

Abstract
Full-length transient receptor potential (TRP) cation channel TRPC4α and shorter TRPC4β lacking 84 amino acids in the cytosolic C terminus are expressed in smooth muscle and endothelial cells where they regulate membrane potential and Ca2+ influx. In common with other “classical” TRPCs, TRPC4 is activated by Gq/phospholipase C-coupled receptors, but the underlying mechanism remains elusive. Little is also known about any isoform-specific channel regulation. Here we show that TRPC4α but not TRPC4β was strongly inhibited by intracellularly applied phosphatidylinositol 4,5-bisphosphate (PIP2). In contrast, several other phosphoinositides (PI), including PI(3,4)P2, PI(3,5)P2, and PI(3,4,5)P3, had no effect or even potentiated TRPC4α indicating that PIP2 inhibits TRPC4α in a highly selective manner. We show that PIP2 binds to the C terminus of TRPC4α but not that of TRPC4β in vitro. Its inhibitory action was dependent on the association of TRPC4α with actin cytoskeleton as it was prevented by cytochalasin D treatment or by the deletion of the C-terminal PDZ-binding motif (Thr-Thr-Arg-Leu) that links TRPC4 to F-actin through the sodium-hydrogen exchanger regulatory factor and ezrin. PIP2 breakdown appears to be a required step in TRPC4α channel activation as PIP2 depletion alone was insufficient for channel opening, which additionally required Ca2+ and pertussis toxin-sensitive Gi/o proteins. Thus, TRPC4 channels integrate a variety of G-protein-dependent stimuli, including a PIP2/cytoskeleton dependence reminiscent of the TRPC4-like muscarinic agonist-activated cation channels in ileal myocytes.
The seven members of the “classical” family of mammalian TRP4 (TRPC) proteins are closely related to the prototypical Drosophila TRP and TRPL (dTRPs) both structurally and functionally as they are commonly gated by phospholipase C (PLC) activation (1). However, the molecular scenarios of TRPC activation downstream of PLC are diverse and may involve relevant lipids such as diacylglycerol (DAG) in the case of TRPC2, -C3, -C6, and -C7, inositol 1,4,5-trisphosphate formation, and/or Ca2+ store depletion in the case of TRPC1 and -C3 (1). The activation mechanism for the TRPC4/5 subgroup remains most elusive as none of the above appears to be involved (2). Notably, TRPC4/5 are most closely related to dTRPs, and recombinant dTRPL is known to be inhibited by the PLC substrate phosphatidylinositol 4,5-bisphosphate (PIP2), although PIP2 hydrolysis alone cannot account for dTRPL activation (3). Moreover, in trp mutants PIP2 depletion in vivo causes inhibition, rather than potentiation, of the dTRPL channels, which then remain insensitive to light until PIP2 is resynthesized (4). Among mammalian TRPs, several members of the TRPV (V1, V5) and TRPM (M4, M5, M7, and M8) families have recently been shown to be regulated by PIP2 (5, 6), whereas the C termini of several TRPCs (C1, C5, C6, and C7) and TRPV1 bind to anionic lipids directly (7). In TRPC6, this binding, in competition with calmodulin (CaM), removes its inhibitory action and thus facilitates channel activation (7).
Here we tested the hypothesis that PIP2 can regulate TRPC4 channel activity. TRPC4 is widely expressed in different tissues, particularly in brain, vascular endothelium, and gastrointestinal smooth muscles (8). Native functions of TRPC4 include its role in the endothelium-dependent vasorelaxation and control of endothelial permeability (9, 10). Its biophysical properties also resemble muscarinic cation current (mICAT) of the gastrointestinal tract, which is similarly regulated by the PLC system via M3 muscarinic receptors (11). Indeed, evidence is growing that TRPC4 is a critical part of mICAT ((12) and our observations5 showing the lack of mICAT in TRPC4-deficient mouse ileal myocytes).
Two most abundant TRPC4 variants are a “full-length” TRPC4α and shorter TRPC4β (or TRPC4Δ) lacking a stretch of 84 amino acids (781-864 in the case of mouse TRPC4) in the cytosolic C terminus (Δ84AA). The functional implications of their differential tissue expression (13, 14) are intriguing, and they call for further investigations into the differential regulation of TRPC4 isoforms. They form heteromultimers whereby TRPC4α exerts a dominant negative effect ascribed to an autoinhibitory function of the Δ84AA region (15). However, the nature of this inhibition remains unknown.
In this study we found that PIP2 inhibited TRPC4α, but not TRPC4β, suggesting a role of the Δ84AA region in PIP2 interaction with the channel. This was revealed by direct in vitro binding assay. Furthermore, we show that this PIP2-dependent regulation involves cytoskeleton and TRPC4 C-terminal PDZ-interacting domain through which TRPC4/5 are known to associate with NHERF/ezrin-radixin-moesin (ERM)/actin cytoskeleton (16, 17). Importantly, we found that several other phosphoinositides, which bind to the C termini in other TRPCs (7), including PI(3,4)P2, PI(3,5)P2, and PI(3,4,5)P3, as well as highly negatively charged inositol hexaphosphate (IP6) had no effect or even potentiated TRPC4α indicating that the inhibitory action of PIP2 was highly specific and was not simply a negative charge effect. Overall, PIP2 depletion appears to be a required step in the activation of TRPC4α but not TRPC4β. Additional requirement of TRPC4 channel activation include intracellular Ca2+, pertussis toxin (PTX)-sensitive Gi/o proteins, and perhaps other undefined components of PLC signaling. The multifaceted gating requirement suggests that TRPC4 channels are capable of integrating a variety of G-protein-dependent stimuli, including a PIP2/cytoskeleton dependence much like the TRPC4-like muscarinic acetylcholine receptor (mAChR)-activated cation channels in ileal myocytes. Integration of these stimuli might enable fine-tuning of channel activation as required for coordinated functions of the endothelium or smooth muscles in the intestine and the bladder, as well as neural signaling in the nervous system.
EXPERIMENTAL PROCEDURES
Cells and Treatments—HEK293 cells stably expressing mouse TRPC4 (mTRPC4) isoforms were grown under culture conditions as described (16) and seeded in 35-mm dishes 2 days prior to patch clamp recordings. For cells coexpressing M5AChR, the receptor cDNA in pIREShyg2 vector (BD Biosciences) was transfected to the stable TRPC4 cell lines, and transformants were selected and maintained in the culture medium supplemented with 100 μg/ml hygromycin B. Male guinea pigs (300-400 g) were killed by exposure to carbon dioxide (Schedule One of the UK Animals Scientific Procedures Act 1986). Single smooth muscle myocytes from the longitudinal muscle layer of the guinea pig ileum were isolated after collagenase (type 1A, 1 mg ml-1 at 36 °C for 25 min) treatment (18). Cells were treated with cytochalasin D (5 μm) for 2 h at 37 °C, PTX (100 ng/ml) for 16-18 h at 37 °C, and wortmannin (20 or 30 μm) for 10-60 min at 20-23 °C.
Total RNA Isolation and Reverse Transcription-PCR—Purified single guinea pig ileal myocytes were used for protein and RNA analysis. Myenteric ganglia were removed from cell suspension using cell filtration through 80-μm nylon net filters (Millipore). Total RNA was extracted from ileal myocytes using TRIzol reagent (Invitrogen). Reverse transcription was performed using SuperScript II reverse transcriptase (Invitrogen) and random hexamers. PCR was performed using sense primer 5′-CTGCAAATATCTCTGGGAAG-3′ and antisense primer 5′-CTACAATCTTGTGGTCACGTAGT-3′, for predicted products encompassing guinea pig TRPC4 cDNA derived from exons 5 to 9 and the coding sequence for Δ84AA with predicted products of 1422 and 1170 bp for the α and β isoforms, respectively.
