

Abstract
Partially modified retro–inverso, retro, and inverso isomers of hydrazide linked bifunctional peptides were designed, synthesized, and evaluated for bioactivities at δ/μ opioid receptors and CCK-1/CCK-2 receptors. All modifications of the CCK pharmacophore moiety affected bioactivities for the CCK-1 and CCK-2 receptors (up to 180-fold increase in the binding affinity with higher selectivity) and for the δ and μ opioid receptors. The results indicate that the opioid and CCK pharmacophores in one molecule interact with each other to induce topographical changes for both pharmacophores.
Introduction
Endogenous cholecystokinin (CCKa) has been shown to give an antiopioid effect, particularly in the spinal cord.1-5 CCK attenuates morphine antinociception in several assays, whereas CCK antagonists potentiate morphine-induced antinociception in vivo and electrophysiologically.6-8 In earlier studies we hypothesized that novel ligands that interact with the opioid receptors as agonists and with the CCK receptors as antagonists may enhance analgesic effects in chronic pain states without the development of tolerance.9-12 We demonstrated that a potent opioid receptor agonist with CCK receptor antagonist activity has good potency in an in vivo model of neuropatic pain.13 One type of novel ligand we reported recently was the structure in which opioid (Tyr1-d-Ala2-Gly3-Phe4-) and CCK (d-Trp1′-Nle2′-Asp3′-Phe4′-) pharmacophores were combined in one molecule through a hydrazide linker.14 Some of these types of ligands showed very potent δ and μ opioid bioactivities and moderate but balanced CCK-1/CCK-2 binding affinities. On the basis of the bioassay results, the hydrazide linker was considered to play a role in making favorable conformations for opioid receptors more so than for CCK receptors. To obtain structures that are more potent for CCK receptors, retro–inverso, retro, and inverso isomers of the CCK pharmacophore have been designed and synthesized. The modifications examine the roles of the direction of peptide backbone and the topology of side chain positions in molecular recognition at opioid and CCK receptors.
Total or partial retro–inverso modifications have been applied to many families of biologically active peptides, and many examples have been found to retain recognition properties or biological activity as good as the parent peptide in many cases.15-18 Since the retro–inverso modification leads to similar topographies, differing primarily by the reversed direction of amide bonds, it is a useful approach to elucidate the role of direction of the peptide backbone in molecular recognition. Ligands 1 (or 2) and 3 (or 4) are partial retro–inverso isomers where the structures are same in the opioid region but different in the CCK region (Figure 1). The same relationship exists between 5 (or 6) and 7 (or 8), since inverso isomers are correlated to retro isomers of the parent peptide for the same reason. The relation of 1 (or 2) to 5 (or 6) is that of a partial retro isomer in which the direction of the amide bonds are reversed but the chiralities of the four amino acid residues in the CCK region are retained, which results in noncomplementary side chain topography.
Figure 1.
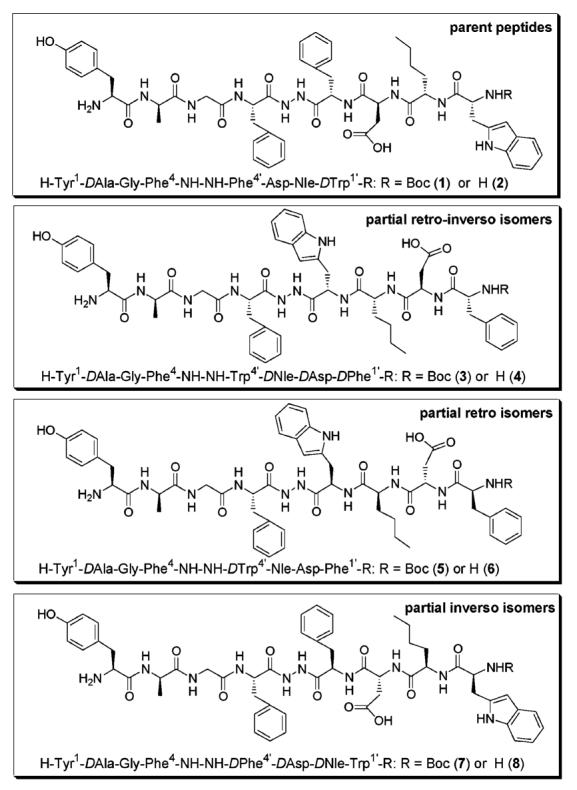
Structures of partially retro–inverso (3 and 4), retro (5 and 6), and inverso (7 and 8) modified bifunctional peptides.
