

Abstract
Mammalian antimicrobial peptides, including β-defensins, represent an ancient arm of innate immunity designed to directly neutralize invading microbes. Previously, we demonstrated that murine β-defensin 2 (mDF2β) also acted as an endogenous ligand for TLR-4-activating maturation of dendritic cells (DCs). Herein, we report that this TLR-4 -dependent activation leads to induction of an atypical cell death that is unexpectedly exaggerated by the inhibition of caspases. Experiments using APCs with nonfunctional TNF-α or its receptors suggest that this is a NF-κB- and TNF-α-dependent process that does not require TNFR1. We demonstrate that mDF2β triggers a TNFR2-mediated signaling cascade of “self-destruction” through up-regulation of membrane-bound TNF-α and TNFR2. This appears not to be an isolated phenomenon, as human synthetic β-defenisn 3 was also able to activate and kill DCs. We propose that β-defenins may play an important immunoregulatory role as controllers of the natural process of elimination of activated APCs.
Keywords: antimicrobial peptide, DC maturation, immunomodulation
INTRODUCTION
Antimicrobial peptides such as defensins are primarily designed to directly neutralize the invading microbes. However, some of them appear to also have immunomodulatory activities (see review, refs. [1-3]). For example, human α-defensins 1-3 activate production of proinflammatory cytokines, including TNF-α and IL-1, and enhance expression of adhesion molecules such as ICAM-1, CD11b, and CD11c by neutrophils [4-7]. β-Defensins activate mast cell degranulation and release of histamine and PGD2 [8-10], augment expression of CXCL8 and CXCL5 [11, 12], and even stimulate odontoblast differentiation [13]. Furthermore, β-defensins chemoattract immature dendritic cells (iDCs) and other effector cells acting through chemokine receptor CCR6 [14-16]. We have recently reported that mouse β-defensin 2 (mDF2β/MBD2) also acts as an endogenous danger signal to activate maturation of DCs using TLR-4 [17]. Functionally, mDF2β-activated DCs exhibit Th1-polarized responses and produce a number of proinflammatory cytokines, including IL-12, IL-1α, IL-1β, and IL-6 [17].
Activation of pattern recognition receptors (PRRs), such as the TLR, is mostly mediated by foreign factors, including microbial products [18]. TLR-4 uses its cytoplasmic Toll/IL-1R (TIR) domain to recruit a TIR domain-containing adaptor MyD88, the main component responsible for recruitment of other factors and induction of a downstream signaling cascade [18, 19]. As a result, it leads to production of a milieu of proinflammatory cytokines, chemokines, and large quantities of small, antimicrobial peptides, such as defensins, and various pro- and antiapoptotic proteins, including Bcl-XL, TNFR-associated factor 2 (TRAF2), A20, and others [20-22]. Although LPS activates TLR through MyD88-independent and -dependent ways, the latter is essential for production of proinflammatory cytokines, such as TNF-α and IL-12, through activation of NF-κB [19]. The pleiotropic effects of TNF-α are transmitted by two receptors, TNFR1 and -2, and its cytotoxicity is usually associated with TNFR1, which corresponds to type I transmembrane TNFR superfamily proteins that contain a socalled death domain (DD) in the cytoplasmic tail. Activation of the DD-containing receptors can lead to recruitment of intracellular DD-containing adaptors, such as TNFR-associated DD, which activates the caspase cascade, leading to apoptosis [23-25]. In contrast, TNFR2 does not have DD, and it belongs to a group of receptors that contain a TRAF-interacting motif in their cytoplasmic tails and are presumed not to be associated with TNF-α-mediated cytotoxicity.
A serendipitous observation that the murine Langerhans cell (LC)/DC cell line XS52 exhibited distinct responses to mDF2β and LPS led us to detect an abnormally elevated death rate in cells treated with mDF2β. Although mDF2β and LPS were able to induce DC maturation, mDF2β was a superior activator of cell death. This was a NF-κB- and TNF-α-dependent process that manifested in an atypical cell death, as it was exaggerated by inhibition of caspases. We demonstrate that the role of mDF2β was to initiate the process by up-regulating expression of membrane-bound TNF-α and TNFR2 on APCs, thus presumably facilitating their engagement to trigger a signaling cascade of self-destruction. Paradoxically, this TNFR2-dependent process did not require TNFR1. It is tempting to speculate that mDF2β may also play a role in bringing inflammatory responses to an end by promoting the elimination of activated APCs.
MATERIALS AND METHODS
Production of recombinant proteins
Human synthetic human β-defensin 1 (PDF-4337 s) and β-defensin 2 (PDF-4382 s) were used at 20 μg/ml (Peptide International, Inc., Louisville, KY, USA). The synthetic β-defensin 2 peptides (Abcam, Cambridge, MA, USA) were also able to activate APCs but at a weaker but significant efficacy, presumably as a result of their improper folding [17]. To circumvent this, we produced from Escherichia coli in vitro refolded recombinant mDF2β fused with irrelevant protein tags (a nonimmunogenic murine self-tumor antigen, sFv315 or sFv38) [26]. Control proteins consisted of sFv alone (sFv315) or fused with functionally active mDF3β or murine MIP-3α/CCL20 (MIP-3α) or with a naturally inactive murine pro-β-defensin 2 (mproDF2β). All samples were more than 95% pure, and residual endotoxin was removed by repeated purification on Acticlean columns (Sterogene Bioseparations, Inc., Carlsbad, CA, USA). The final endotoxin content of all samples was below 0.5 U per μg protein, as assayed by the Limulus amoebocyte lysate kit (BioWhittaker, Walkersville, MD, USA).
Mammalian cell-derived proteins were produced as fusion with human immunoglobulin constant region Fc (hFc). Briefly, fragments encoding mature sequences for mDF2β or β-defensin, such as molecule EP2c, were cloned in-frame at the N terminus of the IgG1 hFc fragment in the hFc-pcDNA4 plasmid (gift of Dr. Michael Daws, VA Medical Center, San Francisco, CA, USA). mDF2β-hFc and EP2-hFc were purified from conditioned media of human embryo kidney (HEK)293-transfected cells grown in 4% ultra-low Ig FCS (Invitrogen, Carlsbad, CA, USA) using G- or A-protein-coupled resins (Amersham-Pharmacia Biotech, Piscataway, NJ, USA). Purity of purified proteins was more than 95%, as assessed by Coomasie blue-stained PAAG gels and by Western blot hybridization with anti-human Fab antibody-HRP (Jackson ImmunoLabs, West Grove, PA, USA).
Production of DC
DCs were prepared from bone marrow of 4- to 6-month-old mice using published protocols [17]. After 4-5 days of culture, half of the medium was gently removed from the wells and replaced with an equal volume of fusion protein-containing DC medium and incubated for 18 h. Cells were stained using mAb (CD11c-allophycocyanin, B7.2-PE, CD40-FITC, or isotype-matched control mAb, BD Biosciences/PharMingen, San Diego, CA, USA) in buffer with mouse IgG, 25 μg/tube. Samples were analyzed on a FACSCalibur (Becton Dickinson, San Jose, CA, USA) using CellQuest software. DCs generated from various mouse strains retained immature phenotype, and the typical preparation of iDC was, in general, CD11+ (69%), B7.2+ and I-Ab+ (21%), B7.2- and I-Ab+(18%), and CD40+ (27%). Upon maturation, the DCs were CD11c+ (87%), B7.2+ and I-Ab+ (62%), B7.2- and I-Ab+ (3%), and CD40+ (87%).
