

SUMMARY
Mice with a malignant hyperthermia mutation (Y522S) in the ryanodine receptor (RyR1) display muscle contractures, rhabdomyolysis, and death in response to elevated environmental temperatures. We demonstrate that this mutation in RyR1 causes Ca2+ leak which drives increases generation of reactive nitrogen species (RNS). Subsequent S-nitrosylation of the mutant RyR1 increases its temperature sensitivity for activation, producing muscle contractures upon exposure to elevated temperatures. The Y522S mutation in humans is associated with central core disease. Many mitochondria in the muscle of heterozygous Y522S mice are swollen and misshapen. The mutant muscle displays decreased force production and increased mitochondrial lipid peroxidation with aging. Chronic treatment with N-acetylcysteine protects against mitochondrial oxidative damage and the decline in force generation. We propose a feed forward cyclic mechanism that increases the temperature sensitivity of RyR1 activation and underlies heat stroke and sudden death. The cycle eventually produces a myopathy with damaged mitochondria.
INTRODUCTION
Heatstroke is a life-threatening illness characterized by elevated core body temperature (>40 °C) leading to central nervous system and multiple organ dysfunction. Exertional or environmental heat stroke (EHS) is triggered by strenuous exercise performed under hot and humid environmental conditions. However, some individuals are much more sensitive to EHS/sudden death, experiencing episodes under relatively mild environmental conditions. Sudden death in response to exertion or high environmental temperature in young, apparently fit, adults such as athletes and military recruits can arise from pre-existing cardiac abnormalities (Thompson, 2007) or the acute onset of organ failure (e.g., heart, kidney, liver) (Bouchama, 2002). However, death during exercise in warm environmental conditions could also arise from organ failure secondary to rhabdomyolysis of skeletal muscle. Consistent with this, exercise induced sudden death has been reported in young healthy males with family histories of Malignant Hyperthermia (MH), a condition that predisposes individuals to rhabdomyolysis (Ellis, 1988; Pamukcoglu, 1988; Ryan, 1997). MH is a life-threatening pharmacogenetic disorder caused by mutations in the skeletal muscle Ca2+ release channel (or ryanodine receptor, RyR1) characterized by episodes of uncontrolled muscle contracture triggered by halogenated anesthetics such as isoflurane or halothane (Jurkat-Rott, 2000; Lichtman, 2006; Treves, 2005).
Many similarities exist between EHS and MH. Two human RyR1 mutations (R401C and R614C) are associated with MH, EHS and exercise-induced rhabdomyolysis (Davis, 2002; Wappler, 2001). MH and EHS share many common pathological features, including rhabdomyolysis, increases in serum creatine kinase, hyperkalemia, tachycardia, metabolic acidosis, and increased muscle production of inflammatory cytokines (Bouchama, 2002; Ducreux, 2004); effects that can trigger kidney failure and cardiac arrhythmias. In addition, patients who have experienced heat stress are more likely to have MH-positive in vitro contracture tests, in which the contractile sensitivity of a muscle biopsy to triggering agents such as caffeine and halothane is enhanced (Bendahan, 2001; Hackl, 1991; Hopkins, 2000).
We (Chelu, 2006) recently created knock-in mice with a mutation (Y522S) in RyR1, which, in humans, is associated with MH, a high incidence of central cores, and type I fiber type predominance (Quane, 1994). Heterozygous mice (RyR1Y522S/wt) are more sensitive to developing skeletal muscle contractures in response to caffeine treatment in vitro and to isoflurane inhalation in vivo, both hallmarks of MH. In addition, heat alone and/or exercise under warm conditions triggers rhabdomyolysis and death in RyR1Y522S/wt mice (Chelu, 2006). Although the mice undergo sustained whole body contractures upon heat exposure, death also frequently occurs in the absence of detectable sustained contractures. The molecular and cellular mechanisms whereby elevated temperatures with or without exercise leads to death of these mice are unknown. Here we demonstrate that enhanced Ca2+ leak from mutant RyR1 Ca2+ release channels increases oxidative/nitrosative stress, leading to S-nitrosylation of RyR1 that further enhances Ca2+ leak and increases susceptibility to heat-induced sudden death.