Protein Preparation and Western Blot—Microsomal membrane proteins were prepared from wild type mouse brain, TRPC4-deficient mouse brain, and smooth muscle cells isolated from guinea pig ileal longitudinal muscle. Protein samples (150 μg per lane) were separated on 8% SDS-polyacrylamide gels, blotted, and incubated in the presence of the TRPC4 antibody (14). To control protein loading, the filter was stripped and incubated in the presence of a CaVβ2 antibody that recognizes the type 2 β subunit of voltage-activated Ca2+ channels (19). Experiments were repeated twice with identical results.
Coimmunoprecipitation—Cell lysates from untransfected HEK293 cells and stably transfected cell lines were immunoprecipitated using goat anti-actin and goat anti-ezrin antibodies (both were from Santa Cruz Biotechnologies) in a buffer containing 150 mm NaCl, 20 mm Tris-Cl, 1 mm EDTA, and 0.5% Triton X-100, pH 7.4. The precipitated proteins were subjected to SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted using a polyclonal anti-TRPC4 antibody against an N-terminal peptide of TRPC4 (16).
In Vitro PIP2 Binding Assay—The C termini of TRPC4α-(733-974) and TRPC4β-(733-890), as well as Δ84AA-(781-864), were synthesized in vitro as maltose-binding protein fusion proteins using the transcription- and translation-coupled rabbit reticulocyte lysate system (Promega) in the presence of [35S]Met and [35S]Cys. The products were incubated with PIP2-agarose beads (Echelon Inc.) in 260 μl of a binding buffer that contained 120 mm KCl, 20 mm Tris-HCl, pH 7.4, for 1 h at room temperature. Bound proteins were collected by a brief centrifugation at 1,000 × g for 1 min and washed twice with 1 ml of the same binding buffer. Protein separation and exposure to x-ray film are as described previously (16).
Intracellular Ca2+ and Membrane Potential Measurements—Control HEK293 cells and stable cell lines were seeded at 100,000 cells/well in wells of 96-well plates and allowed to grow for 18-24 h. Cells were washed with an extracellular solution and loaded with Fluo4-AM (2 μm) as described (20). [Ca2+]i was measured using a fluid handling integrated fluorescence plate reader, FlexStation (Molecular Devices). Drugs were diluted in extracellular buffer at three times the desired final concentrations and delivered to the sample plate by the integrated robotic 8-channel pipettor at the preprogrammed time points. For membrane potential measurements, the FLIPR membrane potential dye was diluted in the extracellular solution and added to cells following the manufacturer's protocol (Molecular Devices). The excitation/emission wavelengths used for Fluo4 and the membrane potential dye were 494/525 and 530/565 nm, respectively.
Patch Clamp Recordings and Data Analysis—Whole-cell currents were recorded using borosilicate patch pipettes (2-3 megohms) and an Axopatch 200A amplifier (Molecular Devices, Union City, CA) interfaced to Digidata 1322A with the pClamp 9 support. Holding potential was -40 mV. Parallel continuous data acquisition was performed using MiniDigi 1A (Molecular Devices) and AxoScope 9 software. TRPC4 currents in HEK293 cells were activated by carbachol application or, alternatively, by infusing 200 μm GTPγS via pipette. mICAT in ileal myocytes was activated by applying carbachol at a sub-maximally effective concentration of 50 μm.
Identical external and pipette solutions were used for patch clamp recordings in HEK293 and native cells to ensure compatibility of the results. The external solution contained (mm) the following: CsCl 120, glucose 12, HEPES 10, pH 7.4 (CsOH). The pipette solution contained (mm) the following: CsCl 80, MgATP 1, creatine 5, GTPγS 0.2, d-glucose 5, HEPES 10, BAPTA 10, CaCl2 4.6 ([Ca2+]i = 100 nm), pH 7.4 (CsOH). Carbachol-induced currents were recorded with 1 mm GTP instead of GTPγS.
Data were analyzed and plotted using Clampfit 9 (Molecular Devices) and Origin 7 (Microcal, Northampton, MA). Cationic conductance activation curves were obtained from steady-state I-V relationships measured by voltage steps or slow (6-s-long) voltage ramps from 80 to -120 mV (both producing similar I-V curves, e.g. Fig. 1, E and F) and fitted using a modified Boltzmann function as shown in Equation 1,
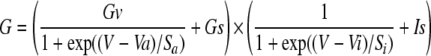 |
(Eq.1) |
where Gv and Gs are voltage-dependent and -independent components, respectively (Gs is typically <15% of Gv); Va and Vi are the potentials of half-maximal activation and inhibition, respectively; sa and si are corresponding slopes; and Is is the noninactivating component. This formalism accounted for both region of negative slope conductance at potentials less than -40 mV and channel inactivation (or block) at potentials more than -20 mV. Fitting was restricted to potentials of <40 mV because at more depolarized potentials the blockade was somewhat relieved, resulting in an overall characteristic doubly rectifying I-V shape or N-shaped conductance curve.
FIGURE 1.

TRPC4 expression and activation in ileal myocytes and HEK293 cells. A, both TRPC4 isoforms are expressed in guinea pig ileal myocytes. Left, reverse transcription-PCR with amplified α-fragment (1422 bp) and β-fragment (1170 bp). Right, Western blot (150 μg of protein per lane). Lane 1, mouse brain wild type; lane 2, mouse brain TRPC4-/-; lane 3, guinea pig ileum. Loading control, CaVβ2. Arrows indicate TRPC4α and -β isoforms. B, Western blot of lysates from control HEK293 cells and stable cell lines expressing TRPC4α or -β. Loading control was actin. The difference between the TRPC4 isoforms is shown schematically in the inset. C and D, carbachol-induced membrane potential and intracellular Ca2+ concentration ([Ca2+]i) changes in cells that expressed TRPC4α. Simultaneous measurement of membrane potential (C) and [Ca2+]i (D) changes in control HEK293 cells (gray lines) and the cell line that stably expressed TRPC4α (black lines). Carbachol (CCh) was added as indicated. Note that the signals represent an average from >100,000 cells. E, typical current response to carbachol application and mICAT I-V relationship in a guinea pig ileal myocyte (marked as GP). For comparison, I-V relationship of the GTPγS-induced current in a TRPC4β-expressing HEK293 cell is shown by the gray line, with superimposed thin black line showing current approximation according to I = G(V - Vrev), where G is determined using Equation 1, and Vrev is the current reversal potential (best fit values were Va =-36.8 mV, and Vi = 0.2 mV, sa =-23.4 mV and si = 8.6 mV). Circles show the I-V relationship measured in the same cell by the voltage-step protocol shown in F. F, superimposed current traces (top) and voltage step protocol used (bottom) illustrating voltage dependence of the GTPγS-induced current in a TRPC4β-expressing HEK293 cell. G, mean normalized GTPγS-induced cation conductance activation curves measured in TRPC4α-(squares, n = 47) and TRPC4β-expressing (circles, n = 37) HEK293 cells. Data points between -120 and 40 mV were approximated by Equation 1. See text for the best fit parameters.
Stock solutions of Me2SO-soluble drugs were made at the following concentrations: 2 mm U-73122 and U-73343, 5 mm cytochalasin D, 10 mm wortmannin. diC8-phosphoinositides (Echelon Research Laboratories, Salt Lake City, UT) were dissolved in deionized and deoxygenated water at 0.4 mm or directly in the pipette solution (if applied at concentrations higher than 20 μm) by a 30-min sonication on ice. All other drugs were from Sigma.
All values are expressed as means ± S.E. A t test (for two groups) or analysis of variance (for multiple comparisons) followed by Dunnett's test to compare all data versus control were used, and differences were considered statistically significant with p < 0.05.