Human DCs were produced using methods described elsewhere [17] from peripheral blood of healthy donors in accordance with Human Subject Protocol #2003054 by the Health Apheresis Unit and the Clinical Core Laboratory of the National Institute on Aging (NIA; Baltimore, MD, USA).
Cell lines and mice
All mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA), including C57BL/6 wild-type (wt) or mice with nonfunctional TLR-4, C57BL10ScNcr, or TNFR1 (p55) knock-out (KO) gene, B6.129-Tnfrsf1atm1Mak. Mice with both TNFRs disabled were a generous gift of Dr. Mark Mattson (NIA). The HEK293 and RAW 264.7 cells were from the American Type Culture Collection (Manassas, VA, USA). Murine CCR6-expressing HEK293 cells (HEK293/CCR6) were a gift of Dr. Joshua Farber [National Institute of Allergy and Infectious Diseases [NIAID; National Institutes of Health (NIH), Bethesda, MD, USA]. Dr. Akira Takashima (University of Texas, Dallas, TX, USA) kindly provided the XS52 and XS106 cell lines [27]. Immortalized macrophage cell lines from B6.129 mice or mice with nonfuctional/deleted TNF-α, NF-κB, or MyD88 genes were established by infecting primary bone marrow cells with the J2 recombinant retrovirus as described [28, 29].
Activation and cell death of APCs
DCs or macrophages were cultivated in 96-well plates and treated with an equal volume of 2× samples, prepared in DC medium and sterile-filtered through a 13-mm 0.45 μ syringe filter (Whatman, UK). Cells were treated up to 6 days with mDF2β and LPS at 5 μg/ml and 10 ng/ml concentrations, respectively, unless otherwise specified. Specific inhibitors of LPS, polymixin B (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA), anti-mouse TNF-α-neutralizing antibody (AF-410-NA, R&D Systems, Inc., Minneapolis, MN, USA), anti-mouse TNFR2 (p75/CD120b)-neutralizing mAb (BD Biosciences/PharMingen), or isotype-matched control antibodies were used at 10 μg/ml concentrations at the start of mDF2β or control treatments. The experiment was performed in triplicate wells in parallel to assess for cell activation [expression of surface molecules after staining with CD11c-allophycocyanin, B7.2-PE, CD40-FITC, anti-mouse TNF mAb (MP6-XT22), CD120b-PE (TR75-89), or isotype-matched control mAb (BD Biosciences/PharMingen)] and induction of cell death (Annexin-V-Fluos staining kit, Roche Diagnostic Corp., Indianapolis, IN, USA) and then analyzed by FACS.
Effect of signal transduction inhibitors
The effect of signal transduction inhibitors was analyzed by incubating DCs with 5 μg/ml mDF2β or 10 ng/ml LPS alone or in the presence of Z-Val-Ala-Asp-fluoromethylketone (ZVAD-fmk), the control Z-phenyl-alanyl (ZFA)-fmk (50 μM, Sigma-Aldrich), or PD98059 (20 μM), LY294002 (10 μM), SB203580 (2 μM), and PP2 (1 μM, all from Calbiochem) and TPCK (20 μM, Sigma-Aldrich). Control cells were treated with mDF2β or LPS with comparable amounts of DMSO. Inhibition percent = 100 (%) × (data from cells treated with inhibitors/data from cells treated without inhibitors).
The multiprobe RNase protection assay
The multiprobe RNase protection assay was performed according to the manufacturer’s (BD Biosciences/PharMingen) directions with the following modifications: 33P UTP (70-80 μCi/reaction) was used to synthesize the probe, and 0.5-1.0 × 106 cpm was added to each hybridization reaction.
Electron microscopy
Electron microscropy was performed using XS52 cells cultured in 24-well plates and processed in situ for the electron microscopy analysis described previously [30]. Briefly, cells were fixed in 2% glutaraldehyde, followed by 1% osmium tetroxide (Electron Microscopy Sciences, Ft. Washington, PA, USA), dehydrated in a series of graded ethanol, and embedded in epoxy resin (Embed 812, Electron Microscopy Sciences). The resin was thin-sectioned in a parallel direction to the growth of cells using a diamond knife and ultramicrotome. Thin sections were stained in uranyl acetate and lead citrate and then stabilized by carbon evaporation. Thin-sectioned cells were examined and imaged with an Hitachi H7000 electron microscrope (Tokyo, Japan), equipped with a charged coupled device camera (Gatan, Pleasantton, CA, USA).
RESULTS
mDF2β induces activation of APCs
We previously reported that among the murine β-defensins tested, only mDF2β acted as an endogenous ligand of TLR-4, activating maturation of murine bone marrow-derived iDCs [17]. However, mDF2β also activates other types of APCs, including XS52 cells, an iDC/LC cell line (Fig. 1A), the iDC cell line XS106 (data not shown), the macrophage cell line RAW264.7 (Fig. 1B), and immortalized B6/129 macrophages (see Fig. 4C). Cell activation was judged by up-regulation of surface markers, such as CD40 and CD86 costimulatory molecules and MHC class II (Fig. 1B) or by production of proinflammatory cytokines (Fig. 1C and Supplemental Fig. 1, A and B). The synthetic β-defensin 2 peptides were also able to activate APCs but at a weaker efficacy, presumably as a result of their improper folding [17]. Although APCs can be readily activated by contaminations such as LPS, in concordance with our previous report [17], the activity was specifically associated with the mature form of mDF2β. The final endotoxin content (≤0.5 U/μg) of the proteins was at least tenfold lower than the minimum LPS concentration required for activation of APCs. Moreover, the presence of specific inhibitors, such as polymixin B (Fig. 1B, and see Figs. 3C and 4C) and Rhodobacter sphaeroides diphosphoryl lipid A (data not shown [17]), only completely abrogated the LPS but not mDF2β-induced maturation of APCs. Furthermore, the same way produced control proteins, such as sFv315 (Fig. 1), mproDF2β (see Fig. 5B and ref. [17]), or mDF3β (Supplemental Fig. 2, B-D), failed to activate APCs. In addition, mDF2β that was produced from mammalian cells as fusion protein with Ig heavy chain hFc (mDF2β-Fc) was also able to activate maturation of murine bone marrow-derived iDCs (mDF2β-Fc, Supplemental Fig. 1B) and XS52 cells (data not shown). In contrast, a control fusion protein encoding human defensin-like peptide, fused with the same Ig heavy chain Fc, failed to affect these cells (EP2-Fc, Supplemental Fig. 1B).
Fig. 1.