RESULTS
Heat Sensitivity of RyR1Y522S/wt mice
RyR1Y522S/wt mice exposed to elevated environmental temperatures undergo an MH-like response characterized by rapid rhabdomyolysis and death (Chelu, 2006). Upon exposure to a 41ºC heat challenge, the rectal temperature of anesthetized (nontriggering anesthetic etomidate) RyR1Y522S/wt mice increases more rapidly than in RyR1wt/wt mice (Figure 1A and B), suggesting that the mutant mice display enhanced metabolism and/or heat-induced muscle tension rapidly during heat exposure. Both enhanced metabolism and heat-induced muscle tension are likely to occur since: 1) RyR1Y522S/wt mice exhibit a higher metabolic rate at 32ºC than wildtype mice (Figure 1C) and 2) solei from RyR1Y522S/wt mice display increased basal stress at much lower temperatures than solei from wild type animals (Figure 1D and E). Both enhanced metabolic rate and increased basal stress are likely to increase skeletal muscle production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). To assess the role of ROS and RNS in the response of RyR1Y522S/wt mice to elevated temperatures, we tested the effects of treating mice for 3–5 days with either the antioxidant N-acetylcysteine (NAC, a precursor for glutathione synthesis) or the nitric oxide synthase (NOS) inhibitor, N (Ω)-nitro-L- arginine methyl ester (L-NAME). Both NAC and L-NAME delayed the rapid rise in core temperature in mutant mice (Figure 1B) and decreased the temperature sensitivity of basal tension of isolated solei (Figure 1E). NAC and L-NAME administered together did not improve the temperature response above either agent alone and, therefore, combined treatment was not pursued further. NAC treatment reduces both ROS and RNS and L-NAME prevents both RNS and ROS production by NOS (Clark, 2004; Pou, 1999), suggesting that either or both may be involved in the sensitization of RyR1Y522S/wt mice to temperature.
Figure 1. Effects of warming on RyR1Y522S/wt mice.
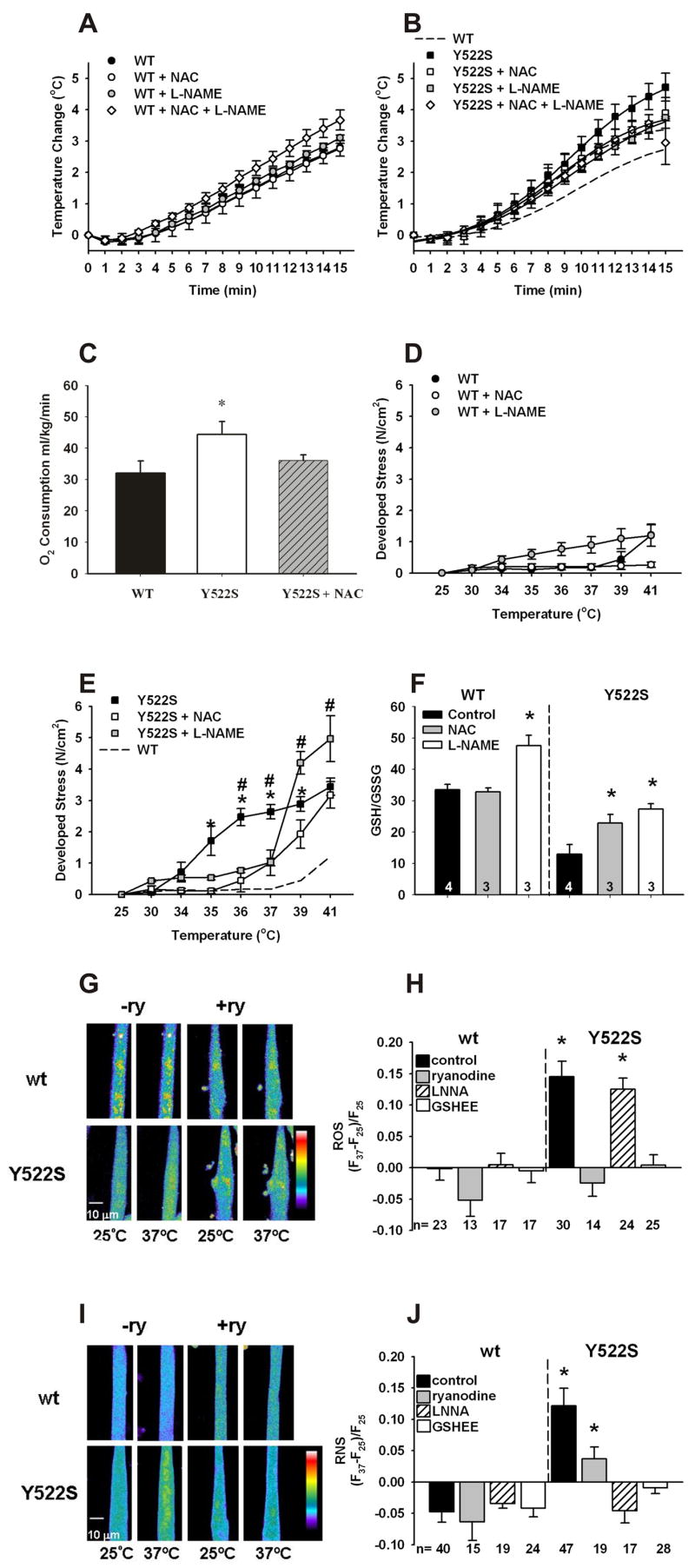
A. Rectal temperatures in RyR1wt/wt mice. Mice were anesthetized with etomidate (i.p.) and placed in an environmental chamber (41°C) and rectal temperatures were measured as a function of time in RyR1wt/wt mice (●, n=10) and in mice treated for 3–5 days with NAC (○, n=6), L-NAME (gray circles, n=4) or combined NAC and L-NAME (◇, n=4). B. Rectal temperatures in RyR1Y522S/wt mice. Similar measurements were made with RyR1Y522S/wt mice (■, n=7) and mice treated with NAC (□, n=4), L-NAME (gray squares, n=4), and combined NAC and L-NAME (◇, n=4). The dashed line represents the WT curve from Figure 1A for comparison. When the curves were compared by F-tests, NAC (p<0.05), L-NAME (p<0.001) and NAC + L-NAME (p<0.001) significantly attenuated the response versus no treatment in the RyR1Y522S/wt mice. C. VO2 is increased at thermoneutral conditions in RyR1Y522S/wt mice. VO2 was measured at 32°C using 4–5 mice in each group *p<0.05 vs. RyR1wt/wt. D. Temperature sensitivity of basal stress in RyR1wt/wtsolei. Solei from RyR1wt/wt (with and without prior treatment with NAC or L-NAME) were isolated, attached to force transducers, and heated progressively from an initial temperature of 25°C to 41°C. n=3–8 mice/group. E. Temperature sensitivity of basal stress in RyR1Y522S/wt solei. Group, temperature, and interaction effects were significant (p<0.0001) when data were analyzed by 2-way ANOVA. *p<0.05 to p<0.001 RyR1Y522S/wt vs RyR1Y522S/wt NAC, #p<0.05 to p<0.01 RyR1Y522S/wt vs. RyR1Y522S/wt L-NAME. For clarity, significant differences between L-NAME and NAC treated groups at 39°C and 41°C are not indicated