RESULTS
Activation of TRPC4 Is Dependent on Voltage, PLC, and PIP2—TRPC4 channel currents and mICAT in native gastrointestinal myocytes share many biophysical and regulatory properties (2, 11). By examining the expression of TRPC4 in single guinea pig ileal myocytes with myenteric ganglia removed by cell filtration, we found that both TRPC4α and -β isoforms were expressed at the mRNA and protein levels (Fig. 1A). Importantly, Western blotting showed comparable levels of expression of the two isoforms. Thus, in this study we first investigated regulation of TRPC4α and -β channels stably expressed in HEK293 cells. We then tested relevance of our findings under identical conditions for functional regulation of mICAT, which we used as a native counterpart.
We established HEK293 cells stably expressing murine TRPC4α or TRPC4β (Fig. 1B). These isoforms differ in the Δ84AA stretch in the C terminus, which is present in TRPC4α only and which contains two CaM-binding sites as shown schematically in Fig. 1B, bottom inset. In fluorescence membrane potential or intracellular Ca2+ concentration measurements, TRPC4 activity was readily detectable as membrane depolarization in response to carbachol (Fig. 1C) or a more prolonged agonist-induced Ca2+ signal indicative of an enhanced Ca2+ entry (Fig. 1D). This occurred through an endogenous Gq/11-coupled mAChR, most likely the M3 type as an M3 antagonist, p-F-HHSiD, inhibited the responses with a pA2 value of 7.4 (derived from Schild regression analysis; data not shown).
In ileal myocytes, carbachol induces robust current responses (Fig. 1E, top inset), which have been shown to occur through synergistic activation of Gi/o-coupled M2 and Gq/11-coupled M3 mAChR (21-23). This current shows a characteristic doubly rectifying I-V relationship (Fig. 1E, black curve), which is similar to that of TRPC4 current, especially in case of the TRPC4β isoform (e.g. Fig. 1E, gray line), which shows lesser inactivation at positive potentials (see below).
Gating of several TRPVs and TRPMs is efficiently regulated by both membrane potential and phosphoinositides (PIs) (24), whereas voltage-dependent aspects of TRPC gatings are less well appreciated. Although the origin of TRPC4 voltage dependence is not known, the current shows prominent regulation by membrane potential. This was particularly evident in the voltage-step experiments (Fig. 1F). Thus, stepping the membrane potential negatively from the holding potential of -40 mV resulted in an instantaneous current increase because of increased driving force followed by rapid current relaxations to levels that were smaller than the holding current at -40 mV. Tail currents were also prominent as shown in Fig. 1F (magnified inset) suggesting channel deactivation at negative potentials as well as additional activation at positive potentials. In the same cell, an identical I-V relationship could be obtained by applying a 6-s duration voltage ramp (Fig. 1E, compare gray line, ramp protocol, and circles, voltage step protocol). This justified the use of the slow voltage ramps for evaluation of the steady-state voltage-dependent properties of TRPC4. In this and in the majority of other experiments we mainly analyzed TRPC4 currents induced by intracellular GTPγS because carbachol was rather inefficient (see below).
We quantitatively analyzed this voltage dependence by generating TRPC4 conductance curves and fitting them to Equation 1, which can simultaneously approximate either channel activation (between -120 and about -20 mV) and block or activation (positive to about -20 mV) (Fig. 1G). In one well studied case, TRPV6, strong inward rectification occurs because of intracellular Mg2+ channel block (25), but its origin in TRPC4 remains unclear.
The two TRPC4 isoforms differed (p < 0.0001) in the potentials of half-maximal activation (-55.0 ± 1.6 mV for α-isoform and -38.7 ± 1.9 mV for β-isoform) and the extent of channel inactivation at positive potentials (92.5 ± 0.9% for α-isoform and 76.9 ± 3.9% for β-isoform). The slopes for activation (-22.4 ± 0.5 mV versus -21.8 ± 0.6 mV) and inhibition (8.2 ± 0.1 mV versus 8.0 ± 0.2 mV), as well as the potentials of half-maximal inactivation (-9.5 ± 0.7 mV versus -8.0 ± 0.2 mV) were similar (all values are for α-isoform (n = 47) and β-isoforms (n = 37), respectively). As will be shown later, some of PIs tested in this study produced substantial alteration of this apparently intrinsic TRPC4 voltage dependence raising a possibility that TRPC4 similarly to some other TRPs (24) is dually regulated by membrane potential and PIs.
Both heterologously expressed TRPC4 channels and native mICAT are activated downstream of PLC, but the prime activator in the PLC pathway remains unknown in both cases. The main products of PLC activation, inositol 1,4,5-trisphosphate and DAG (as well as DAG metabolites), do not activate TRPC4 or mICAT to any appreciable extent (2, 11). Therefore, in this study, we focused on the role of the PLC substrate, PIP2, and tested the hypothesis that PIP2 may represent a physiologically important brake on TRPC4 activity.
In the fluorescence membrane potential assay, both TRPC4α and -β cell lines displayed similar sensitivity to carbachol with EC50 values of ∼7 μm (Fig. 2, A and B). However, in whole-cell recordings, carbachol failed to induce any significant current in TRPC4β-expressing cells (not shown). By contrast, about two-thirds of TRPC4α-expressing cells responded to carbachol by generating inward currents of variable amplitudes (Fig. 2, C and D). This discrepancy between fluorescence membrane potential assay and the whole-cell recording could be due to the fact that the undisturbed cytosolic environment in the former assay allowed some positive feedback mechanisms, which could be missing in the whole-cell configuration (e.g. limited Ca2+-dependent TRPC4 potentiation because of [Ca2+]i buffering), to amplify channel activation and that small and noisy current responses recorded in voltage clamp experiments were nevertheless sufficient to produce significant membrane depolarization owning to a very high input resistance of these cells.
FIGURE 2.

Enhanced sensitivity of membrane depolarization responses to carbachol in M5AChR coexpressing cells and PIP2 sensitivity of agonist-induced TRPC4α currents. A and B, membrane depolarization elicited by carbachol in stable cell lines that expressed TRPC4α (A) or TRPC4β (B) without (open symbols) or with coexpressed M5AChR (closed symbols) expressed as a relative maximum change in FLIPR membrane potential dye signal (ΔF/F0). Concentration-effect data are means ± S.E. (n = 6) and are fitted by the Hill equation with the corresponding EC50 values indicated near each trace. C, representative cation current responses to carbachol (100 μm) application in control HEK293 cell expressing TRPC4α (top) and in the presence of 20 μm diC8-PIP2 in the pipette solution (bottom). The horizontal bar indicating duration of agonist application applies to both traces. Dashed lines show zero current. D, left, mean current density measured at -60 mV under control conditions (n = 9) and with PIP2 in the pipette solution (n = 12). Right, data points measured in individual cells in control (squares) and with PIP2 (triangles).
To study the effect of PIP2 on agonist-induced TRPC4α current, we included 20 μm dioctanoyl analog of PIP2 (diC8-PIP2), the more water-soluble short form of PIP2, in the recording pipettes. Under these conditions, none of the 12 tested TRPC4α-expressing cells produced any substantial response to carbachol (Fig. 2, C, bottom trace, and B). Mean current density with PIP2 was significantly lower than without PIP2 (p = 0.015). This finding is in support of the above hypothesis that PLC-mediated breakdown of PIP2 is involved in TRPC4 activation. However, the fact that not all control cells were responsive to the agonist prompted us to establish more appropriate recording conditions to study TRPC4 activation.
Intriguingly, overexpression of the Gq-coupled M5AChR failed to improve activation by carbachol of both TRPC4 isoforms in the whole-cell experiments. In fluorescence membrane potential assay, the EC50 to carbachol was reduced 15- and 51-fold for TRPC4α and -β, respectively, without a change in the maximal response (Fig. 2, A and B), showing that the receptor is expressed and can be coupled to TRPC4 activation. The fact that it did not exert an effect on eliciting TRPC4 current in whole-cell recording indicated that PLC activation may not be the sole mechanism of TRPC4 gating (see below), at least under the whole-cell conditions used for recording mICAT. The above mentioned reasons would, of course, make the membrane potential assay more sensitive compared with voltage clamp recording conditions.