Results and Discussion
Partial retro, inverso, and retro–inverso modified bifunctional peptides were synthesized by the stepwise solution-phase method using Boc/Bzl chemistry.14 The C terminal of Phe in position 4 was blocked as an ethyl ester group and converted to a hydrazide after completing the chain elongation steps for the opioid part. The hydrazide linked tetrapeptide was coupled stepwise with subsequent residues for the CCK part. Peptide couplings were done using the DCC/HOBt/NMM or BOP/HOBt/NMM methods, and Nα-Boc groups were deprotected by 100% TFA at 0 °C except for Trp-contained peptide intermediates, which needed a scavenger to avoid unwanted tert-butylation. During the deprotection of the Nα-Boc group of the Trp residue, a tert-butylated side product was detected. Nonetheless the correct final product could be isolated from the side product by crystallization from EtOAc. Continuous deprotections of the Nα-Boc groups of the other residues did not lead to tert-butylation. It is likely that this is because the Trp residue was not as exposed to the surface of the molecule to be attacked by the carbocation. After the chain elongations, the Z/Bzl groups were removed from the peptides by hydrogenation to give the target peptides in more than 90% purity. The crude peptides were isolated by preparative RP-HPLC to afford more than 95% pure peptides.
CCK binding affinities were evaluated by competition assay using [125I]CCK-8(SO3) to label membranes from HEK293 cells that express the human CCK-1 or human CCK-2 receptors (Table 1). As intended in this work, the modifications improved the binding affinities at the CCK-1 and CCK-2 receptors. Interestingly from a comparison of ligands 1 and 2 to the most modified ligands, the modified ligands lost their balance of selectivity for the CCK-1 and CCK-2 receptors (more than 140-fold) while gaining selective binding affinities at one receptor up to 180-fold. (Ligands 1 and 2 have low but well balanced binding affinities at CCK-1 and CCK-2 receptors in the micromolar range.) The best CCK binding affinities at both receptors occurred when the backbone of the CCK pharmacophore was reversed, with inversion of chirality of all amino acids, and the amino function of the Phe4′ was protected with a Nα-Boc group to give 3. Removal of the Nα-Boc group of ligand 3 resulted in conversion to ligand 4, which had reduced binding affinities for the CCK-1 receptor (15-fold) and for the CCK-2 receptor (more than 9-fold). Considering the fact that the N-terminal protecting group plays an important role in CCK binding and that hydrophobic protecting groups such as an adamantyloxy (Adoc) increase the CCK binding affinity,19 the result was very interesting because in this case the presence of a free amine group at the N-terminal of the CCK pharmacophore, contrary to expectations, had increased bioactivities for the μ opioid receptor (Table 2).
Table 1.
Bioactivities of the Modified Bifunctional Peptides at CCK-1 and CCK-2 Receptors
| PI assayd |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| hCCK-1a [125I]CCK-8(SO3)b |
hCCK-2a [125I]CCK-8(SO3)c |
hCCK-1a |
hCCK-2a |
|||||||
| log IC50e | Ki,f nM | log IC50e | Ki,f nM | log EC50e,h | EC50,nM | Emaxg | log EC50e,h | EC50,nM | Emaxg | |
| 1 | −4.45 ± 0.11 | >10000 | −5.00 ± 0.19 | >10000 | nr | −7.80 ± 0.17 | 16 | 11 ± 0.5 | ||
| 2 | −4.99 ± 0.27 | 10000 | −5.03 ± 0.23 | 9300 | nr | −8.34 ± 0.22 | 4.6 | 22 ± 0.4 | ||
| 3 | −6.88 ± 0.27 | 130 | −5.96 ± 0.38 | 1100 | nc | nc | ||||
| 4 | −5.72 ± 0.27 | 1900 | −4.87 ± 0.27 | >10000 | nc | nc | ||||
| 5 | −2.93 ± 0.58 | >10000 | −6.77 ± 0.24 | 71 | nr | nr | ||||
| 6 | −5.15 ± 0.16 | 7000 | −5.11 ± 0.07 | 7700 | nc | nc | ||||
| 7 | −5.05 ± 0.20 | 8300 | −1.18 ± 0.87 | >10000 | nc | nc | ||||
| CCK-8(SO3) | −7.55 ± 0.15 | 28 | 59 ± 3.0 | −7.57 ± 0.24 | 27 | 16 ± 1.1 | ||||
Competition analyses against radiolabeled ligand were carried out using whole cell lysate preparation from transfected HEK293 cells that constitutively expressed the respective receptor types.