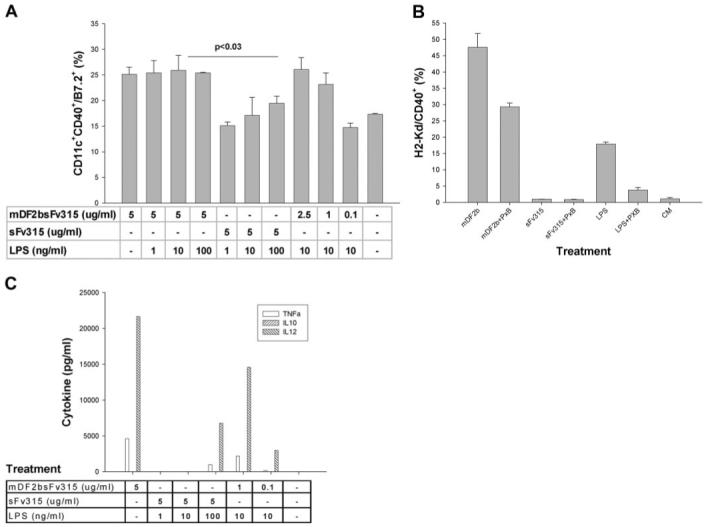
MDF2β activates APCs. (A) Maturation of XS52 cells was judged by up-regulation of CD11c+/CD40+/B7.2 cells after overnight incubation with indicated amounts (μg/ml) of mDF2β fusion protein (mDF2bsFv315). Control cells were treated with 5 μg/ml sFv315 in the presence or absence of indicated amounts of LPS (ng/ml). Comparable results were reproduced at least five times, and the mean of the representative triplicate experiment is shown ± sem. P < 0.03 is for comparison between treatments with mDF2β and sFv315. (B) Surface expression of H2Kd and CD40 on overnight-treated macrophage cell RAW264.7. Cells were treated with mDF2β-sFv315 (mDF2b; 5 μg/ml), sFv315 (5 μg/ml), or LPS (10 ng/ml) in the presence or absence of polymixin B (PxB; 5 μg/ml). Shown are representative data from four independent experiments with similar results ± sem. CM, Conditioned media. (C) ELISA results of cytokine production (pg/ml) in culture media of XS52 cells overnight treated with 5 μg/ml proteins or 10 ng/ml LPS in the presence or absence of 5 μg/ml polymixin B. The mean of the single representative triplicate experiment is shown.
Fig. 4.
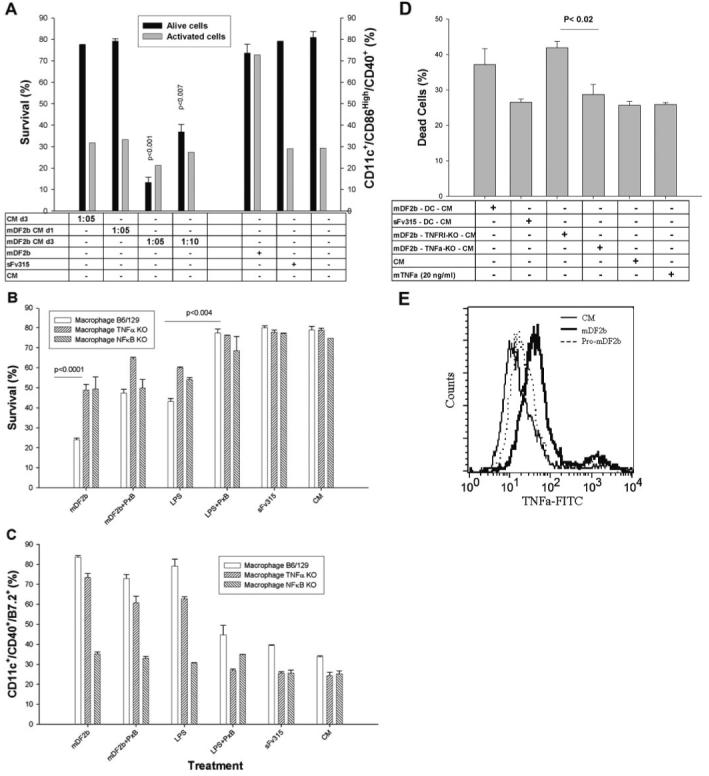
mDF2β induces death of APCs via activation of signaling cascades and secretion of cytotoxic factors. (A) Conditioned media from XS52 cells treated for 3 days with mDF2β (mDF2b CM d3) but not sFv315 are cytotoxic for naïve XS52 cells. In contrast, overnight conditioned media from overnight, mDF2β-treated XS52 cells are not toxic (mDF2b CM d1). Conditioned media are used at 1:5 and 1:10 dilutions. Cell survival (left y-axis) and activation (right y-axis) were measured as in Figure 3. The data were reproduced three times in triplicate experiments. (B and C) Experiments with macrophage cells with nonfunctional NF-κB (NFκB KO) and TNF-α (TNFα KO). mDF2β cannot kill (B) or activate (C) when cells do not express NF-κB. Moreover, cytotoxicity of mDF2β (B) is significantly abrogated when cells do not express TNF-α. The ability to be activated with mDf2β is not affected (C). Used, immortalized macrophage cells from B6/129 mice with KO p50 NF-κB or TNF-α genes, respectively. Controls were immortalized macrophage cells from B6/129 mice. Cell were treated for 3 days and stained in parallel to assess cell survival (B) or cell activation (C), as described in Figure 3. (D) Survival of naïve DCs incubated overnight with conditioned media (at dilutions 1:10) from wt DCs (mDF2b-DC-CM) or with nonfunctional genes TNFR1 (mDF2b-TNFR1-KO-CM) and TNF-α (mDF2b-TNFα-KO-CM) treated with 5 μg/ml mDF2β for 3 days. Control DCs were treated with 20 ng/ml TNF-α, or conditioned media were from sFv315-treated wt DCs (sFv315-DC-CM). Data were compared with effects of CM from XS52 cells treated with mDF2β (mDF2β-XS52-CM). (E) mDF2β (solid line) but not mproDF2β (dotted line) induces expression of cell-anchored TNF-α on the surface of the B6/129 macrophage. The black line is for untreated cells. Cells were stained with FITC-labeled anti-mouse TNF-α mAb after overnight incubation with proteins. No specific staining was detected with FITC-labeled, isotype-matched control antibody (data not shown). All results shown (A-E) were reproduced at least three times, and the mean of the representative triplicate experiment is shown ± sem. P values are for comparisons of between Day 1 and Day 3 conditioned media (A); wt and TNF-α KO or NF-κB KO macrophages treated with mDF2β or LPS (B and C); or between conditioned media of TNFR1 KO and TNF-α KO (D).
Fig. 3.
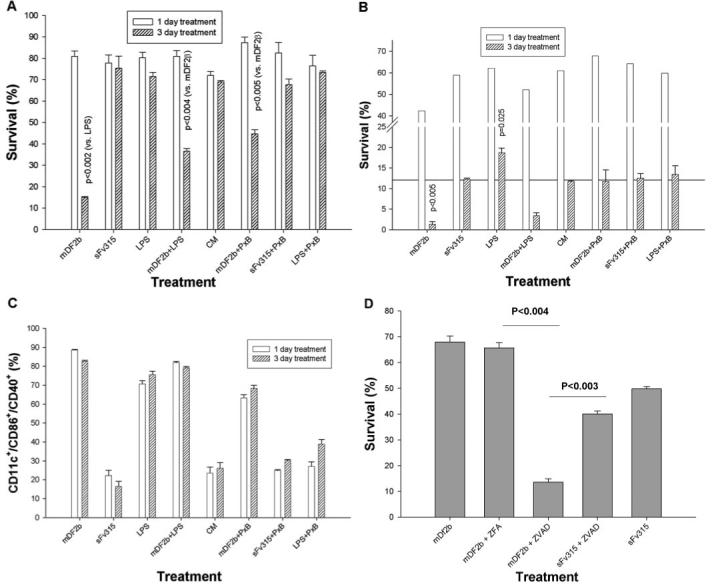
Treatment with mDF2β reduces survival of XS52 cells (A and D) and bone marrow-derived DCs (BMDCs; B and C). Cells, treated for the indicated days with 5 μg/ml mDF2β-sFv38 (mDF2b), sFv315, or 10 ng/ml LPS, with or without 5 μg/ml polymixin B. To assess cell survival, cells were stained with Annexin V/propidium iodide (PI; A, B, and D). In parallel, cells were stained with antibodies for CD11c+/CD40+/CD86+ to assess cell activation (C). Inhibition of pan-caspases using ZVAD exaggerates the mDF2β-induced killing, leading to the cell death after only overnight incubation (D). XS52 cells were incubated overnight with 5 μg/ml mDF2β or sFv315 in the presence or absence of ZVAD-fmk or control ZFA-fmk. All results shown (A-D) were independently reproduced at least three times, and the mean of the representative triplicate experiment is shown ± sem. P values are for comparisons of pooled mDF2β data with LPS, mDF2β + LPS, or mDF2β + polymixin B (A) or between groups indicated by solid lines (D).