on the graph. F. GSH/GSSG ratios. The data (n=3–4) were compared between untreated mice of each genotype and the mice receiving the indicated treatment. *p<0.05. G. Fluorescence imaging of myotubes loaded with DCF. All imaging data were obtained from multiple cells across at least three independent myotube preparations. Representative images obtained with DCF-loaded RyR1wt/wt and RyR1Y522S/wt myotubes at 25ºC and 37ºC in the presence and absence of ryanodine (20 μM). H. Changes in DCF fluorescence with temperature. ROS production was detected with DCF in the presence absence of ryanodine (20μM), L-NNA (50 μM) and GSHEE (5mM). I. Fluorescence imaging of myotubes loaded with DAF-AM. Representative images obtained with DAF-AM loaded RyR1wt/wt and RyR1Y522S/wt myotubes at 25ºC and 37ºC in the presence and absence of ryanodine (20 μM). J. Changes in DAF fluorescence with temperature. RNS production was detected with DAF in the presence or absence of ryanodine (20μM), L-NNA (50 μM) and GSHEE (5mM). All data in this figure are shown as mean ± S.E.M.
Oxidative/Nitrosative stress in RyR1Y522S/wt muscle
To assess oxidative/nitrosative stress in the muscle of the RyR1Y522S/wt mice, we measured levels of the primary intracellular antioxidant buffer glutathione (GSH), its oxidized form, glutathione disulfide (GSSG), and the GSH/GSSG ratio in skeletal muscle homogenates from mice not exposed to elevated temperatures. RyR1Y522S/wt muscle exhibits profound basal oxidative stress, with approximately a 50% reduction in both total GSH content (Supplemental Figure 1) and the ratio GSH/GSSG (Figure 1F). A decreased GSH/GGSG ratio could reflect changes in ROS and/or RNS levels in muscle. Pretreatment of RyR1Y522S/wt mice with either NAC or L-NAME partially restores GSH levels and GSH/GSSG (Figure 1F), suggesting that part of the effect is due to RNS production.
To assess ROS and RNS production in myotubes derived from RyR1Y522S/wt mice, we used confocal imaging to compare, respectively, the fluorescence of 5-carboxy-2’,7’-dichlorodihydrofluorescein (DCF) and 4-amino-5-methylamino-2',7'-difluorofluorescein (DAF) at room temperature and 37ºC. In RyR1Y522S/wt myotubes, we found a significant temperature dependent increase in ROS (Figures 1G and H) and RNS (Figures 1I and J). Both increases in RNS and ROS are blocked by treatment with ryanodine and GSH ethyl ester (GSHEE), a membrane permeant form of glutathione (Figures 1G–J). The ability of GSHEE to block temperature dependent increases in DAF fluorescence suggests that GSHEE treatment also reduces RNS. Importantly, L-NNA (N(Ω)-nitro-L-arginine), an inhibitor of nitric oxide synthase (NOS), blocks temperature dependent increases in DAF fluorescence but does not significantly alter ROS production as assessed by DCF fluorescence (Figures 1G–J). While L-NNA inhibits superoxide production by NOS (Clark, 2004), the absence of an inhibition of DCF fluorescence by L-NNA in our experiments suggests that DAF fluorescence in myotubes specifically reflects RNS production. Overall, these data demonstrate a temperature dependent increase in both ROS and RNS production in RyR1Y522S/wt myotubes. Inhibition by ryanodine of the temperature dependent increase in DCF and DAF fluorescence suggests that both ROS and RNS production are stimulated by RyR1-mediated Ca2+ release from the sarcoplasmic reticulum (SR). Two obvious questions arise from these studies: Why is there increased ROS/RNS production with temperature in RyR1Y522S/wt myotubes and how do increased ROS/RNS levels relate to the enhanced temperature sensitivity of these mice?