Kd = 1.9 ± 0.2 nM.
Kd = 1.3 ± 0.4 nM.
The [3H]phosphotidylinositol hydrolysis assays were carried out on hCCK-1 or hCCK-2 expressing cells cultured in 24-well titer plates.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments.
Antilogarithmic value of the respective IC50.
The Emax value is the saturable, maximum level of 3[H]inositol phosphates produced in the cells upon incubation with the peptide, expressed as a ratio ± standard error of the amount of 3[H]inositol phosphates produced in the presence of peptide over the basel level of 3[H]inositol phosphates in untreated cells.
nr: no stimulatory (or inhibitory) response at the highest concentration. nc: incomplete competition of the radioligand at the highest test concentration of the peptide. EC50 value cannot be determined.
Table 2.
Bioactivities of the Partially Modified Bifunctional Peptides at the δ and μ Opioid Receptors
| [35S]GTP-γ-S binding |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hDORa [3H]DPDPEb |
rMORa [3H]DAMGOc |
hDORd |
rMORd |
IC50,e nM |
||||||||
| log IC50f,g | Ki,h nM | log IC50f,g | Ki,h nM | log EC50f | EC50, nM | Emax,i % | log EC50f | EC50, nM | Emax,i % | MVD(δ) | GPI(μ) | |
| 1 | −7.03 ± 0.11 | 45 | −7.19 ± 0.30 | 31 | −7.13 ± 0.27 | 74 | 100 | −6.69 ± 0.11 | 210 | 170 | 24 ± 5 | 460 ± 130 |
| 2 | −8.49 ± 0.11 | 1.4 | −8.92 ± 0.06 | 0.5 | −8.02 ± 0.16 | 9.4 | 64 | −8.02 ± 0.11 | 96 | 82 | 26 ± 5 | 81 ± 21 |
| 3 | −8.04 ± 0.11 | 3.9 | −6.88 ± 0.09 | 56 | −7.91 ± 0.36 | 13 | 99 | −6.76 ± 0.31 | 17 | 71 | 18 ± 4 | 530 ± 100 |
| 4 | −8.29 ± 0.12 | 2.2 | −8.10 ± 0.08 | 3.4 | −7.83 ± 0.14 | 15 | 64 | −8.11 ± 0.14 | 7.7 | 82 | 25 ± 7 | 76 ± 9 |
| 5 | −8.86 ± 0.16 | 0.63 | −6.92 ± 0.08 | 52 | −8.23 ± 0.51 | 5.9 | 120 | −6.20 ± 0.37 | 630 | 63 | 16 ± 2 | 240 ± 40 |
| 6 | −8.59 ± 0.18 | 1.1 | −7.54 ± 0.16 | 13 | −7.7 ± 0.18 | 19 | 89 | −7.13 ± 0.26 | 75 | 85 | 12 ± 2 | 93 ± 6 |
| 7 | −7.79 ± 0.09 | 6.9 | −7.10 ± 0.14 | 34 | −8.08 ± 0.07 | 8.4 | 93 | −8.90 ± 0.18 | 1.3 | 106 | 31 ± 5 | 210 ± 50 |
| 8 | −8.78 ± 0.10 | 0.8 | −8.16 ± 0.10 | 3.0 | −7.68 ± 0.19 | 21 | 48 | −8.73 ± 0.43 | 1.9 | 20 | 15 ± 3 | 97 ± 9 |
| biphalin | −8.95 ± 0.17 | 1.1 | 84 | 2.7 ± 1.5 | 8.8 ± 0.3 | |||||||
Competition analyses were carried out using membrane preparations from transfected HN9.10 cells that constitutively expressed the respective receptor types.
Kd = 0.50 ± 0.1 nM.