Fig. 5.
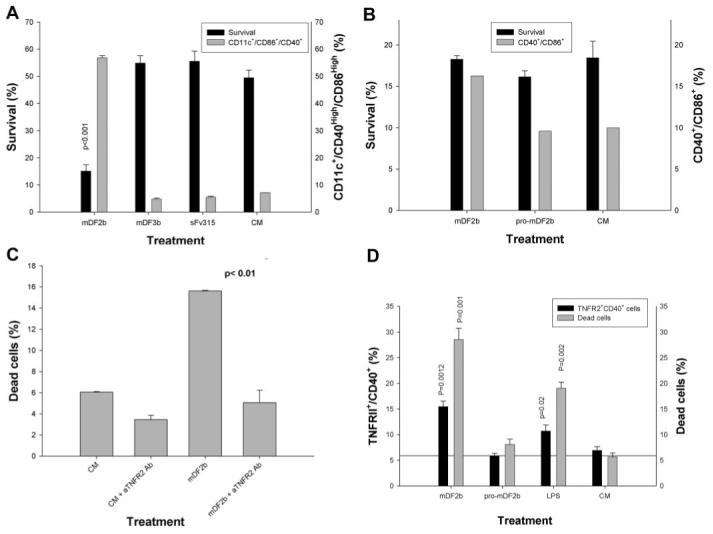
The mDF2β-induced cell death is mediated through TNF-α and its receptor TNFR2 but not TNFR1. BMDCs from nonfunctional TNFR1 (A) or TNFR1 and TNFR2 mice (B) were treated with mDF2β, mDF3β, and sFv315 or mock-treated for 3 days to assess cell survival (solid bars, left y-axis) or DC maturation (shaded bars, right y-axis), as described in Figure 3. (C) The mDF2β-induced death of B6/129 macrophage cells is abrogated in the presence of anti-mouse TNFR2-neutralizing mAb (10 μg/ml) but not isotype-matched IgG (10 μg/ml). The y-axis is in arbitrary proportion of Annexin V/PI-positive cells (dead cells). (D) TNFR2 is up-regulated on the surface of immortalized B6/129 macrophages treated overnight with mDF2β or LPS but not with mproDF2β. The cells were stained for TNFR2 and CD40 (solid bars, left y-axis) and Annexin/PI (shaded bars, right y-axis). All results shown (A-D) were reproduced at least two times, and the mean of the representative triplicate experiment is shown ± sem. P values are for comparisons of viability of cells treated with mDF2β and control protein sFv315 (A) or mproDF2β (D); between two groups as depicted by horizontal lines (C); or between mDF2β or LPS with mproDF2β (D). (B) The survival difference between mDF2β- and mproDF2β-treated groups was not insignificant.
Interestingly, compared with LPS, mDF2β was a stronger inducer of maturation of the immature LC/iDC cell line XS52 (mDF2β, Fig. 1A), which was only partially responsive to LPS at even higher concentrations (100 ng/ml sFv315+LPS, Fig. 1A). Similarly, mDF2β induced higher levels of proinflammatory cytokines, such as IL-12, TNF-α (Fig. 1C), and IL-1α/β (Supplemental Fig. 1A). We do not have an explanation for this differential response, but at least it is not a result of differences in CD14 expression, a coreceptor of LPS, as XS52 cells expressed CD14 (data not shown). Moreover, LPS and mDF2β did not act synergistically (mDF2+LPS, Fig. 1A), and pretreatment with LPS did not affect maturation of XS52 cells in response to subsequent treatment with mDF2β (data not shown) nor did it increase responses upon coincubation with suboptimal doses of mDF2β (0.1 μg/ml). Taken together and in concordance with our previous report [17], these data clearly indicate that mDF2β induces maturation and activation of APCs, including LC/DCs and macrophages.
mDF2β induces caspase-independent cell death
XS52 cells treated for 3 days with mDF2β exhibited significant reduction of cell viability (mDF2β, Fig. 2A) regardless of the presence of polymixin B (mDF2β+PxB, Fig. 2D), ruling out effects of endotoxin. In support, viability of XS52 cells was not affected by the treatment with LPS (Fig. 2B, and LPS+PxB, Fig. 2E). Moreover, the killing required an active mDF2β, as its inactive form mproDF2β failed to kill XS52 cells (Pro-mDF2β, Fig. 2F). β-Defensins have been suggested to have some cytotoxicity for mammalian cells [27], but homologous peptide mDF3β was not able to induce cell death even at higher doses (30 μg/ml, data not shown; also see Fig. 5A). However, paradoxically, no killing of XS52 cells was detected when they were treated with mDF2β for less than 2 days. Electron microscope analysis (Fig. 2G) and the staining with Annexin V and PI (open bars, Fig. 3A) failed to detect any increased abnormalities and cell death when the samples were examined after overnight incubation with mDF2β. In contrast, starting from 3 days of incubation with mDF2β, the cells exhibited characteristic features of apoptotic and necrotic death, such as reduced cell size, condensed nucleus and chromatin, and degraded intracellular organelles, and contained numerous large bubbles and blebs in the cytoplasm (might be dilated rough endoplasmic reticulum) and on the surface (Fig. 2K). They have had drastically reduced cell survival (15±0.38% compared with 75±5.5% viability of the sFv315-treated cells, P<0.002, Fig. 3A). Throughout the manuscript, to diminish any factors that might be caused by the natural variability of the experimental system, each cell viability experiment was analyzed in parallel with its side-by-side-tested cell activation. The data were considered to be true if they were reproduced in at least three independent, triplicate experiments with P values <0.05.
Fig. 2.
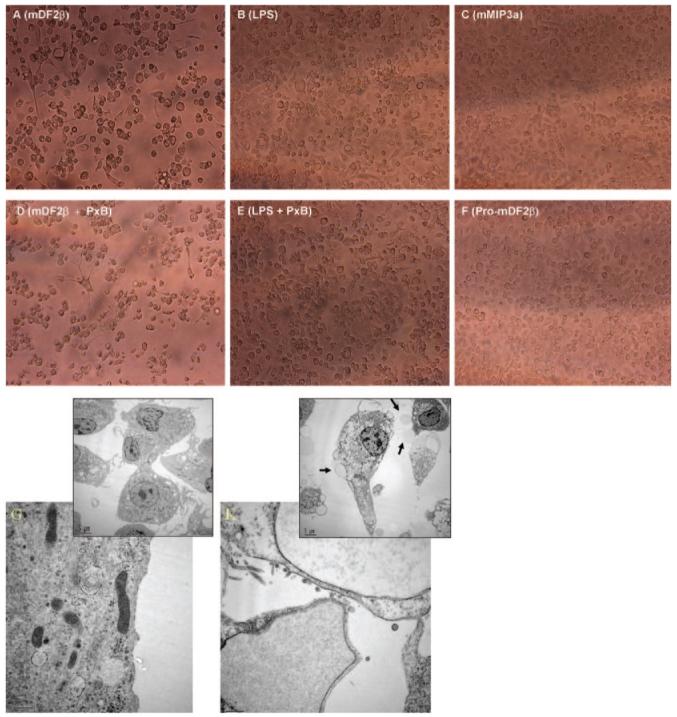
mDF2β kills XS52 cells, as detected by light microscope (A-F) and scattered electron microscopy (G and K). Cells were treated for 3 days with 5 μg/ml mDF2β-sFv315 (A) or 10 ng/ml LPS (B) alone and with 5 μg/ml polymixin B (D and E, respectively). Control treatments were 5 μg/ml murine MIP-3α-sFv315 (mMIP3a; C) and mproDF2β-sFv38 (Pro-mDF2β; F). Overnight treatment with mDF2β did not show signs of abnormalities and death (G), and numerous large blebs (arrows) and loss of intact mitochondria could only be detected after 3 days of treatment (K). The experiment was independently reproduced twice.