Temperature dependent increases in resting Ca2+ levels in muscle of RyR1Y522S/wt mice
To define the relationship between the mutation in RyR1 and increased oxidative/nitrosative stress, we determined the effects of temperature and antioxidants on resting Ca2+ levels in myotubes from RyR1Y522S/wt and RyR1wt/wt mice. We previously (Chelu, 2006) found enhanced caffeine sensitivity in RyR1Y522S/wt myotubes in the absence of a change in global resting Ca2+ levels at room temperature. However, the mutation in RyR1 is highly likely to produce local changes in Ca2+ that are rapidly sequestered back into the SR. We re-examined the effects of the Y522S mutation on Ca2+ homeostasis in myotubes (Figure 2A–C) and muscle fibers (Figure 2D) at more physiological temperatures. Cytosolic Ca2+ levels increase with temperature to a much greater extent in RyR1Y522S/wt than in wild type myotubes and muscle fibers (Figure 2A–D). Figure 2C quantifies temperature dependent changes in the intracellular concentration of free Ca2+ (in nM) in RyR1wt/wt and RyR1Y522S/wt myotubes. The lack of a statistically significant difference in cytosolic Ca2+ concentration at 23ºC likely reflects the ability of SERCA to adequately resequester local increases in Ca2+ due to leak from the mutant RyR1 (i.e., compensated leak). Temperature dependent increases in cytosolic Ca2+ are blocked by GSHEE (Figure 2A). Elevations in resting Ca2+ increases are also blocked by both L-NNA (Figure 2B) and L-NAME (Supplemental Figure 2), suggesting that the increases in cytosolic Ca2+ at higher temperatures are primarily attributable to increased RNS rather than ROS since L-NNA only prevents temperature dependent increases in RNS (Figure 1H and J).
Figure 2. Temperature dependent increases cytosolic Ca2+ levels in RyR1Y522S/wt myotubes and solei.

A. Temperature dependent increases in cytosolic Ca2+ levels measured with fura-2. Myotubes loaded with fura-2AM were warmed to the indicated temperatures in the presence and absence of 5mM GSHEE. Values are mean ± SEM for 3 independent cultures for each group: RyR1wt/wt (●, n=27), RyR1wt/wt + GSHEE (○, n=31), RyR1Y522S/wt (■, n=29), RyR1Y522S/wt + GSHEE (□, n=32) (*p < 0.001, one-way ANOVA followed by Scheffe's comparison). B. Effect of L-NNA on temperature dependent increase in resting Ca2+. Myotubes loaded with fura-2AM were warmed to the indicated temperatures in the presence or absence of 50μM L-NNA. RyR1Y522S/wt (■, n= 27) RyR1Y522S/wt + L-NNA (□, n= 31), RyR1wt/wt (●, n= 28) and RyR1wt/wt + L-NNA (○, n= 33). *p<0.05, one way ANOVA followed by Scheffe’s comparison. C. Temperature dependent increases in cytosolic free Ca2+ concentration in RyR1Y522S/wt myotubes. Indo-1-loaded myotubes were warmed to the indicated temperatures and indo-1 ratios were calibrated as described in Methods. *p<0.05 compared to RyR1wt/wt at 23°C. D. Temperature dependent increases in cytosolic Ca2+ in solei fibers. Solei fibers of RyR1Y522S/wt mice were loaded with fura-2 and resting Ca2+ was measured in the presence or absence of ryanodine (20μM). RyR1Y522S/wt (■, n= 17), RyR1Y522S/wt + 20μM ryanodine (□, n=10), RyR1wt/wt (●, n= 12) and RyR1wt/wt + 20μM ryanodine (○, n= 4). *p<0.05, one-way ANOVA. E. Effects of GSHEE on the voltage dependence of L-type Ca2+ currents. Voltage dependence of average (± SEM) peak L-currents at room temperature in RyR1wt/wt myotubes (●), RyR1Y522S/wt myotubes (■), and RyR1Y522S/wt myotubes preincubated with 5 mM GSHEE (□). F. The effects of GSHEE on the voltage dependence of intracellular Ca2+ release. Ca2+ transients at room temperature were measured in RyR1wt/wt myotubes (●), RyR1Y522S/wt myotubes (■), and RyR1Y522S/wt myotubes preincubated with 5 mM GSHEE (□). G. Temperature dependence of L-type Ca2+ currents in RyR1wt/wt and RyR1Y522S/wt myotubes. Voltage dependence of average (± SEM) peak L-currents at 23°C (closed symbols) and 37°C (open symbols) in RyR1wt/wt myotubes (circles) and RyR1Y522S/wt (squares) myotubes. Each dataset was fit (smooth solid lines) using equations described previously (Chelu, 2006) in order to determine Gmax, VG1/2, kG, and Vrev at both 23°C (167 nS/nF, 7.8 mV, 8.6 mV, and 72.6 mV for RyR1wt/wt and 242 nS/nF, 17.3 mV, 7.1 mV, and 73.0 mV for RyR1Y522S/wt, respectively) and 37°C (309 nS/nF, −3.0 mV, 7.0 mV, and 72.5 mV for RyR1wt/wt and 442 nS/nF, −0.6 mV, 4.5 mV, and 78.2 mV for RyR1Y522S/wt, respectively). H. Temperature dependence of intracellular Ca2+ transients in RyR1wt/wt and RyR1Y522S/wt myotubes. Voltage dependence of average (± SEM) peak Ca2+ transient amplitude at 23°C (closed symbols) and 37°C (open symbols) in RyR1wt/wt myotubes (circles) and RyR1Y522S/wt (squares) myotubes Each dataset was fit (smooth solid lines) using equations described previously (Chelu, 2006) in order to determine Fmax, VF1/2, and kF at both 23°C (3.1, −16.8 mV, and 6.2 mV for RyR1wt/wt and 2.7, −23.1 mV, and 7.5 mV for RyR1Y522S/wt, respectively) and 37°C (1.4, −31.8 mV, and 5.7 mV for RyR1wt/wt and 1.0, −35.6 mV, and 8.2 mV for RyR1Y522S/wt, respectively for RyR1Y522S/wt, respectively). All data in this figure are shown as mean ± S.E.M.
