (30). Studies involving homologous deletion of genomic sequences in mice revealed that the insulin-like growth factor 2 gene is regulated by a pair of enhancers ∼90 kb away from the gene (31). Although the insulin-like growth factor 2 and β-globin genes are regulated by imprinting and developmental cues, respectively, the distant enhancers and the locus control region are constitutively active in adult tissues. In contrast, the SMIT TonEs are regulated by ever-changing fluctuations in tonicity. In this regard, the SMIT TonEs are a rare and important model system that provides an unusual opportunity to study the role of chromatin structure in regulation of gene expression in response to physiological and pathological signals.
Acknowledgments
We thank S. K. Woo for providing nuclear extracts of HeLa cells.
Footnotes
This work was supported by National Institutes of Health Grant DK42479 (to H. M. K.), a fellowship from the Juvenile Diabetes Foundation International (to J. S. R.), and National Research Service Awards Grants DK07712 and DK09469 (to M. G. A.).
The abbreviations used are: SMIT, the sodium/myo-inositol cotransporter; MI, myo-inositol; BGT1, the sodium- and chloride-coupled betaine/γ-aminobutyrate transporter; AR, aldose reductase; TonE, tonicity-responsive enhancer; TonEBP, TonE-binding protein; EMSA, electrophoretic mobility shift assay; MDCK, Madin-Darby canine kidney; PCR, polymerase chain reaction; bp, base pair(s); kb, kilobase pair(s).
H. Miyakawa, J. S. Rim, and H. M. Kwon, unpublished observations.
H. Miyakawa, S. K. Woo, and H. M. Kwon, unpublished observations.
References
- 1.Kwon HM, Yamauchi A, Uchida S, Preston AS, Garcia-Perez A, Burg MB, Handler JS. J Biol Chem. 1992;267:6297–6301. [PubMed] [Google Scholar]
- 2.Hager K, Hazama A, Kwon HM, Loo DDF, Handler JS, Wright EM. J Membr Biol. 1995;143:103–113. doi: 10.1007/BF00234656. [DOI] [PubMed] [Google Scholar]
- 3.Yamauchi A, Miyai A, Shimada S, Minami Y, Tohyama M, Imai E, Kamada T. J Clin Invest. 1995;96:1195–1201. doi: 10.1172/JCI118151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamauchi A, Nakanishi T, Takamitsu Y, Sugita M, Imai E, Noguchi T, Fujiwara Y, Kamada T, Ueda N. J Am Soc Nephrol. 1994;5:62–67. doi: 10.1681/ASN.V5162. [DOI] [PubMed] [Google Scholar]
- 5.Yamauchi A, Uchida S, Preston AS, Kwon HM, Handler JS. Am J Physiol. 1993;264:F20–F23. doi: 10.1152/ajprenal.1993.264.1.F20. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura H, Yamauchi A, Nakanishi T, Takamitsu Y, Sugiura T, Akagi A, Moriyama T, Horio M, Imai E. Am J Physiol. 1997;272:F267–F272. doi: 10.1152/ajprenal.1997.272.2.F267. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura H, Yamauchi A, Sugiura T, Matsuoka Y, Horio M, Tohyama M, Shimada S, Imai E, Hori M. Kidney Int. 1998;53:146–153. doi: 10.1046/j.1523-1755.1998.00747.x. [DOI] [PubMed] [Google Scholar]
- 8.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 9.Uchida S, Kwon HM, Yamauchi A, Preston AS, Marumo F, Handler JS. J Clin Invest. 1991;88:656–662. doi: 10.1172/JCI115350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Perez A, Burg MB. Physiol Rev. 1991;71:1081–1115. doi: 10.1152/physrev.1991.71.4.1081. [DOI] [PubMed] [Google Scholar]