Kd = 0.85 ± 0.2 nM.
Expressed from CHO cells.
Concentration at 50% inhibition of muscle contraction at electrically stimulated isolated tissues.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments.
Competition against radiolabeled ligand.
Antilogarithmic value of the respective IC50.
Net total bound/basal binding × 100 ± SEM.
In our earlier work,14 we suggested that the CCK and opioid pharmacophores have a common backbone conformational requirement for both receptors and that substitution of d-Trp1′-NMeNle2′ is not tolerated by both receptors. While the chirality of the 1′ position does not affect CCK binding affinities and activities by itself, the backbone conformation achieved by combination with the 2′ position residue seems to affect the CCK binding affinity and selectivity. It is possible that the 1′ and 2′ residues with the same chirality could be responsible for the loss of balance for both CCK receptors relative to the other ligands. The finding of increased binding affinities for the retro and retro–inverso modifications suggests that the shorter distance between Phe4 in the opioid part and Trp4′ in the CCK part corresponds more closely to the requirements for the CCK receptors, though this consideration could be dependent on conformational factors. All of the modified ligands showed no CCK agonist activity in the unstimulated GPI/LMMP assay and the PI assay.
Opioid binding affinities of these peptides for the human δ opioid receptor (hDOR) or the rat μ opioid receptor (rMOR) were determined by radioligand competition assay using [3H]DPDPE to label the δ opioid receptor and using [3H]DAMGO to label the μ opioid receptor (Table 2). Opioid binding affinity results showed that these kinds of modifications in the CCK region also affected the opioid binding affinities. Comparing the parent compound 1, which has good opioid binding at δ and μ opioid receptors (Ki = 45 and 31 nM, respectively), all ligands increased their selectivities (μ/δ = 5- to 83-fold) for the δ opioid receptor by enhancing the binding affinities (Ki = 6.9−0.63 nM). In our earlier study,14 it was shown that structural modifications in the 1′ position of the CCK part play an important role in δ opioid activity while μ opioid activity depends on the presence of an N-terminal protecting group in the same region. With these earlier results, the broad range of binding affinities at the δ opioid receptor was predicted because here the modifications changed the amino acid residue in the 1′ position from Phe to Trp and in some cases with inversion of their chiralities. As mentioned before, comparisons between appropriate analogue pairs (1 vs 2, 3 vs 4, 5 vs 6, and 7 vs 8) showed that the free amine group results in greater activity for μ opioid receptor, up to 110-fold. The structure–activity relationships (SAR) results for the opioid receptors coincided well with our earlier study. The binding affinity results correlated well with the GTP-γ-S binding and the functional assays. These assays showed the distinct function of the N-terminal free amine in the CCK region for the μ opioid receptor. All ligands retained better agonist activity for the δ opioid receptor in the MVD assay than for the μ opioid receptor in the GPI assay.
The novel observation in this work was that modification of the CCK pharmacophore affected not only CCK activities but also opioid activities in binding and functional assays. It can be seen from Tables 1 and 2 that the modifications in the CCK region resulted in better binding affinities at opioid and CCK receptors in most cases. It also showed different biological profiles at both receptors. This indicates that the structural modifications in the comparatively separated CCK pharmacophore could change the topography of the opioid pharmacophore structure, which resulted in a different biological profile.
As a result of this observation, we were interested in finding out how the structural modifications in the CCK part affected the opioid activities. To gain insights into the conformational profiles of the modified peptides, molecular modeling experiments were performed using MacroModel 8.1.20 As observed in these experiments, hydrogen bonds between Tyr of the opioid part and the Trp and/or Asp residues of CCK part dominate the conformational structures of the ligands, which generally give loop structures. On the basis of these observations, it is likely that the free amine of the CCK part participate in hydrogen bonding as a counterpart of the hydroxyl group of Tyr or the carboxylic acid group of Asp, and as a result, the N-terminal amine of the Tyr residue in the opioid region is exposed to the molecular surface and is recognized well by the μ opioid receptor. It is interesting that the retro modified CCK pharmacophore linked ligand 5, which uniquely showed the characteristics of a well-folded structure in molecular modeling experiments, exhibited the best δ opioid activity with high selectivity over μ opioid receptor in binding (Ki = 0.63, μ/δ = 83) and GTP-γ-S assays (EC50 = 5.9, μ/δ = 110).