Next, to rule out that this is not a result of cell line-related abnormalities, we treated BMDCs and macrophages with mDF2β. Three days of culturing alone without any treatments usually reduces viability of BMDCs (11.7±0.27%, conditioned media, Fig. 3B) at the same extent as BMDCs incubated with control protein (12.3±0.28%, sFv315, Fig. 3B). However, the proportion of viable BMDCs was further and significantly reduced with 3 days of treatment with mDF2β (1.3±0.68%, P≤0.005, Fig. 3B). Unlike XS52 cells, BMDCs can also be killed with LPS. Interestingly, it appears that effects of LPS are biphasic, as cell death was observed at its higher concentrations (>100 ng/ml), and lower concentrations of LPS (10 ng/ml, P<0.025) exhibited prosurvival properties (data not shown). In addition, LPS at lower doses acted as a partial inhibitor of the mDF2β-induced cytotoxicity on both cell types tested, XS52 cells (P<0.004, Fig. 3A) and BMDCs (Fig. 3B).
As mDF2β-treated cells exhibited apoptotic and necrotic features (Fig. 2), and apoptosis depends on activation of caspases, cells were treated with mDF2β in the presence of pan-caspase inhibitor ZVAD-fmk or control ZFA-fmk. Despite a slight toxicity, ZVAD-fmk did not affect mDF2β-induced maturation of APCs (Supplemental Fig. 2B) nor viability of APCs treated with control proteins (XS52, Fig. 3D, and BMDCs, Supplemental Fig. 2A). In contrast and unexpectedly, the presence of ZVAD-fmk drastically reduced viability of mDF2β-treated APCs (mDF2b+ZVAD, Fig. 3D, and Supplemental Fig. 2A). The synergistic effects of ZVAD-fmk and mDF2β were so strong that significant cell death can be detected now after only overnight incubation (68±2.4% vs. 13±5.2%, P<0.004, Fig. 3D), instead of the required 3 days. Thus, these results indicate that mDF2β-induced cell death is a “nonclassical” and caspase-independent apoptosis/necrosis process that is exaggerated by inhibition of caspase-mediated, prosurvival functions.
NF-κB-dependent cell activation is required for mDF2β-induced cytotoxicity
Our data indicate that mDF2β is not a cytotoxic protein for APCs per se, but rather, it activates some kind of signaling process that leads to cell death after 3 days. However, mDF2β is required to initiate the process, presumably by triggering TLR-4 signaling, as DCs from C57BL10ScNcr mice that do not express TLR-4 or immortalized macrophages from MyD88 (-/-) mice, a key accessory factor for TLR-4 signaling, cannot be activated (data not shown; see ref. [17] and Supplemental Fig. 2E) or killed with mDF2β (Table 1). The mDF2β-activated wt APCs secreted a number of potentially cytotoxic, proinflammatory cytokines, including IL-12, TNF-α, and MIP-1α (Supplemental Fig. 2, C-E and Fig. 1C), which may mediate mDF2β-induced cell killing. Thus, to test this, BMDCs were incubated with conditioned media from cells treated with mDF2β overnight (when cells are viable) or for 3 days (when the cell death is detected). Conditioned media from untreated or overnight-treated XS52 cells did not induce any cell death (solid bars, Fig. 4A) or activation (shaded bars, Fig. 4A), indicating that mDF2β was consumed or neutralized after overnight incubations with DCs. In concordance, death of overnight mDF2β-treated cells was not affected by replacement of the cell culture media with one without mDF2β. In contrast, the 3-day conditioned media of XS52 cells treated with mDF2β were cytotoxic for DCs (mDF2b CM d3, solid bars, Fig. 4A). Interestingly, despite this, the 3-day conditioned media were not able to activate maturation of DCs (shaded bars, Fig. 4A). This is not a result of some artifacts of the system, as DCs were readily activated but not killed after overnight treatment with mDF2β (shaded and solid bars, respectively, Fig. 4A). Thus, mDF2β-induced activation and death of APCs are two clearly separated by time processes, and the role of mDF2β is to initiate cell death through production of cytotoxic factor(s).
TABLE 1.
Macrophages from MyD88 KO Mice Are Not Affected by Treatment with mDF2β
| Treatment | MyD88 KO Alive cells (%) | B6/129 MΦ Alive cells (%) | MyD88 KO B7.2+CD40High (%) | B6/129 MΦ B7.2+CD40High (%) |
|---|---|---|---|---|
| mDF2β | 83 ± 1.84 | 49.3 ± 0.8a | 27.7 ± 1.7 | 37.04 |
| LPS 10 ng/ml | 86.2 ± 0.1 | 67.3 ± 0.2 | 22.6 ± 0.8 | 27.91 |
| sFv315 | 80.7 ± 0.5 | 84.3 ± 4.2 | 19.7 ± 0.5 | 16.1 |
| CM | 80.6 ± 0.1 | 80.6 ± 0.3 | 21.2 ± 0.9 | 15.4 |
Immortalized macrophage cells from wt mice B6/129 [B6/129 macrophage (MΦ)] or MyD88 KO mice, treated with mDF2β, sFv315, or LPS, were stained for cell survival and maturation (B7.2+CD40High markers) as in Figure 3.
P < 0.001 is for comparisons of pooled data of mDF2β with sFv315. Data are from two independent, triplicate experiments.
This process involves NF-κB, a key signaling molecule that controls expression of various pro- and antiapoptotic factors [19], as mDF2β failed to kill or activate APCs derived from p50 NF-κB KO mice (Fig. 4, B and C, respectively). Similarly, mDF2β-induced XS52 cell maturation and death were inhibited by more than 60% in the presence of a specific IκB activity (TPCK), and PI-3Ks (LY294002) were able to inhibit (Supplemental Fig. 3, B and C).