To bypass the undefined components required for TRPC4 gating, we used GTPγS, which nonselectively activates all heterotrimeric G-proteins (i.e. bypassing receptors). Infusion of 200 μm GTPγS produced robust long lasting currents (termed ITRPC4) in cells expressing both TRPC4α (n = 14) and -β (n = 7) (43.5 ± 5.8 pA/pF at -40 mV, n = 21), but not in control cells (2.3 ± 0.7 pA/pF, n = 5) (Fig. 3A). The current peaked, on average, at 336 ± 39 s (variations seen in individual cells were from 120 to 720 s) and showed slight desensitization during the next 10 min. Thus, in all subsequent experiments GTPγS was used to activate ITRPC4 as this approach allowed us to investigate both TRPC4 isoforms in the most reliable manner. To account for variations in cell size (CM 8-41 pF, on average 19.0 ± 1.3 pF in TRPC4α- and 16.6 ± 1.1 pF in TRPC4β-expressing cells, n = 19-28), as well as time-dependent variations in culture, we normalized ITRPC4 by CM and performed all test and relevant control measurements by alternating them, usually on the same day. Steady-state I-V relationships were established by applying 6-s voltage ramps from 80 to -120 mV seen as vertical current deflections at 30-s intervals, e.g. Fig. 3, B-E, left panels.
FIGURE 3.

GTPγS activates TRPC4 in a PLC-dependent manner. A, averaged current amplitude induced at -40 mV by 200 μm GTPγS infusion in control nontransfected cells (n = 5) and TRPC4-expressing cells (TRPC4α, n = 14 and TRPC4β, n = 7). Time 0 corresponds to the moment when the whole-cell configuration has been achieved. B-D, both TRPC4α and -β currents were inhibited by U-73122 (2.5 μm) (B and D) but not affected by U-73343 (2.5 μm) (C and E). Corresponding I-V relationships are shown on the right with times of their measurements indicated on the continuous recordings (a and b). Residual currents shown in B and D were similar to those seen in control nontransfected HEK293 cells.
Consistent with previous reports for both ITRPC4 and mICAT (22, 23, 26), the PLC blocker U-73122 strongly suppressed both TRPC4α and -β currents as representative examples in Fig. 3, B and D, show (n = 4; p < 0.02 or better). Its inactive analog U-73343 had no detectable effect (Fig. 3, C and E; n = 4). However, it has more recently been shown that TRPC5, and perhaps TRPC4, can be activated by breaking a disulfide bridge near the selectivity filter (27). Given that the pyrrole-2,5-dione moiety of U-73122 is highly thiol-reactive, whereas the corresponding pyrrolidin-2,5-dione of U-73343 is not (28), it cannot be excluded that U-73122 may act more directly on the TRPC4 channel rather than through PLC.
Isoform-specific Inhibition of TRPC4α but Not TRPC4β by PIP2—Similarly to the above-described experiments with carbachol, diC8-PIP2 (20 μm) was applied via pipette together with GTPγS. This almost completely prevented GTPγS-induced activation of TRPC4α (Fig. 4A; p < 0.02). In controls, infusion of diC8-PIP2 alone had no effect. Notably, the small remaining current showed more deactivation at -120 mV as its I-V relationship at negative potentials became more U-shaped (see amplified I-V curve shown in the inset of Fig. 4C).
FIGURE 4.

PIP2 selectively inhibits TRPC4α isoform and binds to its C terminus. A and B, representative TRPC4α and -β GTPγS-induced current responses in control and with 20 μm diC8-PIP2 added to the pipette solution, as indicated. C and D, mean I-V relationships measured at maximal response to GTPγS in control and in the presence of diC8-PIP2 for the α and β isoforms of TRPC4 (n = 7-11). The inset in C shows I-V relation recorded with diC8-PIP2 on an extended current scale. E, in vitro binding study showing that the C terminus of TRPC4α-(733-974), but not that of TRPC4β-(733-890), binds to PIP2. Δ84AA contains the extra 84 amino acids in TRPC4α-(781-864) and shows weaker binding. PLCδ-PH was used as a positive control. MBP, maltose-binding protein.
In contrast, in TRPC4β-expressing cells diC8-PIP2 had no effect (Fig. 4, B and D), immediately suggesting that the Δ84AA stretch is an important determinant of PIP2 interaction with TRPC4α. We therefore hypothesized that this domain may bind to PIP2, leading to channel closure as depicted by the mechanistic model shown later in Fig. 5D. We tested the interaction of TRPC4α C terminus (Ct, 733-974), TRPC4β-Ct-(733-890), and the Δ84AA region with PIP2. As a positive control, we used the PH domain of PLCδ (PLCδ-PH), which binds PIP2 with a high affinity. The binding of TRPC4α-Ct, but not TRPC4β-Ct, to PIP2 agarose was clearly detectable, although it was much weaker compared with PLCδ-PH (Fig. 4E). Δ84AA fragment showed weak binding compared with TRPC4α-Ct suggesting that this domain may be only a part of a larger PIP2-binding domain. Plausibly, such relatively low PIP2 affinity permits TRPC4α sensing a moderate PIP2 change, as opposed to a high affinity regulatory site which would require a more severe PIP2 depletion.
FIGURE 5.

The association of TRPC4α with actin cytoskeleton is critical for PIP2-dependent channel inhibition. A, disruption of F-actin by cytochalasin D completely prevented TRPC4α inhibition by PIP2 (n = 9). B, in ΔTTRL mutant, the effect of PIP2 was also completely prevented (n = 10). C, coimmunoprecipitation of TRPC4α with ERMs and actin. Cell lysates from untransfected control HEK293 cells and stable lines expressing human TRPC3, C-terminal hemagglutinin-tagged murine TRPC4α (C4α-HAc), wild type TRPC4α, or ΔTTRL mutant were immunoprecipitated using goat anti-actin (middle) and goat anti-ezrin (lower) antibodies and then immunoblotted using a polyclonal anti-TRPC4 antibody. TRPC4α but not C4α-HAc or ΔTTRL was precipitated by the anti-actin and anti-ezrin antibodies. Upper panel is a Western blot of total cell lysates performed using the anti-TRPC4 antibody. D, proposed mechanistic model showing that PIP2 binding to the Δ84AA stretch (and likely to adjacent sites) present only in TRPC4α stabilizes its closed conformation and that interactions of TRPC4α with NHERF/ERM/F-actin are required for PIP2-dependent channel inactivation.
The PIP2-mediated Inhibition Is Dependent on the Association of TRPC4 with Actin Cytoskeleton—PIP2 interacts with cytoskeletal proteins and anchors various signaling molecules to the plasma membrane (29, 30). Proteins of cytoskeletal remodeling represent important targets of PIP2 signaling. On the other hand, TRPC4, along with PLCβ, is associated with actin cytoskeleton through binding to a PDZ domain protein NHERF (also known as EBP50), which in turn interacts with the actin-binding ERM proteins (16, 17). In related TRPC5, deleting the PDZ-binding motif did not have any effect on channel activation or biophysical properties, but overexpressing NHERF introduced a significant delay in current activation of the wild type channel (31).
We therefore asked whether the association with actin cytoskeleton has a role in PIP2 regulation of TRPC4α. First, cells were treated with cytochalasin D, a potent cell-permeable fungal toxin that inhibits actin polymerization and thus disrupts actin microfilaments. This completely prevented diC8-PIP2 inhibition of TRPC4α (Fig. 5A). Second, the last four amino acids of TRPC4α, which constitute the PDZ recognition sequence, were deleted (ΔTTRL mutant); the mutant was stably expressed in HEK293 cells, and the current was studied as for the wild type TRPC4α. The current of ΔTTRL mutant channel showed identical biophysical properties to the wild type channel (compare Figs. 5B and 4C, and also to Ref. 31 in case of TRPC5), but, again, its PIP2 sensitivity was completely lost (Fig. 5B).