We also examined the effects of the mutation on the magnitude, kinetics, and voltage dependence of L-type Ca2+ channel activity and SR Ca2+ release that occurs during excitation-contraction (E–C) coupling. For these experiments, myotubes derived from either RyR1wt/wt or RyR1Y522S/wt mice were incubated for 30 minutes in either vehicle or 5 mM GSHEE prior to perforated voltage clamp recordings at room temperature (Figure 2E, 2F and Supplemental Figure 3A). Additional experiments were conducted to compare E–C coupling at room temperature (23°C) and 37°C (Figure 2G, 2H and Supplemental Figure 3B). Consistent with previous findings (Chelu, 2006), at 23°C L-current density is slightly larger in RyR1Y522S/wt myotubes (Figure 2E and Table 1) and the magnitude of Ca2+ release reduced and its voltage-dependence shifted to more negative voltages in RyR1Y522S/wt myotubes (Figure 2F and Table 1). Preincubation of RyR1Y522S/wt myotubes with 5 mM GSHEE normalized peak L-current density and voltage-gated Ca2+ release without normalizing the voltage dependence of release (Figure 2E, 2F and Table 1). These results are consistent with increased oxidative/nitrosative stress in RyR1Y522S/wt myotubes reducing Ca2+ release during E–C coupling, but not in modulating the sensitivity of RyR1 to activation by the voltage sensor. The temperature dependence of representative L-type Ca2+ currents and intracellular Ca2+ transients in RyR1wt/wt and RyR1Y522S/wt myotubes are shown in Supplemental Figure 3B. At 37°C, L-current magnitude is similarly increased and its kinetics of channel activation and inactivation accelerated in RyR1wt/wt and RyR1Y522S/wt myotubes. In addition, a similar reduction in voltage-gated Ca2+ release and hyperpolarization in the sensitivity of release is also observed in RyR1wt/wt and RyR1Y522S/wt myotubes at 37°C. The average voltage dependence of L-current density and intracellular Ca2+ transients are summarized in Figures 2G and H, respectively. Together, these data indicate that temperature has a similar effect on maximal Ca2+ channel conductance (Gmax is increased about 180%) and voltage sensitivity of release (VF1/2 is left-shifted ~13 mV) in both RyR1wt/wt and RyR1Y522S/wt myotubes indicating that the EC coupling process in RyR1wt/wt and RyR1Y522S/wt myotubes is similarly affected by temperature. Thus, the increased temperature sensitivity observed RyR1Y522S/wt mice represents effects of temperature on the resting RyR1 channel and not the EC coupling process per se.
Table 1. Effects of GSHEE on E-C coupling.
Values represent mean ± SEM for n number of experiments. Parameters for the voltage dependence of Ca2+ conductance and Ca2+ transients were obtained as previously described (Chelu, 2006). Gmax, maximal L-channel conductance; (ΔF/F)max, maximal relative change in fluo-4 fluorescence; Vrev, L-channel reversal potential; VG1/2 and VF1/2, potential at which G and F are half-maximal, respectively; kG and kF, slope factors for IV and FV, respectively.
| Genotype | Gmax (nS/nF) | kG (mV) | VG1/2 (mV) | Vrev (mV) | (ΔF/F)max | kF (mV) | VF1/2 (mV) |
|---|---|---|---|---|---|---|---|
| RyR1wt/wt | 264 ± 13
n=6 |
6.4 ± 0.2
n=6 |
12.8 ± 0.8
n=6 |
76.8 ± 1.7
n=6 |
4.5 ± 0.4
n=6 |
5.4 ± 0.84
n=6 |
−16.9 ± 1.4
n=6 |
| RyR1Y522S/wt | 302 ± 22
n=7 |
4.7 ± 0.4
n=7 |
7.2 ± 0.7*
n=7 |
76.8 ± 2.9
n=7 |
2.9 ± 0.3*
n=7 |
8.5 ± 1.1
n=7 |
−29.0 ± 1.6*
n=7 |
| RyR1Y522S/wt + 5mM GSHEE | 266 ± 13
n=5 |
4.8 ± 0.2
n=5 |
1.3 ± 4.8
n=5 |
70.5 ± 4.6
n=5 |
3.9 ± 0.1
n=5 |
7.6 ± 1.6
n=5 |
−29.2 ± 1.4*
n=5 |
p<0.01 compared to RyR1wt/wt. Data are mean ± S.D.