- 11.Kwon HM, Handler JS. Curr Opin Cell Biol. 1995;7:465–471. doi: 10.1016/0955-0674(95)80002-6. [DOI] [PubMed] [Google Scholar]
- 12.Uchida S, Garcia-Perez A, Murphy H, Burg MB. Am J Physiol. 1989;256:C614–C620. doi: 10.1152/ajpcell.1989.256.3.C614. [DOI] [PubMed] [Google Scholar]
- 13.Ferraris JD, Burg MB, Williams CK, Peters EM, Garcia-Perez A. Am J Physiol. 1996;270:C650–C6514. doi: 10.1152/ajpcell.1996.270.2.C650. [DOI] [PubMed] [Google Scholar]
- 14.Kwon HM. In: Cellular and Molecular Physiology of Cell Volume Regulation. Strange K, editor. CRC Press; Boca Raton, FL: 1993. pp. 383–394. [Google Scholar]
- 15.Gullans SR, Verbalis JG. Annu Rev Med. 1993;44:289–301. doi: 10.1146/annurev.me.44.020193.001445. [DOI] [PubMed] [Google Scholar]
- 16.Zhou C, Cammarata P. Exp Eye Res. 1997;35:349–363. doi: 10.1006/exer.1997.0335. [DOI] [PubMed] [Google Scholar]
- 17.Minami Y, Inoue K, Shimada S, Morimura H, Miyai A, Yamauchi A, Matsunaga T, Tohyama M. Mol Brain Res. 1996;40:64–70. doi: 10.1016/0169-328x(96)00034-4. [DOI] [PubMed] [Google Scholar]
- 18.Morimura H, Shimada S, Otori Y, Yamauchi A, Minami Y, Inoue K, Miyai A, Ishimoto I, Tano Y, Tohyama M. Mol Brain Res. 1996;35:333–338. doi: 10.1016/0169-328x(95)00245-n. [DOI] [PubMed] [Google Scholar]
- 19.Ibsen L, Strange K. Am J Physiol. 1996;271:F877–F885. doi: 10.1152/ajprenal.1996.271.4.F877. [DOI] [PubMed] [Google Scholar]
- 20.Takenaka M, Preston AS, Kwon HM, Handler JS. J Biol Chem. 1994;269:29379–29381. [PubMed] [Google Scholar]
- 21.Ko BCB, Ruepp B, Bohren KM, Gabbay KH, Chung SSM. J Biol Chem. 1997;272:16431–16437. doi: 10.1074/jbc.272.26.16431. [DOI] [PubMed] [Google Scholar]
- 22.Miyakawa H, Woo SK, Chen CP, Dahl SC, Handler JH, Kwon HM. Am J Physiol. 1998;274:F753–F761. doi: 10.1152/ajprenal.1998.274.4.F753. [DOI] [PubMed] [Google Scholar]
- 23.Mallee JJ, Atta MG, Lorica VL, Rim JS, Kwon HM, Lucente AD, Wang Y, Berry GT. Genomics. 1997;46:459–465. doi: 10.1006/geno.1997.5055. [DOI] [PubMed] [Google Scholar]
- 24.Rim JS, Tana S, Takenaka M, Handler JS, Kwon HM. Arch Biochem Biophys. 1997;341:193–199. doi: 10.1006/abbi.1997.9950. [DOI] [PubMed] [Google Scholar]
- 25.Uchida S, Yamauchi A, Preston AS, Kwon HM, Handler JS. J Clin Invest. 1993;91:1604–1607. doi: 10.1172/JCI116367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smardo FL, Burg MB, Garcia-Perez A. Am J Physiol. 1992;262:C776–C782. doi: 10.1152/ajpcell.1992.262.3.C776. [DOI] [PubMed] [Google Scholar]
- 27.Lucht JM, Dersch P, Kempf B, Bremer E. J Biol Chem. 1994;269:6578–6586. [PubMed] [Google Scholar]
- 28.Albertyn J, Hohmann S, Thevelein JM, Prior BA. Mol Cell Biol. 1994;14:4135–4144. doi: 10.1128/mcb.14.6.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishitani M, Nakamura T, Han SY, Takabe T. Plant Mol Physiol. 1995;27:307–315. doi: 10.1007/BF00020185. [DOI] [PubMed] [Google Scholar]
- 30.Wijgerde M, Grosveld F, Fraser P. Nature. 1995;377:209–213. doi: 10.1038/377209a0. [DOI] [PubMed] [Google Scholar]
- 31.Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilgman SM. Genes Dev. 1995;9:2079–2089. doi: 10.1101/gad.9.17.2079. [DOI] [PubMed] [Google Scholar]