These results, taken together with the coimmunoprecipitation results showing that wild type but not C-terminal hemagglutinin-tagged or ΔTTRL mutant channel interacts with actin and ezrin (Fig. 5C), strongly imply that an intact multiprotein TRPC4α·NHERF·ERM·actin complex, in addition to the Δ84AA stretch, is required for PIP2 inhibition of TRPC4α. Thus, our mechanistic model explaining PIP2-mediated channel closure includes both the Δ84AA stretch and these important protein-protein interactions (Fig. 5D).
Despite the complete loss of PIP2 inhibition, no current or little spontaneous current was observed in both cases. Thus, relief from the PIP2-blocked state may not be the sole mechanism of TRPC4α activation (see below).
PIP2 Inhibits mICAT Activation in an F-actin-dependent Manner—Because mICAT appears to be largely mediated by TRPC4, it was interesting to see whether the PIP2 dependence of TRPC4α gating could be interposed between the M3/PLC system and mICAT activation. When carbachol was applied 4-5 min after breakthrough with patch pipettes containing 20 μm diC8-PIP2, no or small mICAT was detectable. Therefore, to follow the time course of the PIP2 effect, carbachol was applied shortly after establishing whole-cell access (<1 min as required for the series resistance compensation and background I-V curve measurement). In this case, mICAT was normally activated by the agonist but then rapidly declined to a small steady-state level (Fig. 6A, compare with control response shown in gray). Similar to TRPC4α, F-actin disruption by cytochalasin D prevented PIP2 inhibition (Fig. 6B, n = 5).
FIGURE 6.

PIP2 inhibits native mICAT current in guinea pig ileal myocytes in an actin cytoskeleton-dependent manner. A, shortly after breakthrough with 20 μm diC8-PIP2-containing patch pipette mICAT could be activated by carbachol but rapidly desensitized (gray trace is a representative control trace). B, in cytochalasin D-treated myocytes (n = 5) desensitization rate was similar or even slower than in control cells (compare with Ref. 18) despite the presence of 20 μm diC8-PIP2 in the pipette solution. C, I-V relationships measured at times indicated in A. D, mean I-V relationships of mICAT in control cells or shortly after breakthrough with diC8-PIP2 in the pipette (n = 15) and at the steady-state inhibition by diC8-PIP2 (n = 9).
Similarly to PIP2 action on TRPC4α, the I-V relationships acquired during diC8-PIP2 action became pronouncedly more U-shaped (Fig. 6, C and D, compare with Fig. 4C, inset) resulting in almost complete current inhibition at -120 mV.
PIP2 Depletion Is Not Sufficient for Full TRPC4α Activation; Some Additional Mechanisms of Channel Gating—Results presented so far revealed that although PIP2 is an important determinant of TRPC4 gating, its depletion may not be the sole mechanism of the channel activation. Indeed, no or little spontaneous current was observed in all cases when PIP2 inhibition was lacking (e.g. TRPC4β isoform, or disrupted cytoskeleton interaction; Fig. 4B and Fig. 5, A and B).
As an additional test, cells were treated with wortmannin, which at micromolar concentrations inhibits phosphatidylinositol 4-kinase thus resulting in PIP2 depletion. For PIP2-activated TRP channels, this usually has profound effects on current activation and desensitization (32-35).
Control unstimulated (e.g. no GTPγS in the pipette) TRPC4α-expressing cells showed small currents characterized by a linear I-V relationship (Fig. 7A). Wortmannin-treated cells showed larger noisy currents with a characteristic TRPC4 I-V relationship (Fig. 7, A and B, squares), yet the amplitude of these responses (e.g. 2.0 ± 0.3 pA at -40 mV; n = 12) was <2% of GTPγS-induced currents. In contrast, TRPC4β-expressing cells did not show any significant increase of the background currents after wortmannin treatment (n = 4, p = 0.15).
FIGURE 7.

Effects of PIP2 depletion and additional mechanisms of TRPC4 activation. A, PIP2 depletion by wortmannin or poly-l-lysine application induced relatively small currents. Top, TRPC4α-expressing cells were recorded in control with or without 0.2-0.3% Me2SO in the external solution and showed small background currents. Middle and bottom traces show currents recorded in wortmannin-treated cells or with 50 μg/ml PLL in the pipette solution, respectively. B, mean I-V relationships measured in wortmannin-treated cells (closed squares, n = 12) and with PLL in the pipette solution (open circles, n = 4). C, activation of both TRPC4 isoforms requires intracellular Ca2+ as evident from the lack of ITRPC4 under conditions of abnormally low intracellular Ca2+ level (Ca2+-free solution with 10 mm BAPTA, n = 6-8). D, PTX treatment abolished current responses both in TRPC4α- and β-expressing cells (n = 8-9).
Poly-l-lysine (PLL) was used as a PIP2 scavenger inhibiting the activity of the PIP2-activated TRPM4 and TRPM8 channels (33, 35). However, applied at 50 μg/ml via pipette (without GTPγS), PLL did not cause any significant TRPC4α current activation even during prolonged, up to 20 min, recordings (Fig. 7A). The I-V relationship recorded with PLL in the pipette solution was again reminiscent of TRPC4 (Fig. 7B, circles), but current density at -40 mV was only 2.4 ± 0.9 pA (n = 4).
Taken together, these data demonstrate that releasing TRPC4α from PIP2 block is necessary but not sufficient for its full activation. On the other hand, intracellular Ca2+ is required for TRPC4 activation (2). Although this mechanism was unlikely to make any significant contribution under conditions of strongly buffered [Ca2+]i, as used in our experiments, we nevertheless studied its implications for TRPC4α and -β isoforms. This was especially interesting because the isoforms differ in two CaM-binding sites located within the Δ84AA sequence (Fig. 1B). For both isoforms, ITRPC4 was abolished by Ca2+-free internal solution with 10 mm BAPTA (Fig. 7C). This is similar to mICAT for which internal Ca2+ is also required (36).
Furthermore, mICAT is blocked by PTX treatment (37, 38), an effect ascribed to Go protein involvement (39, 40). We therefore hypothesized that the failure of carbachol to reliably induce ITRPC4 could be due to the lack of a Go-coupled mAChR in HEK293 cells. Consistent with this, treatment with PTX abolished GTPγS-induced ITRPC4 (Fig. 7D). The effect was again similar for both TRPC4 isoforms. All the above described effects were statistically significant (p < 0.02 or better). Nearly complete current inhibition observed in all cases (Fig. 7, C and D) indicated that functional Gi/o proteins are necessary for ITRPC4 activation. In a separate study, we show that coexpression of M2AChR resulted in activation of large and sustained TRPC4 currents by carbachol under whole-cell conditions for all cells. Further investigation of the pathways linking Gi/o-coupled receptors to TRPC4 activation is undertaken and is beyond the scope of this study.
Other PIs and TRPC4α Gating—Montell and co-workers (7) have recently demonstrated direct binding of several PIs to other members of the TRP superfamily, including TRPC1, - 5, -6, and -7. In some cases this binding displaced inhibitory CaM, but in a related TRPC5 neither the PIs nor IP6 displaced CaM. We therefore asked whether other PIs could also inhibit TRPC4α. The effect of PIP2 on TRP and other ion channels is often interpreted in terms of electrostatic interactions between the negatively charged headgroup of PIP2 and basic amino acid residues, often found in the C termini of various ion channels (6, 41, 42). Eight common PIs exist, which differ in the number and position of phosphorylation of the inositol ring and which can convert into each other through the physiological action of various lipid kinases and phosphatases (41). From the point of view of the role of the negative charge density and specificity of interactions with TRPC4α, of particular interest were the two similarly charged isoforms of PIP2, PI(3,4)P2 and PI(3,5)P2, as well as IP6 with a higher negative charge resulting from the six phosphates.