Effects of ROS/RNS stress on RyR1 channel function in RyR1Y522S/wt mice
Since RyR1 channel activation is increased by both oxidative/nitrosative modifications (Aghdasi, 1997; Marengo, 1998; Oba, 2002; Sun, 2003, 2001b), we compared the extent of RyR1 oxidative/nitrosative modifications using antibodies that are specific for either S-nitrosylation or S-glutathionylation in skeletal muscle SR membranes of RyR1wt/wt and RyR1Y522S/wt mice (Figure 3A–B). To normalize for differences in membrane preparation, we also western blotted with antibodies to RyR1. The use of antibodies to detect S-nitrosylation and S-glutathionylation with western blots was previously validated with the biotin switch in our laboratory (Aracena-Parks, 2006) and by mass spectrometric analyses of peptides labeled using the redox switch technology. We find significant increases in S-nitrosylation and S-glutathionylation of RyR1 in skeletal muscles of RyR1Y522S/wt mice (Figure 3A and B) that are eliminated by DTT. Ascorbic acid (AA) significantly reduces S-nitrosylation (Figure 3B) but not S-glutathionylation of RyR1 (Figure 3C). Oxidation of disulfides is also likely to occur and we have previously detected a single intersubunit disulfide formed between subunits of the RyR1 tetramer (Zhang, 2003). We find no significant difference in the formation of this intersubunit crosslink in RyR1 between wild type and mutant muscle (data not shown). We also compared the extent of S-nitrosylation and S-glutathionylation of RyR1 in mice treated for several days with either NAC or L-NAME. Both NAC and L-NAME prevent the increase in S-nitrosylation of the RyR1Y522S/wt (Figure 3B), while increased S-glutathionylation is decreased by NAC and abolished by L-NAME.
Figure 3. Redox modifications of RyR1 and functional consequences.

A. Redox modifications of RyR1. Representative blots obtained with 3 independent microsomal preparations were obtained from RyR1wt/wt (top) and RyR1Y522S/wt (bottom) mice. Density of the bands corresponding to the redox modifications of RyR1 were obtained under control conditions (middle) or in the presence of either ascorbic acid (left) or DTT (right). B. Fluorescence signals for S-nitrosylation normalized to the Coomassie stain of each band. Data (mean ± SD, n=3–5) are presented as the ratio to untreated microsomes. *p<0.05 compared to control. C. Fluorescence signals for S-glutathionylation normalized to the Coomassie stain of each band. Data (mean ± SD, n=3–5) are presented as the ratio to untreated microsomes. *p<0.05 compared to control. D–F. Equilibrium [3H]ryanodine binding. Scatchard plot analysis (see Supplemental Figure 4) determination of KD values for [3H]ryanodine binding to microsomes from RyR1wt/wt and RyR1Y522S/wt muscle (D). [3H]ryanodine binding was titrated at different Ca2+concentrations to calculate EC50 (E) and IC50 (F) values from traces as those shown in Supplemental Figure 5. *p<0.05 compared to RyR1wt/wt or untreated controls. G. Temperature dependence of the association kinetics of [3H]ryanodine binding. Microsomes from untreated mice were pre-incubated in vitro with buffer (untreated), AA or DTT as in (A). [3H]ryanodine binding was assessed at different time points (1–90min) and kobs values (mean ± SD) were determined from 3–4 independent experiments. Statistical significance for all panels was obtained by two-way ANOVA. *p<0.05 compared to RyR1wt/wt or untreated controls. H. Rate of Ca2+ efflux from SR vesicles. Ca2+-induced Ca2+ release in the presence of 1mM free ATP and 9–10 μM free Ca2+ was measured using stopped-flow spectrofluorometry. Ca2+ release was measured in RyR1wt/wt and RyR1Y522S/wt vesicles using extravesicular Calcium Green-5N under control conditions or following treatment with AA. Release rate constant values (k) were obtained by peak differential analysis of fluorescence data (representative traces shown in Supplemental Figure 7). All data in this figure are shown as mean ± S.E.M.