Conclusions
We have modified a chimeric bifunctional peptide that had been previously identified as potent μ/δ opioid agonists and CCK-1/CCK-2 antagonists. This initial SAR study has examined the effect of backbone directions and chiralities, and we have shown that all modifications in the CCK region examined here increase binding affinities selectively for the CCK-1 or CCK-2 receptors. This result indicates that the direction of the amide bond alone does not appear to be important for the biological activity. However, it is noteworthy that the modifications in the CCK region resulted in changes of bioactivities not only at the CCK receptors but also at the opioid receptors. These results clearly demonstrate that the two different pharmacophores can be combined in one single molecule and can interact with each other to modify their own structures, which also can result in complementary topographies for both receptors.
Experimental Section
All amino acid derivatives were purchased from ChemImpex (Woodale, NJ), Novabiochem (San Diego, CA), and Bachem (Torrance, CA). The materials for bioassays were purchased from Perkin-Elmer, American Peptide Company Inc., Gibco, and Sigma. Peptides were synthesized by stepwise solution-phase synthesis using Boc/Bzl methodology. Analytical HPLC was performed on a Hewlett-Packard 1090 (C-18, Vydac, 4.6 mm × 250 mm, 5 μm), and preparative RP-HPLC was performed on a Hewlett-Packard 1100 (C-18, Vydac, 10 mm × 250 mm, 10 μm). Mass spectra were taken in the positive ion mode under ESA methods. Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH2 was synthesized by the same method described previously.14
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-Trp-d-Nle-d-Asp-d-Phe-Boc (3)
Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH2 (1.25 g, 1.8 mmol) and Boc-d-Trp (0.66 g, 2.16 mmol) were coupled in DMF using the BOP/HOBt procedure to afford 1.60 g of pure Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH-Trp-Boc [MS (M − TFA + H)+ 1009.1; analytical HPLC tR 30.6 min, purity >95%] in quantitative yield. The Nα-Boc-pentapeptide (0.98 g) was dissolved in 10 mL of a solution of TFA/DCM/anisole (10:10:1) and stirred for 40 min at 0 °C. The mixture was evaporated and crystallized from EtOAc to give pure Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH-Trp-H·TFA [MS (M − TFA + H)+ 909.5; analytical HPLC tR 24.9 min, purity >95%] in 86% yield. The pentapeptide was coupled stepwise with Boc-d-Nle, Boc-d-Asp(OBzl), and Boc-d-Phe using the same BOP/HOBt method and hydrogenated using Pd–C as a catalyst for 2 days at room temperature to afford 3 in overall 75% yield in greater than 80% purity. The crude peptide was purified by preparative RP-HPLC (20–50% of acetonitrile within 20 min) to give pure 3 as a white powder.
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-Trp-d-Nle-d-Asp-d-Phe-H·TFA (4)
Crude 3 was stirred in TFA at 0 °C for 20 min and evaporated under vacuum to an oil. The residual oil was triturated with diethyl ether and purified by preparative RP-HPLC (20–50% of acetonitrile within 30 min) to give pure 4 as a white powder in 76% yield.
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-d-Trp-Nle-Asp-Phe-Boc (5)
Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH-d-Trp-Nle-Asp-Phe-Boc (analytical HPLC tR 35.8 min, purity >80%) was prepared by the same method as described above and hydrogenated using Pd–C to give crude 5 in overall 73% yield in 84% purity. The crude peptide was purified by preparative RP-HPLC (20–65% of acetonitrile within 25 min) to give pure 5 as a white powder.
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-d-Trp-Nle-Asp-Phe-H·TFA (6)
The Nα-Boc group of crude 5 was deprotected by the same method as described above and purified by preparative RP-HPLC (10–50% of acetonitrile within 20 min) to give pure 6 as a white powder in 68% yield.
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-d-Phe-d-Asp-d-Nle-Trp-Boc (7)
Z-Tyr(Bzl)-d-Ala-Gly-Phe-NH-NH-d-Phe-d-Asp-d-Nle-Trp-Boc was prepared by a method described previously13 and hydrogenated using Pd–C to give crude 7 in overall 61% yield in 82% purity. The crude peptide was purified by preparative RP-HPLC (20–60% of acetonitrile within 20 min) to give pure 7 as a white powder.