Taking into account differences in the binding affinities (7), IP6 and PIs were applied (similarly to PIP2) at the following concentrations: IP6 and PI(3,4,5)P3 at 20 μm, PI(3,4)P2 and PI(3,5)P2 at 100 μm, and PI(4)P at 200 μm. Current densities at three different test potentials (-60, -120, and 80 mV) were compared (Fig. 8A). No ITRPC4 inhibition was seen in these experiments, highlighting the specificity of the inhibitory action of PIP2, which thus appears to be not simply a negative charge effect.
FIGURE 8.

Effects of IP6 and other PIs on TRPC4α currents. A, mean current density at the maximal response to GTPγS infusion measured at three different test potentials in control (n = 7) and with IP6 (20 μm, n = 7), PI(3,4,5)P3 (20 μm, n = 7), PI(3,4)P2 (100 μm, n = 6), PI(3,5)P2 (100 μm, n = 5), or PI(4)P (200 μm, n = 5); * and ** indicate p < 0.05 and p < 0.02, respectively. B-D, examples of TRPC4α I-V relationships measured during the time course of GTPγS-induced current development in control (B) and with IP6 (C) or PI(3,5)P2 (D) in the pipette solution. The insets illustrate the time course of the current development (measured at the holding potential of -40 mV) with triangles (plotted at zero current level) indicating moments when I-V relationships shown in the main plots were measured. Scale bars: 5 min in B and C and 10 min in D. Current scale is omitted because complete I-V relationships are shown. E, normalized and averaged activation curves obtained under these various conditions. Compared with control (circles) significant differences were seen in case of IP6 (squares) and PI(3,5)P2 (triangles); with other PIs the curves are shown by the gray lines overlapping with control.
Intriguingly, in two cases significant potentiation of ITRPC4 occurred in a highly specific voltage-dependent manner. Thus, IP6 increased the current at negative but not at positive potentials, whereas with PI(3,5)P2 a much larger current at positive potentials was observed. The time-dependent changes of I-V relationships of these two examples are further illustrated, in comparison with control conditions, in Fig. 8, B-D.
We also examined the normalized and averaged activation curves obtained under these various conditions (Fig. 8E). Again, compared with control (Fig. 8E, circles) significant differences were seen in the case of IP6 (squares) and PI(3,5)P2 (triangles). The possibility that TRPC4 voltage dependence can be altered depending on the lipid environment shows that TRPC4 is a novel example of the increasing number of TRPs showing important connections between lipid-sensing and voltage-dependent regulation (6, 24, 43).
Interestingly, during the initial TRPC4α activation the I-V relationship was similar to control both with IP6 (Fig. 8C) and with PI(3,5)P2 (Fig. 8D), but the above described characteristic changes developed somewhat later. For example, in Fig. 8D larger outward current developed and the I-V relationship at positive potentials became progressively more linear only after the current at negative potentials fully stabilized. This may suggest that interaction of these ligands with TRPC4α can take place only after substantial PIP2 depletion, or some other components required for TRPC4 activation are fully engaged. Clearly, much further work is needed to define PIP2, PI(3,5)P2, and IP6 binding properties and sites of interactions, but at present we can conclude that PI(3,4,5)P3, PI(3,4)P2, and PI(4)P do not functionally regulate ITRPC4 and are thus unlikely to interact with TRPC4.
DISCUSSION
This study adds TRPC4 to the growing list of TRP and other channels (e.g. Kir, KCNQ1, and P/Q-type Ca2+ channels) regulated by PIP2 (41). Although PIP2 and related phosphoinositides exert positive effects on the activity of many TRP channels, such as TRPV5 (32), TRPM4 (35, 44), TRPM5 (45), TRPM7 (46, 47), TRPM8 (33, 34), and TRPC6 (7, 48), it is inhibitory to few others, such as dTRPL (3) and TRPV1 (42, 49). Ironically, the inhibitory effect of PIP2 on TRPV1 has been challenged by more recent studies showing the activating effect of PIP2 on TRPV1 in inside-out patches (50, 51). Therefore, among mammalian TRP channels, the PIP2-induced inhibition appears to be unique for TRPC4α. The only other example is the closely related Drosophila channel, TRPL (3). Three lines of evidence support the notion that relief from PIP2 inhibition is required but not sufficient for full activation of TRPC4α as follows: (i) cytochalasin D treatment (Fig. 5A), (ii) deletion of the PDZ-binding domain (Fig. 5B) both prevented PIP2 dependent TRPC4α inhibition, whereas (iii) TRPC4β was naturally lacking this inhibition (Fig. 4, B and D), yet in all cases no significant spontaneous channel activity was observed. Wortmannin and PLL also caused a minimal TRPC4α activation (Fig. 7, A and B). On the other hand, PIP2 interaction with TRPC4α in terms of channel gating appears to be strong enough to override all other activating stimuli as under conditions of constant intracellular diC8-PIP2 supply carbachol or GTPγS failed to induce any significant current (Fig. 2C and Fig. 4, A and C).
One of the most interesting and important outcomes of this study is uncovering of a previously unsuspected complexity of PIP2 interaction with an ion channel, which, as we show here, requires integrity of a multiprotein complex rather than a single domain. Furthermore, although PIP2 may be more directly associated with ERMs and actin cytoskeleton (29, 30), the intricate connection between PIP2-induced TRPC4α inhibition and the intact F-actin network is mediated solely through the C-terminal binding of the channel to the PDZ domain scaffolding protein NHERF, indicating that this type of regulation is rather specific. These results have interesting functional implications as actin cytoskeleton is a highly dynamic structure, which can undergo a considerable remodeling during receptor activation. This can potentially relieve TRPC4α from PIP2 inhibition even at a constant PIP2 level. Taken together, our results reveal considerable complexity of TRPC4 activation whereby some factors are required (i.e. permissive) in nature (e.g. PIP2 depletion, a certain minimal [Ca2+]ilevel), whereas others are required for channel gating such as activated Gi/o proteins (Fig. 7, C and D).
Thus, TRPC4 channels can integrate a variety of G-protein-dependent stimuli. In some tissues, such multiple coupling can be naturally realized through the expression of differentially coupled receptors for the same agonist, such as acetylcholine in visceral smooth muscles acting simultaneously on M2 and M3 mAChRs. In this study, we showed that both TRPC4 isoforms were about equally expressed in ileal myocytes (Fig. 1A) and that mICAT was inhibited by PIP2, and in a cytoskeleton-dependent manner (Fig. 6). Because TRPC4β is insensitive to PIP2, the strong inhibition of mICAT may seem puzzling, yet this is as expected if both isoforms form native channels, assuming that even one α-subunit in the tetramer is sufficient for its PIP2 inhibition. If both isoforms are expressed equally and combine randomly, the chance that all four subunits are TRPC4β will only be 1 of 16.
Our present results extend novel insights into the common logic of TRP regulation by PIP2 and other PIs and a voltage connection recently provided by Nilius and co-workers (6, 24, 43) to TRPC4 regulation. Testing the effects of IP6 and other PIs, we found that the inhibition was specific to PIP2, thus suggesting that it was not simply a negative charge effect. Moreover, IP6 with its highest surface negative charge selectively potentiated ITRPC4 at negative potentials, whereas one of the isoforms of PIP2, PI(3,4)P2, was without effect and the other one, PI(3,5)P2, potentiated ITRPC4 at positive potentials (Fig. 8).