Ryanodine binds preferentially to the RyR1 open state and is widely used to assess channel activity (Chu, 1990). We compared [3H]ryanodine binding to membranes derived from RyR1Y522S/wt and RyR1wt/wt mice (Figures 3D–H and Supplemental Figures 4–6). The apparent KD for [3H]ryanodine binding is greatly decreased in SR membranes from RyR1Y522S/wt compared to RyR1wt/wt mice, and this difference is eliminated by DTT (Figure 3D and Supplemental Figure 4). Enhanced caffeine sensitivity, arising from an increased affinity of the activating site on RyR1 for Ca2+ (Pessah, 1987), is an inherent property of the mutant channel (Chelu, 2006). Consistent with this, RyR1 from RyR1Y522S/wt mice exhibits increased affinity for the Ca2+ activation site and this increase in affinity is maintained in the presence of DTT (Figure 3E and Supplemental Figure 5). Thus, the Y522S mutation alters the intrinsic sensitivity of the channel to activators (e.g. caffeine, Ca2+, voltage sensor), while the nitrosative modifications alter the sensitivity of the channel to temperature with little or no additional effect on its sensitivity to activators. The modifications also increase the IC50 for Ca2+ inhibition of [3H]ryanodine binding to membranes from the muscle of RyR1Y522S/wt mice (Figure 3F and Supplemental Figure 5), suggesting that the redox modified mutant channel remains open at Ca2+ concentrations that normally close the channel, thus contributing to increased Ca2+ leak.
The previously described binding assays were performed at room temperature. RyR1 is, however, not stable for extended periods of time at higher temperatures (Carroll, 1991), making equilibrium binding studies at physiologic temperatures difficult. To circumvent this problem, we assessed the rate of association of [3H]ryanodine to skeletal muscle membranes from RyR1Y522S/wt and RyR1wt/wt mice at different temperatures and in the presence or absence of either DTT or AA. Representative association curves are shown in Supplemental Figure 6 and the kobs values are shown in Figure 3G. [3H]ryanodine associates much more rapidly to muscle membranes from RyR1Y522S/wt mice than from RyR1wt/wt mice at 37°C. This difference is eliminated by DTT and significantly reduced by AA. Since AA does not significantly alter S-glutathionylation but reverses S-nitrosylation (Figure 3A–C), these findings indicate that S-nitrosylation enhances the temperature sensitivity of RyR1. Our data further suggest that Ca2+ leak from the SR, arising from the Y522S mutation, increases RNS production that leads to subsequent S-nitrosylation of RyR1, which, in turn, further enhances Ca2+ leak and increases RyR1 sensitivity to activation by temperature. This results in a vicious feed-forward cycle in RyR1Y522S/wt mice whereby Ca2+ leak increases RNS production and RNS production in turn potentiates increased Ca2+ leak at permissive temperatures.
To further demonstrate that the temperature dependent effect on Ca2+ levels arises from S-nitrosylation of RyR1, we measured the rates of Ca2+ efflux from SR vesicles from RyR1Y522S/wt and RyR1wt/wt mice using stopped flow and Ca2+ Green 5N (Donoso, 2000), in the presence and absence of AA (Figure 3H). Supplemental Figure 7 shows representative curves using SR vesicles from RyR1Y522S/wt and RyR1wt/wt membranes in the presence or absence of AA. Consistent with increased activity of the mutant channel, we found that the rate of Ca2+-induced Ca2+ release (in the presence of 1mM free ATP and 10μM free Ca2+) from RyR1Y522S/wt microsomes at 37°C was increased compared to RyR1wt/wt (Supplemental Figure 7). The observed rate constants for Ca2+ efflux are shown in Figure 3G. The rate of Ca2+ efflux is significantly greater for RyR1Y522S/wt membranes at 37ºC compared to that of RyR1wt/wt membranes and this difference is eliminated by AA, indicating that the increased rate is due to RyR1 S-nitrosylation. Ryanodine (100μM) completely blocks efflux in all conditions (data not shown).