TFA·H-Tyr-d-Ala-Gly-Phe-NH-NH-d-Phe-d-Asp-d-Nle-Trp-H·TFA (8)
Crude 7 was dissolved in a solution (TFA/DCM/anisole = 10:10:1) and stirred for 30 min at 0 °C and evaporated under vacuum to an oil. The residual oil was triturated with diethyl ether and purified by preparative RP-HPLC (10–50% of acetonitrile within 20 min) to give pure 8 as a white powder in 20% yield.
Bioassays
All bioassays for the δ/μ opioid and CCK-1/CCK-2 receptors were performed by the same method as described previously.14
Acknowledgment
The work was supported by grants from the USDHS, National Institute on Drug Abuse (Grants DA-12394 and DA-06284). We thank Margie Colie for assistance with the manuscript.
Footnotes
Abbreviations: Adoc, adamantyloxycarbonyl; Boc, tert-butyloxycarbonyl; BOP, (benzotriazole-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; Bzl, benzyl; CCK, cholecystokinin; CHO, Chinese hamster ovary; DCC, N,N-dicyclohexylcarbodiimide; DMF, N,N-dimethylformamide; DMSO, dimethylsulfoxide; hDOR, human δ opioid receptor; DPDPE, c[d-Pen2,d-Pen5]-enkephalin; DAMGO, [d-Ala2,NMePhe4,Gly5-ol]enkephalin; GPCR, G-protein-coupled receptor; GPI, guinea pig isolated ileum; HEK, human embryonic kidney; HOBt, 1-hydroxybenzotriazole; LMMP, longitudinal muscle with myenteric plexus; rMOR, rat μ opioid receptor; MVD, mouse vas deferens; NMM, N-methylmorpholine; SAR, structure–activity relationship; TFA, trifluoroacetic acid; Z, benzyloxycarbonyl.
Supporting Information Available: 1H NMR, MS, HRMS, TLC, HPLC, and purity data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Noble F, Roques BP. CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog. Neurobiol. 1999;58:349–379. doi: 10.1016/s0301-0082(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 2.Wiesenfeld-Hallin Z, Lucas GA, Alster P, Xu X-J, Hokfelt T. Cholecystokinin/opioid interaction. Brain Res. 1999;848:78–89. doi: 10.1016/s0006-8993(99)01978-2. [DOI] [PubMed] [Google Scholar]
- 3.Itoh S, Katsuura G, Maeda Y. Caerulein and cholecystokinin suppress β-endomorphin-induced analgesia in the rat. Eur. J. Pharmacol. 1982;80:421–425. doi: 10.1016/0014-2999(82)90089-9. [DOI] [PubMed] [Google Scholar]
- 4.Faris PL, Komisaruk BP, Watkins LR, Mayer DJ. Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia. Science. 1983;219:310–312. doi: 10.1126/science.6294831. [DOI] [PubMed] [Google Scholar]
- 5.Heinricher MM, Neubert MJ. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J. Neurophysiol. 2004;92:1982–1989. doi: 10.1152/jn.00411.2004. [DOI] [PubMed] [Google Scholar]
- 6.Lu L, Huang M, Liu A, Ma L. Cholecystokinin-B receptor antagonists attenuate morphine dependence and withdrawal in rats. NeuroReport. 2000;11:829–832. doi: 10.1097/00001756-200003200-00034. [DOI] [PubMed] [Google Scholar]
- 7.Suberg SN, Culhane ES, Carstens E, Watkins LR. The potentiation of morphine-induced inhibition of spinal transmission by proglumide, a putative cholecystokinin antagonist. Ann. N. Y. Acad. Sci. 1985;448:660–662. [Google Scholar]
- 8.Watkins LR, Kinscheck IB, Mayer DJ. Potentiation of opiate analgesia and apparent reversal of morphine tolerance by proglumide. Science. 1984;224:395–396. doi: 10.1126/science.6546809. [DOI] [PubMed] [Google Scholar]
- 9.Lee YS, Agnes RS, Davis P, Ma S-W, Lai J, Porreca F, Hruby VJ. Hydrazide Linked Bifunctional Peptides for the Treatment of Pain. In: Chorev M, Sawyer TK, editors. Peptide Revolution: Genomics, Proteomics & Therapeutics. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2004. pp. 615–616. [Google Scholar]