Interestingly, the Δ84AA segment displayed weaker binding to PIP2 than the entire C terminus of TRPC4α (Ct, 733-974) (Fig. 4E), suggesting that residues outside of the Δ84AA region also contribute to the lipid binding. Together with four amino acids upstream from the beginning of the Δ84AA region is a fragment, 776KSQSEGNGKDKRK788, that confers the (R/K)X(3,11)(R/K)X(R/K)(R/K) (X is any amino acid) role of the TRPM4 PIP2-binding site, believed to represent the “pleckstrin homology” (PH) domain (32). However, of the two maltose-binding fusion proteins that contain this fragment, Ala765-Ala804 and Glu775-Pro801, neither showed detectable binding to PIP2-agarose in the in vitro binding assay (data not shown). Therefore, PIP2 binding most likely involves other regions of the Δ84AA segment, where there are two clusters of positive charges at the C-terminal half. In other TRPs, a modular C-terminal domain containing eight positively charged residues in TRPV1 (42), three positive charges in the conserved TRP box and TRP domain in TRPM8 (34), and the C-terminal PH domain in TRPM4 (35) have been demonstrated to be PIP2-interacting sites. For TRPC6, the PI-binding motif overlaps with the previously identified CaM/inositol 1,4,5-trisphosphate receptor binding (CIRB) site (53), and it contains six positively charged residues with three being important for binding to PIs (7). In case of TRPC4, PIP2 binding could involve discontinuous regions because the Δ84AA segment showed weaker binding than the full C terminus. There are additional positively charged residues, 6 upstream and 14 downstream, from the Δ84AA region, with some clustering on either side. Exactly how the TRPC4α C terminus folds to form the PIP2-binding pocket is unknown. However, based on what is known about PIP2 binding to PH domains, positive charges and some hydrophobic residues are important (54). These residues may not be all contained within the Δ84AA region, and they do not have to be continuous. It is possible that deletion of the Δ84AA segment, such as in the case of TRPC4β, changes the conformation of the whole C terminus which eliminates its ability to bind PIP2.
PH domains are commonly known to bind PIs. These motifs are about 100 residues long with minimal sequence homology but conserved three-dimensional structures of widely variable affinities and selectivities for PIs. For example, PLC-δ1 PH domain binds PI(3,4,5)P3 even somewhat stronger than PIP2 or PI(3,4)P2, whereas pleckstrin PH domain binds PI(3,4)P2 only weakly, and the PH domain of Sos does not bind PI(3,4,5)P3 (52). The Δ84AA region does not confer the main feature of a typical PH domain in that it contains mostly α-helices. However, there exist two CaM-binding sites, implicating that PIP2 may compete with CaM for channel regulation like in the case of TRPC6 (7). On the other hand, the site of CaM and PIs competition for TRPC6 is at the CIRB site, which is more upstream of the Δ84AA region and common to all TRPCs (53) (Fig. 1B). Unlike the CIRB site, the two CaM-binding sites within the Δ84AA regions are unique to TRPC4α with the more C-terminal one being homologous to a similar site in the closely related TRPC5 (55). Given that CaM binding to TRPC5 C terminus was not competed off by PIs (7), competition between CaM and PIP2 for binding to TRPC4α is also unlikely. Therefore, the exact mechanisms and binding sites for the lipids regulating TRPC4 remain to be established, but at least in case of PIP2 it may be narrowed to the Δ84AA stretch and, very likely, adjacent sites in the cytosolic C terminus because the inhibition, as well as the binding, was absent in TRPC4β lacking this sequence (Fig. 4, B and D).
In conclusion, we suggest a novel mechanistic model of TRPC4 gating, whereby TRPC4 interaction via its C-terminal PDZ-binding domain with the adaptor NHERF, ERM proteins, and cortical actin is necessary for keeping the Δ84AA stretch close to the inner surface of the plasma membrane, thus stabilizing its binding with PIP2 (Fig. 5D). According to the model, these multiple interactions stabilize the inactive conformation of TRPC4α, whereas depletion of PIP2 and/or cytoskeleton rearrangement relieve the channel from this inactivation. However, this alone is not sufficient for TRPC4α activation as other factors (e.g. Gi/o and intracellular Ca2+, or additional components of PLC signaling) need to synergize with PIP2 removal for an efficient channel opening.
Acknowledgments
We thank Dr. E. R. Liman for the PLCδ-PH cDNA construct and the PIP2 binding reagent and Dr. S. Stokesberry for help with cell culture.
This work was supported by Queen's University Belfast (to A. V. Z.), the Ministry of Education, Culture, Sports, Science and Technology (Mext), Japan (to S. I., and K. O.), the Deutsche Forschungsgemeinschaft (to V. F.), and National Institutes of Health and American Heart Association grants (to M. X. Z.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TRP, transient receptor potential; dTRP, Drosophila TRP and TRPL; mAChR, muscarinic acetylcholine receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; CaM, calmodulin; IP6, inositol hexaphosphate; PIs, phosphoinositides; NHERF, sodium-hydrogen exchanger regulatory factor; DAG, diacylglycerol; mICAT, muscarinic cation current; ERM, ezrin-radixin-moesin; PTX, pertussis toxin; PLL, poly-l-lysine; CIRB, CaM/inositol 1,4,5-trisphosphate receptor binding; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; GTPγS, guanosine 5′-3-O-(thio)triphosphate; PH, pleckstrin homology; Δ84AA, the 84 amino acids lacking in TRPC4β; Ct, C terminus.
V. Tsvilovskyy, M. Freichel, and V. Flockerzi, unpublished observations.
References
- 1.Ramsey, I. S., Delling, M., and Clapham, D. E. (2006) Annu. Rev. Physiol. 68 619-647 [DOI] [PubMed] [Google Scholar]
- 2.Plant, T. D., and Schaefer, M. (2005) Naunyn-Schmiedeberg's Arch. Pharmacol. 371 266-276 [DOI] [PubMed] [Google Scholar]
- 3.Estacion, M., Sinkins, W. G., and Schilling, W. P. (2001) J. Physiol. (Lond.) 530 1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardie, R. C. (2007) J. Physiol. (Lond.) 578 9-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rohacs, T. (2007) Pfluegers Arch. 453 753-762 [DOI] [PubMed] [Google Scholar]
- 6.Voets, T., and Nilius, B. (2007) J. Physiol. (Lond.) 582 939-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon, Y., Hofmann, T., and Montell, C. (2007) Mol. Cell 25 491-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freichel, M., Vennekens, R., Olausson, J., Stolz, S., Philipp, S., Weissgerber, P., and Flockerzi, V. (2005) J. Physiol. (Lond.) 567 59-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freichel, M., Suh, S. H., Pfeifer, A., Schweig, U., Trost, C., Weissgerber, P., Biel, M., Philipp, S., Freise, D., Droogmans, G., Hofmann, F., Flockerzi, V., and Nilius, B. (2001) Nat. Cell Biol. 3 121-127 [DOI] [PubMed] [Google Scholar]