mDF2β-induced cell death requires expression of membrane-bound TNF-α
Although any of the proinflammatory cytokines could be cytotoxic, several lines of evidence indicate that TNF-α is the primary culprit. It was readily detectable in conditioned media of the treated cells (Fig. 1C and Supplemental Fig. 2, D and E). Moreover, APCs from mice with nonfunctional TNF-α were resistant to mDF2β-induced killing (TNF-α KO, P<0.0001, Fig. 4B). This is not a result of functional problems of TNF KO APCs, as they were able to be activated (Fig. 4C) and to secrete proinflammatory cytokines (except TNF-α; not shown) when stimulated with mDF2β. Moreover, 3-day conditioned media from mDF2β-treated TNF-α KO cells did not kill XS52 cells (mDF2b-TNFa-KO-CM, Fig. 4D), and conditioned media from wt DCs (mDF2β-XS-CM) or DCs with nonfunctional TNFR1 (mDF2b-TNFR1-KO-CM, Fig. 4D) were cytotoxic. Lastly, mDF2β-induced cell death was partially, but significantly, suppressed by coincubations with TNF-α-neutralizing antibodies (Supplemental Fig. 3A). Thus, these data clearly indicate that TNF-α is not required for activation of APCs, but rather, it is necessary to mediate cellular self-destruction. Paradoxically, soluble recombinant TNF-α alone was not cytotoxic for APCs (Fig. 4D and Supplemental Fig. 3C), indicating the possible involvement of cell-anchored TNF-α, as it was shown to render cells sensitive to TNF-mediated cytotoxicity [31, 32]. In support, significant upregulation of the cell-bound TNF-α was detected on the surface of APCs treated with mDF2β but not with inactive mproDF2β (Fig. 4E). The cytotoxicity of conditioned media can presumably be explained by the presence of TNF-α, which was bound on the membranes of dead cell debris.
TNFR2 but not TNFR1 is responsible for the mDF2β-induced cell death
The cell-bound TNF-α usually acts through TNFR2, and cytotoxic effects of soluble TNF-α are often associated with TNFR1, which contains a so-called DD in its cytoplasmic portion [24, 25]. Paradoxically, mDF2β killed DCs, even if TNFR1 were nonfunctional (P<0.001, solid bars, Fig. 5A), as efficiently as DCs from wt mice. Thus, TNFR1 is not a primary receptor that promotes death in our model. In contrast, mDF2β failed to kill DCs from mice with inactive TNFR2 and TNFR1 receptors (TNFR1/2 KO, solid bars, Fig. 5B). These effects are not a result of functional artifacts that could be associated with loss of TNFRs, as mDF2β activated DCs from both types of mice (TNFR1 KO, shaded bars, Fig. 5A; and TNFR1/2 KO, shaded bars, Fig. 5B). Furthermore, mDF2β-induced death of wt APCs was significantly abrogated in the presence of murine TNFR2-neutralizing mAb (Fig. 5C). Furthermore, mDF2β not only killed APCs (shaded bars, Fig. 5D) but also significantly up-regulated TNFR2 on the surface of the treated APCs (solid bars, Fig. 5D). In contrast, controls, such as an inactive mproDF2β (Fig. 5D) or homologous mDF3β and sFv315 (data not shown), failed to up-regulate TNFR2 or kill APCs. Thus, taken together, our data clearly indicate that mDF2β (and LPS, at a lesser extent) not only activates APCs but also induces expression of cell-bound TNF-α and TNFR2 to eventually trigger self-destruction of the cells.
Human β-defensin 3 also activates human DCs and promotes their death
We have reported that mDF2β was not able to induce human APCs [17], and it appears that it is so far the only β-defensin that specifically mediates activation of murine APCs. Attempts to search for its counterpart led to the finding that human β-defensin 3 might also have similar properties in humans. For example, it activated human DC maturation after overnight incubation (Fig. 6A) and induced their death by Day 3 of the treatment (Fig. 6B). In contrast, homologous, synthetic human β-defensin 1 peptide or control mDF2β failed to activate human DCs (Fig. 6). Thus, these data clearly suggest that our findings about mDFβ may not be an isolated, mouse-specific phenomenon; rather, it is an important, immunoregulatory feature of antimicrobial peptides.
Fig. 6.
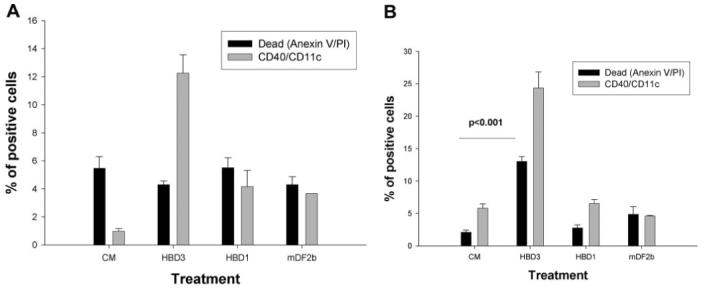
Human β-defensin 3 (HBD3) also induces both maturation death of human DCs. Human PBL-derived iDCs were treated for 1 day (A) and 3 days (B) with human β-defensin 3 or human β-defensin 1 (20 μg/ml) or with 10 μg/ml mDF2β or saline (conditioned media). Cell death (solid bars) and DC maturation (shaded bars) were assessed as described in Figure 3. Shown are data of a triplicate experiment using individual human donor PBL ± sem. The results were reproduced using DCs from two donors. P values are for comparisons of maturation (A) and survival (B) between two groups as depicted by horizontal lines.
DISCUSSION
Current understanding of PRRs is that they are primarily designed to recognize external ligands to promote tailored immune responses against invading pathogens [18, 20, 33]. However, the fact that some of them, specifically TLR-4, can also be activated by endogenous antigens, such as heat shock antigens heat shock protein 60 (HSP)60 and HSP70 [34, 35] and the high-mobility group box 1 alarmin [34-36], questions this simplistic view. Here, expanding our previous observation that mDF2β acts as an endogenous TLR-4 ligand [17], we report that mDF2β also plays a unique role in induction of self-destructive processes of activated APCs. Interestingly, these activities were exerted at significantly lower concentrations of mDF2β and human β-defensin 3 (10-50 nM) than ones shown to be produced at inflammatory sites (≤50 μM) [37]. Overall, our finding is in concordance with the growing list of other activities of β-defensins reported to date, such as production of histamine and PGD2 [10], induction of IL-18 secretion from keratinocytes [38, 39], and control of cell migration acting via chemokine receptor CCR6 [14, 15].