- 10.Hruby VJ, Agnes RS, Davis P, Ma S-W, Lee YS, Vanderah TW, Lai J, Porreca F. Design of novel peptide ligands which have opioid agonist activity and cck antagonist activity for the treatment of pain. Life Sci. 2003;73:699–704. doi: 10.1016/s0024-3205(03)00390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hruby VJ, Fang SN, Kramer TH, Davis P, Parkhurst D, Nikiforovich G, Boteju LW, Slaninova J, Yamamura HI, Burks TF. Analogues of Cholecystokinin26–33 Selective for B-Type CCK Receptors Possess Opioid Receptor Agonist Activity in Vitro and in Vivo: Evidence for Similarities in CCK-B and Opioid Receptor Requirements. In: Hodges RS, Smith JA, editors. Peptides, Chemistry, Sturucture and Biology. Escom; Leiden, The Netherlands: 1993. pp. 669–671. [Google Scholar]
- 12.Agnes RS, Lee YS, Badghisi H, Davis P, Ma S-W, Lai J, Porreca F, Hruby VJ. Structure–activity relationships of bifunctional peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. J. Med. Chem. 2006;49:2868–2875. doi: 10.1021/jm050921q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hruby VJ, Agnes RS, Vagner J, Lee YS, Handl H, Ma S-W, Davis P, Herman DS, Vanderah TW, Yamamura HI, Lai J, Gillies RJ, Porreca F. Utilizing Genomics and Proteomics for Peptide Based Drug Design. Drug Design for Disease Reconsidered. In: Flegel M, Fridkin M, Gilon C, Slaninova J, editors. Peptide 2004. Kenes International; Geneva, Switzerland: 2004. pp. 77–79. [Google Scholar]
- 14.Lee YS, Agnes RS, Badghisi H, Davis P, Ma S-W, Lai J, Porreca F, Hruby VJ. Design and synthesis of novel hydrazide linked bifunctional peptides as δ/μ opioid receptor agonists and CCK-1/CCK-2 receptor antagonists. J. Med. Chem. 2006;49:1773–1780. doi: 10.1021/jm05085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chorev M, Goodman M. A dozen years of retro-inverso peptidomimetics. Acc. Chem. Res. 1993;26:266–273. [Google Scholar]
- 16.Verdini AS, Viscomi GC. Synthesis, resolution, and assignment of configuration of potent hypotensive retro-inverso bradykinin potentiating peptide 5a (BPP5a) anlogues. J. Chem. Soc., Perkin Trans. 1. 1985:697–701. [Google Scholar]
- 17.Fuller WD, Goodman M, Verlander MS. Model for the sweet taste of stereoisomeric retro-inverso and dipeptide amides. J. Am. Chem. Soc. 1987;109:4712–4714. [Google Scholar]
- 18.Verdini AS, Dilvestri S, Becherucci C, Longobardi MG, Parente L, Peppoloni S, Perretti M, Pileri P, Pinori M, Viscomi GC, Nencioni L. Immunostimulation by a partially modified retro-inverso-tuftsin analogue containing Thr1Ψ[NHCO](R,S)Lys2 modification. J. Med. Chem. 1991;34:3372–3379. doi: 10.1021/jm00116a005. [DOI] [PubMed] [Google Scholar]
- 19.Elliott RL, Kopecka H, Bennett MJ, Shue Y-K, Craig R, Lin C-W, Bianchi BR, Miller TR, Witte DG, Stashko MA, Asin KE, Nikkel A, Bednarz L, Nadzan AM. Tetrapeptide CCK agonist: structure–activity studies on modifications at the N-terminus. J. Med. Chem. 1994;37:309–313. doi: 10.1021/jm00028a015. [DOI] [PubMed] [Google Scholar]
- 20.Moledular modeling experiments were performed on MacroModel 8.1 equipped with the Maestro 5.0 graphical interface installed on a Linux RedHat 8.0. Molecular structures were minimized using OPLS-AA force field and Polak–Ribiere conjugate gradient (PRCG). Optimizations were converged to a gradient rmsd of less than 0.05 kJ Å−1 mol−1 or continued until a limit of 50 000 iterations was reached.