- 10.Tiruppathi, C., Freichel, M., Vogel, S. M., Paria, B. C., Mehta, D., Flockerzi, V., and Malik, A. B. (2002) Circ. Res. 91 70-76 [DOI] [PubMed] [Google Scholar]
- 11.Zholos, A. V. (2006) Acta Pharmacol. Sin. 27 833-842 [DOI] [PubMed] [Google Scholar]
- 12.Lee, K. P., Jun, J. Y., Chang, I. Y., Suh, S. H., So, I., and Kim, K. W. (2005) Mol. Cells 20 435-441 [PubMed] [Google Scholar]
- 13.Walker, R. L., Hume, J. R., and Horowitz, B. (2001) Am. J. Physiol. 280 C1184-C1192 [DOI] [PubMed] [Google Scholar]
- 14.Flockerzi, V., Jung, C., Aberle, T., Meissner, M., Freichel, M., Philipp, S., Nastainczyk, W., Maurer, P., and Zimmermann, R. (2005) Pfluegers Arch. 451 81-86 [DOI] [PubMed] [Google Scholar]
- 15.Schaefer, M., Plant, T. D., Stresow, N., Albrecht, N., and Schultz, G. (2002) J. Biol. Chem. 277 3752-3759 [DOI] [PubMed] [Google Scholar]
- 16.Tang, Y., Tang, J., Chen, Z., Trost, C., Flockerzi, V., Li, M., Ramesh, V., and Zhu, M. X. (2000) J. Biol. Chem. 275 37559-37564 [DOI] [PubMed] [Google Scholar]
- 17.Mery, L., Strauss, B., Dufour, J. F., Krause, K. H., and Hoth, M. (2002) J. Cell Sci. 115 3497-3508 [DOI] [PubMed] [Google Scholar]
- 18.Zholos, A. V., and Bolton, T. B. (1996) Br. J. Pharmacol. 119 997-1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weissgerber, P., Held, B., Bloch, W., Kaestner, L., Chien, K. R., Fleischmann, B. K., Lipp, P., Flockerzi, V., and Freichel, M. (2006) Circ. Res. 99 749-757 [DOI] [PubMed] [Google Scholar]
- 20.Hu, H. Z., Gu, Q., Wang, C., Colton, C. K., Tang, J., Kinoshita-Kawada, M., Lee, L. Y., Wood, J. D., and Zhu, M. X. (2004) J. Biol. Chem. 279 35741-35748 [DOI] [PubMed] [Google Scholar]
- 21.Zholos, A. V., and Bolton, T. B. (1997) Br. J. Pharmacol. 122 885-893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zholos, A. V., Tsytsyura, Y. D., Gordienko, D. V., Tsvilovskyy, V. V., and Bolton, T. B. (2004) Br. J. Pharmacol. 141 23-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okamoto, H., Unno, T., Arima, D., Suzuki, M., Yan, H. D., Matsuyama, H., Nishimura, M., and Komori, S. (2004) J. Pharmacol. Sci. 95 203-213 [DOI] [PubMed] [Google Scholar]
- 24.Nilius, B., Mahieu, F., Karashima, Y., and Voets, T. (2007) Biochem. Soc. Trans. 35 105-108 [DOI] [PubMed] [Google Scholar]
- 25.Voets, T., Janssens, A., Prenen, J., Droogmans, G., and Nilius, B. (2003) J. Gen. Physiol. 121 245-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer, M., Plant, T. D., Obukhov, A. G., Hofmann, T., Gudermann, T., and Schultz, G. (2000) J. Biol. Chem. 275 17517-17526 [DOI] [PubMed] [Google Scholar]
- 27.Xu, S. Z., Sukumar, P., Zeng, F., Li, J., Jairaman, A., English, A., Naylor, J., Ciurtin, C., Majeed, Y., Milligan, C. J., Bahnasi, Y. M., Al-Shawaf, E., Porter, K. E., Jiang, L. H., Emery, P., Sivaprasadarao, A., and Beech, D. J. (2008) Nature 451 69-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feisst, C., Albert, D., Steinhilber, D., and Werz, O. (2005) Mol. Pharmacol. 67 1751-1757 [DOI] [PubMed] [Google Scholar]
- 29.Bretscher, A., Chambers, D., Nguyen, R., and Reczek, D. (2000) Annu. Rev. Cell Dev. Biol. 16 113-143 [DOI] [PubMed] [Google Scholar]
- 30.McLaughlin, S., Wang, J., Gambhir, A., and Murray, D. (2002) Annu. Rev. Biophys. Biomol. Struct. 31 151-175 [DOI] [PubMed] [Google Scholar]
- 31.Obukhov, A. G., and Nowycky, M. C. (2004) J. Cell. Physiol. 201 227-235 [DOI] [PubMed] [Google Scholar]
- 32.Lee, J., Cha, S. K., Sun, T. J., and Huang, C. L. (2005) J. Gen. Physiol. 126 439-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, B., and Qin, F. (2005) J. Neurosci. 25 1674-1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohacs, T., Lopes, C. M., Michailidis, I., and Logothetis, D. E. (2005) Nat. Neurosci. 8 626-634 [DOI] [PubMed] [Google Scholar]
- 35.Nilius, B., Mahieu, F. F., Prenen, J., Janssens, A., Owsianik, G., Vennekens, R. F., and Voets, T. (2006) EMBO J. 25 467-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue, R., and Isenberg, G. (1990) J. Physiol. (Lond.) 424 73-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue, R., and Isenberg, G. (1990) Am. J. Physiol. 258 C1173-C1178 [DOI] [PubMed] [Google Scholar]
- 38.Komori, S., Kawai, M., Takewaki, T., and Ohashi, H. (1992) J. Physiol. (Lond.) 450 105-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim, Y. C., Kim, S. J., Sim, J. H., Cho, C. H., Juhnn, Y. S., Suh, S. H., So, I., and Kim, K. W. (1998) Pfluegers Arch. 436 494-496 [DOI] [PubMed] [Google Scholar]
- 40.Yan, H. D., Okamoto, H., Unno, T., Tsytsyura, Y. D., Prestwich, S. A., Komori, S., Zholos, A. V., and Bolton, T. B. (2003) Br. J. Pharmacol. 139 605-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suh, B. C., and Hille, B. (2005) Curr. Opin. Neurobiol. 15 370-378 [DOI] [PubMed] [Google Scholar]
- 42.Prescott, E. D., and Julius, D. (2003) Science 300 1284-1288 [DOI] [PubMed] [Google Scholar]
- 43.Rohacs, T., and Nilius, B. (2007) Pfluegers Arch. 455 157-168 [DOI] [PubMed] [Google Scholar]
- 44.Zhang, Z., Okawa, H., Wang, Y., and Liman, E. R. (2005) J. Biol. Chem. 280 39185-39192 [DOI] [PubMed] [Google Scholar]
- 45.Liu, D., and Liman, E. R. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 15160-15165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Runnels, L. W., Yue, L., and Clapham, D. E. (2002) Nat. Cell Biol. 4 329-336 [DOI] [PubMed] [Google Scholar]
- 47.Gwanyanya, A., Sipido, K., Vereecke, J., and Mubagwa, K. (2006) Am. J. Physiol. 291 C627-C635 [DOI] [PubMed] [Google Scholar]
- 48.Tseng, P. H., Lin, H. P., Hu, H., Wang, C., Zhu, M. X., and Chen, C. S. (2004) Biochemistry 43 11701-11708 [DOI] [PubMed] [Google Scholar]
- 49.Chuang, H. H., Prescott, E. D., Kong, H., Shields, S., Jordt, S. E., Basbaum, A. I., Chao, M. V., and Julius, D. (2001) Nature 411 957-962 [DOI] [PubMed] [Google Scholar]
- 50.Stein, A. T., Ufret-Vincenty, C. A., Hua, L., Santana, L. F., and Gordon, S. E. (2006) J. Gen. Physiol. 128 509-522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukacs, V., Thyagarajan, B., Varnai, P., Balla, A., Balla, T., and Rohacs, T. (2007) J. Neurosci. 27 7070-7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rebecchi, M. J., and Scarlata, S. (1998) Annu. Rev. Biophys. Biomol. Struct. 27 503-528 [DOI] [PubMed] [Google Scholar]
- 53.Tang, J., Lin, Y., Zhang, Z., Tikunova, S., Birnbaumer, L., and Zhu, M. X. (2001) J. Biol. Chem. 276 21301-21310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harlan, J. E., Hajduk, P. J., Yoon, H. S., and Fesik, S. W. (1994) Nature 371 168-170 [DOI] [PubMed] [Google Scholar]
- 55.Zhu, M. X. (2005) Pflugers Arch. 451 105-115 [DOI] [PubMed] [Google Scholar]