Although β-defensins can have some cytotoxicity for mammalian cells [27], the data indicate that mDF2β is not a cytotoxic protein per se, but rather, it activates a specific signaling process that eventually leads to cell death. This is also clearly separated by time process, as the cell death can be only detected after at least 3 days of treatment with mDF2β. Using pharmacological inhibitors and APCs with KO genes, we have demonstrated that the role of mDF2β is to initiate killing via TLR-4/MyD88-mediated activation of NF-κB. Furthermore, although mDF2β induces production of a number of potentially cytotoxic proinflammatory cytokines (IL-1α/β and IL-12), several lines of evidence clearly indicate that TNF-α is a primary culprit. First, mDF2β failed to kill APCs with nonfunctional TNF-α, despite being able to activate their maturation and induce production of the cytokines. Moreover, the cell death was abrogated in the presence of TNF-neutralizing antibodies. Paradoxically, soluble TNF-α (20 ng/ml) did not have any effects on cell viability. Others observed apoptosis of PC60 hybridoma cells at significantly higher TNF concentrations (>100 ng/ml TNF-α) [23], indicating that higher concentrations of soluble TNF-α may be cytotoxic for APCs. On the other hand, we think that TNF-α presumably needs to be membrane- and cell-anchored to be cytotoxic. The cytotoxicity elicited by mDF2β-treated conditioned media from the wt and TNFR KO does not contradict this possibility, as TNF-α could be present on membranous debris of dead cells. In support, significant cell-anchored TNF-α expression was found on the surface of DCs treated with mDF2β but not with inactive mproDF2β. The biological activity of cell-anchored TNF-α is thought to require TNFR2 [25, 31], which was also up-regulated after mDF2β stimulation. Thus, it is tempting to speculate that this up-regulation of cell-anchored TNF and its receptor TNFR2 would enable their engagement, leading to the transmission of proapoptotic signals. In concordance, mDF2β failed to kill APCs with nonfunctional TNFR1/TNFR2, and its cytotoxicity against APCs with nonfunctional TNFR1 was not affected at all. Thus, these results clearly indicate that activation of TNFR2 alone is essential for the mDF2β-induced cell death, which is in striking contrast with the main cytotoxic pathway induced by soluble TNF-α that requires signaling through TNFR1 [40]. Others also observed similar roles of TNFR2 in the TNF-induced death [23, 32, 41], suggesting that mDF2β may render cells sensitive to TNF-mediated cytotoxicity [31, 32] through up-regulation of the cell-bound TNF-α and TNFR2 and inhibition of NF-κB-dependent antiapoptotic functions of TRAF2 degradation [42]. At present, we cannot rule out that both TNFRs are still required, as for LPS-induced apoptosis of murine macrophages [43]. Although TNFR2 but not TNFR1 was also required for induction of apoptosis of PC60 hybridoma mediated by caspase-8- and Fas-associated, DD-dependent processes [23], the mDF2β-activated cytotoxicity probably uses a different pathway, as the process was paradoxically exaggerated by the pan-caspase inhibitor ZVAD-fmk. Inhibition of caspases was shown to render mice more sensitive to TNF-induced shock, kidney failure, and death [44]. This paradoxical effect of ZVAD-fmk on TNF-mediated cytotoxicity macrophages, DCs, and neutrophils has been attributed to the existence of caspase-dependent, protective feedbacks, for example, on reactive oxygen species formation and phospholipase A2 activation [45-47].
The nature of antimicrobial peptides has been presumably shaped by a never-ending race to control invading pathogens—current and novel ones. Some defensins appear to be evolved to exert a large variety of functions, such as modulation of innate and adaptive immune responses, and may also function to counter suppressive microbial factors designed to evade immunosurveillance. In this respect, it is noteworthy that low amounts of LPS (10 ng/ml, Fig. 3, A and B) and a broad range endotoxin inhibitor polymixin B (a cationic cyclic decapeptide obtained from Bacillus polymixa) reproducibly suppressed (although not completely) cell death induced by mDF2β (see mDF2b+PxB, Fig. 3, A and B), although it did not inhibit its TLR-4/MyD88-dependent DC maturation (Fig. 3C). It is tempting to speculate that polymixin B (as well as low doses of LPS) were counteracting mDF2β by activating an alternative NF-κB-dependent survival pathway, as recently reported, it could activate IκB-α/NF-κB and induce a partial maturation of human DCs [48]. In addition, polymixin B is shown to be cytotoxic to various tumor cells [49] and to sensitize immune cells to extracellular ATP by direct interaction with the pore-forming receptor P2X7 [50]. Similarly, LPS is reported to support survival of proteoclasts independent of TNF-α [51]. Taken together, our data indicate that mDF2β presumably plays an important role in modulation of immune cells, particularly APCs. Besides being a recruiter of APCs, acting via the chemokine receptor CCR6, mDF2β (and higher doses of LPS) elicit two important and sequential responses. First, it directly activates APCs through a TLR-4/MyD88-dependent signaling cascade, leading to the NF-κB-dependent up-regulation of various costimulatory molecules and production of Th1-polarized cytokines and other important immunomodulatory factors [17]. In addition, it leads to up-regulation of the cell-anchored TNF-α and its receptor TNFR2, presumably enabling their association to trigger a caspase-independent, “self-destructive” signaling pathway. Although the biological meaning of our observation remains to be elucidated, we speculate that β-defensins may play an important regulatory role in elimination of “used” APCs to prevent long-lasting, harmful effects of activated DCs and macrophages.
ACKNOWLEDGMENTS
This research was fully supported by the Intramural Research Program of the NIH, NIA. We are grateful to Drs. Dolgor Baatar and Giovanni Almanzar (NIA/NIH) for technical help, Dr. Mark Mattson (NIH/NIA) for the gift of TNFR KO mice, Dr. Joshua Farber (NIAID/NIH) for the gift of CCR6/HEK293 cells, Dr. Akira Takashima (University of Texas) for the gift of XS52 and XS106 LC/DC cell lines, Dr. Michael Daws (VA Medical Center) for the gift of hFc-pcDNA4 plasmid, Karen Madara (Apheresis Unit and the Clinical Core Laboratory, NIA) for providing human blood samples, Dr. Dan Longo (NIA/NIH) for helpful comments and suggestions, and Ana Lustig (NIA/NIH) for critical reading of the manuscript.
REFERENCES
- 1.Biragyn A. Defensins—non-antibiotic use for vaccine development. Curr. Protein Pept. Sci. 2005;6:53–60. doi: 10.2174/1389203053027601. [DOI] [PubMed] [Google Scholar]
- 2.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 3.Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 2004;22:181–215. doi: 10.1146/annurev.immunol.22.012703.104603. [DOI] [PubMed] [Google Scholar]
- 4.Chaly YV, Paleolog EM, Kolesnikova TS, Tikhonov II, Petratchenko EV, Voitenok NN. Neutrophil α-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur. Cytokine Netw. 2000;11:257–266. [PubMed] [Google Scholar]
- 5.van Wetering S, Sterk PJ, Rabe KF, Hiemstra PS. Defensins: key players or bystanders in infection, injury, and repair in the lung? J. Allergy Clin. Immunol. 1999;104:1131–1138. doi: 10.1016/s0091-6749(99)70004-7. [DOI] [PubMed] [Google Scholar]
- 6.Di Nardo A, Vitiello A, Gallo RL. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 2003;170:2274–2278. doi: 10.4049/jimmunol.170.5.2274. [DOI] [PubMed] [Google Scholar]
- 7.Feger F, Varadaradjalou S, Gao Z, Abraham SN, Arock M. The role of mast cells in host defense and their subversion by bacterial pathogens. Trends Immunol. 2002;23:151–158. doi: 10.1016/s1471-4906(01)02156-1. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita T, Saito K. Purification, primary structure, and biological activity of guinea pig neutrophil cationic peptides. Infect. Immun. 1989;57:2405–2409. doi: 10.1128/iai.57.8.2405-2409.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Befus AD, Mowat C, Gilchrist M, Hu J, Solomon S, Bateman A. Neutrophil defensins induce histamine secretion from mast cells: mechanisms of action. J. Immunol. 1999;163:947–953. [PubMed] [Google Scholar]
- 10.Niyonsaba F, Someya A, Hirata M, Ogawa H, Nagaoka I. Evaluation of the effects of peptide antibiotics human β-defensins-1/-2 and LL-37 on histamine release and prostaglandin D(2) production from mast cells. Eur. J. Immunol. 2001;31:1066–1075. doi: 10.1002/1521-4141(200104)31:4<1066::aid-immu1066>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Van Wetering S, Mannesse-Lazeroms SP, Van Sterkenburg MA, Daha MR, Dijkman JH, Hiemstra PS. Effect of defensins on interleukin-8 synthesis in airway epithelial cells. Am. J. Physiol. 1997;272:L888–L896. doi: 10.1152/ajplung.1997.272.5.L888. [DOI] [PubMed] [Google Scholar]
- 12.Van Wetering S, Mannesse-Lazeroms SP, Van Sterkenburg MA, Hiemstra PS. Neutrophil defensins stimulate the release of cytokines by airway epithelial cells: modulation by dexamethasone. Inflamm. Res. 2002;51:8–15. doi: 10.1007/pl00000282. [DOI] [PubMed] [Google Scholar]
- 13.Shiba H, Mouri Y, Komatsuzawa H, Ouhara K, Takeda K, Sugai M, Kinane DF, Kurihara H. Macrophage inflammatory protein-3α and β-defensin-2 stimulate dentin sialophosphoprotein gene expression in human pulp cells. Biochem. Biophys. Res. Commun. 2003;306:867–871. doi: 10.1016/s0006-291x(03)01075-1. [DOI] [PubMed] [Google Scholar]
- 14.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schröder JM, Wang JM, Howard OM, Oppenheim JJ. β-Defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 15.Biragyn A, Surenhu M, Yang D, Ruffini PA, Haines BA, Klyushnenkova E, Oppenheim JJ, Kwak LW. Mediators of innate immunity that target immature, but not mature, dendritic cells induce antitumor immunity when genetically fused with nonimmunogenic tumor antigens. J. Immunol. 2001;167:6644–6653. doi: 10.4049/jimmunol.167.11.6644. [DOI] [PubMed] [Google Scholar]
- 16.Niyonsaba F, Ogawa H, Nagaoka I. Human β-defensin-2 functions as a chemotactic agent for tumor necrosis factor-α-treated human neutrophils. Immunology. 2004;111:273–281. doi: 10.1111/j.0019-2805.2004.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by β-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 18.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 20.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 21.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J. Leukoc. Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 22.Tsutsumi-Ishii Y, Nagaoka I. NF-κ B-mediated transcriptional regulation of human β-defensin-2 gene following lipopolysaccharide stimulation. J. Leukoc. Biol. 2002;71:154–162. [PubMed] [Google Scholar]
- 23.Depuydt B, Van Loo G, Vandenabeele P, Declercq W. Induction of apoptosis by TNF receptor 2 in a T-cell hybridoma is FADD dependent and blocked by caspase-8 inhibitors. J. Cell Sci. 2005;118:497–504. doi: 10.1242/jcs.01640. [DOI] [PubMed] [Google Scholar]
- 24.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-α-induced apoptosis in liver injury. J. Cell. Mol. Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 26.Biragyn A, Tani K, Grimm MC, Weeks SD, Kwak LW. Genetic fusion of chemokines to a self tumor antigen induces protective, T-cell dependent antitumor immunity. Nat. Biotechnol. 1999;17:253–258. doi: 10.1038/6995. [DOI] [PubMed] [Google Scholar]
- 27.Kluver E, Schulz-Maronde S, Scheid S, Meyer B, Forssmann WG, Adermann K. Structure-activity relation of human β-defensin 3: influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry. 2005;44:9804–9816. doi: 10.1021/bi050272k. [DOI] [PubMed] [Google Scholar]
- 28.Blasi E, Radzioch D, Merletti L, Varesio L. Generation of macrophage cell line from fresh bone marrow cells with a myc/raf recombinant retrovirus. Cancer Biochem. Biophys. 1989;10:303–317. [PubMed] [Google Scholar]
- 29.Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by Resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J. Immunol. 2005;175:3339–3346. doi: 10.4049/jimmunol.175.5.3339. [DOI] [PubMed] [Google Scholar]
- 30.Gonda MA, Aaronson SA, Ellmore N, Zeve VH, Nagashima K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning and transmission electron microscopy. J. Natl. Cancer Inst. 1976;56:245–263. doi: 10.1093/jnci/56.2.245. [DOI] [PubMed] [Google Scholar]
- 31.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 32.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 33.Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, Young RA, Lander ES, Hacohen N. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- 34.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll-like receptor-4 complex. J. Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 35.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 36.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 37.Panyutich AV, Panyutich EA, Krapivin VA, Baturevich EA, Ganz T. Plasma defensin concentrations are elevated in patients with septicemia or bacterial meningitis. J. Lab. Clin. Med. 1993;122:202–207. [PubMed] [Google Scholar]
- 38.Sampanthanarak P, Niyonsaba F, Ushio H, Nagaoka I, Ikeda S, Okumura K, Ogawa H. The effect of antibacterial peptide human β-defensin-2 on interleukin-18 secretion by keratinocytes. J. Dermatol. Sci. 2005;37:188–191. doi: 10.1016/j.jdermsci.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 39.Niyonsaba F, Ushio H, Nagaoka I, Okumura K, Ogawa H. The human {β}-defensins (-1, -2, -3, -4) and cathelicidin LL-37 induce IL-18 secretion through p38 and ERK MAPK activation in primary human keratinocytes. J. Immunol. 2005;175:1776–1784. doi: 10.4049/jimmunol.175.3.1776. [DOI] [PubMed] [Google Scholar]
- 40.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erickson SL, de Sauvage FJ, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan KC, Schreiber RD, Goeddel DV, Moore MW. Decreased sensitivity to tumor-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–347. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 43.Xaus J, Comalada M, Valledor AF, Lloberas J, Lopez-Soriano F, Argilés JM, Bogdan C, Celada A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-α. Blood. 2000;95:3823–3831. [PubMed] [Google Scholar]
- 44.Cauwels A, Janssen B, Waeytens A, Cuvelier C, Brouckaert P. Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat. Immunol. 2003;4:387–393. doi: 10.1038/ni914. [DOI] [PubMed] [Google Scholar]
- 45.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SO, Ono K, Han J. Apoptosis by pan-caspase inhibitors in lipopolysaccharide-activated macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L1095–L1105. doi: 10.1152/ajplung.2001.281.5.L1095. [DOI] [PubMed] [Google Scholar]
- 47.Liu CY, Takemasa A, Liles WC, Goodman RB, Jonas M, Rosen H, Chi E, Winn RK, Harlan JM, Chuang PI. Broad-spectrum caspase inhibition paradoxically augments cell death in TNF-α-stimulated neutrophils. Blood. 2003;101:295–304. doi: 10.1182/blood-2001-12-0266. [DOI] [PubMed] [Google Scholar]
- 48.Valentinis B, Bianchi A, Zhou D, Cipponi A, Catalanotti F, Russo V, Traversari C. Direct effects of polymyxin B on human dendritic cells maturation. The role of IκB-α/NF-κB and ERK1/2 pathways and adhesion. J. Biol. Chem. 2005;280:14264–14271. doi: 10.1074/jbc.M410791200. [DOI] [PubMed] [Google Scholar]
- 49.Verstovsek S, Maccubbin DL, Ehrke MJ, Mihich E. Polymyxin B-mediated lysis of tumor cells. Int. Arch. Allergy Immunol. 1993;100:47–52. doi: 10.1159/000236386. [DOI] [PubMed] [Google Scholar]
- 50.Ferrari D, Pizzirani C, Adinolfi E, Forchap S, Sitta B, Turchet L, Falzoni S, Minelli M, Baricordi R, Di Virgilio F. The antibiotic polymyxin B modulates P2X7 receptor function. J. Immunol. 2004;173:4652–4660. doi: 10.4049/jimmunol.173.7.4652. [DOI] [PubMed] [Google Scholar]
- 51.Suda K, Woo JT, Takami M, Sexton PM, Nagai K. Lipopolysaccharide supports survival and fusion of preosteoclasts independent of TNF-α, IL-1, and RANKL. J. Cell. Physiol. 2002;190:101–108. doi: 10.1002/jcp.10041. [DOI] [PubMed] [Google Scholar]