Effects of the Y522S mutation on mitochondrial structure and muscle function
The human Y522S mutation is associated with a myopathy characterized by central cores devoid of mitochondria. Prolonged Ca2+ leak combined with increased ROS/RNS production, such as that observed in RyR1Y522S/wt mice, is likely to impact the structure and function of closely apposed mitochondria, which could lead to altered muscle function. Using tetramethyl rhodamine ethylester (TMRE) to assess mitochondrial membrane potential, we found that TMRE fluorescence increases with temperature in RyR1Y522S/wt myotubes, but not in RyR1wt/wt myotubes (Supplemental Figure 8) indicative of a hyperpolarization of the mitochondrial membrane potential. We also compared the ultrastructure of mitochondria in flexor digitorum brevis (FDB) and soleus muscle fibers from 2–3 month and 1 year old RyR1wt/wt and RyR1Y522S/wt mice. Most mitochondria in FDB fibers of wild type mice are located circumferentially around the myofibrils at either side of the Z line in close proximity to the triads (Rossi, 2006). In sections that cut across the inter-myofibrillar space, the mitochondria in muscle of the RyR1wt/wt mice are rounded or slightly elongated (Figure 4A, panels 1–3). The internal matrix is usually dark and cristae appear well organized and parallel to one another. A very small percentage of mitochondria in the wild type muscle are abnormal with “myelin figures” (Figure 4A, panel 4) or a somewhat disarranged external membrane and internal cristae, but are similar in size to the more typical mitochondria. In contrast, although typical mitochondria are also found in FDB fibers of RyR1Y522S/wt mice (Figure 4B, panel 1), a large number of mitochondria are abnormally shaped, swollen, and sometimes severely altered (Figure 4B, panels 2–4). The most noticeable changes include widening and loss of matrix density, an increase in overall size, loss/disorganization of the internal cristae (Figure 4B, panel 2), disruption of the external membrane (Figure 4B, panel 3 arrows), and vacuolization (stars in Figure 4B, panels 2 and 4). The number of severely disrupted mitochondria (such as those in Figure 4B, panels 2–4) varies significantly from fiber to fiber and sample to sample, but is always much higher in fibers from RyRY522S/wt compared to RyR1wt/wt mice (9.1 vs 1.1%, Table 2, Column A). The minimum mitochondrial diameter in RyRwt/wt and RyRY522S/wt mice is about 30% larger (p<0.0001) in fibers from RyRY522S/wt mice, suggesting increased mitochondrial swelling (Table 2, Column B). The mitochondria appear to be even more damaged at one year, but the nature of the damage varies greatly among muscle groups. In FDB and soleus muscles form 1 year old RyRY522S/wt mice, most mitochondria are severely swollen and disrupted (Figure 4D and F, stars). In contrast, in the muscle of RyRwt/wt mice, mitochondria are identical to those at 2–3 months of age, i.e. dark in appearance and small in size (Figure 4C and E, arrows). To more quantitatively evaluate the mitochondrial damage, we isolated samples enriched in mitochondria from the muscle of both 2 and 12 month old RyRwt/wt and RyRY522S/wt mice. Mitochondrial content of the samples was confirmed as a >100-fold enrichment of succinate dehydrogenase (SDH) activity compared to homogenates (not shown). We measured the level of thiobarbituric acid-reactive substances (TBARS) as a marker of lipid peroxidation (i.e. oxidative damage) of mitochondrial membranes. Consistent with the mitochondrial damage observed in EM analyses, an increase in lipid peroxidation is observed in mitochondria isolated from 2 month old RyR1Y522S/wt mice compared to RyR1wt/wt mitochondria (Figure 4G) and this is substantially greater at 12 months. To determine if this arises from increased oxidative/nitrosative stress, we treated mice with NAC in their water supply for several months prior to sacrifice and mitochondrial isolation from pooled skeletal muscles. As can be seen in Figure 4G, NAC treatment completely reversed the increased mitochondrial lipid peroxidation observed in RyR1Y522S/wt mice.
Figure 4. Effects of the Y522S mutation on mitochondrial structure and muscle function.

A–F. Mitochondrial ultrastructure is altered in muscle of RyR1Y522S/wt mice. A. Mitochondria of RyR1wt/wt fibers at 2–3 months of age are usually regularly shaped, appearing either round (panels 1 and 2) or slightly elongated (panel 3), with a dark/dense internal matrix. Abnormal mitochondria (panel 4) are rare in RyR1wt/wt fibers. B. In RyR1Y522S/wt fibers, some normal mitochondria are present (panel 1), but abnormal mitochondria are frequent (panels 2–4). C–F. At 1 year, a much larger percentage of mitochondria are severely swollen/disrupted than at 2–3 months of age in both in FDB and soleus muscles from RyR1Y522S/wt mice (D and F, stars). In wt FDB (C) and soleus (E) muscles, on the other hand, mitochondria appear similar to those at 2–3 month of age. G. Mitochondrial lipid peroxidation in young and aged mice. Mitochondrial enriched fractions from 2 or 12 month-old mice with or without chronic NAC-treatment (≥ 2 months) were isolated and TBARS were measured in acid supernatants after protein precipitation, as detailed in Methods. Shown are the mean of 3 independent determinations ± SD. H–J. Effect of chronic NAC treatment on skeletal muscle function of aged mice. One year old male RyR1wt/wt and RyR1Y522S/wt mice were treated with NAC (1% w/v) in their drinking water for at least 2 months prior to sacrifice. Solei were removed and stimulated in vitro, as described in Experimental Procedures. Shown are data for stress developed at increasing frequency of electrical stimulation (H) and the maximal tension per cross-sectional area obtained by maximal electrical stimulation (I) or application of 20mM caffeine (J). All data in this figure are shown as mean ± S.E.M.
Table 2. Electron microscopic examination reveals frequent severely disrupted and larger mitochondria in Y522S fibers.
Column A: The relative percentage of severely disrupted mitochondria in FDB fibers from 2 month old RyR1wt/wt and RyR1Y522S/wt mice. n, total number of mitochondria; Column B: Differences in mitochondria mean diameter in FDB fibers from RyR1wt/wt and RyR1Y522S/wt mice. n, number of measurements. (*p<0.0001). Data are mean ± S.E.M
| Genotype/age | Column A | Column B |
|---|---|---|
| % of severely disrupted mitochondria | Average diameter of mito, nm ± SD | |
| RyR1wt/wt (2–3 months) | 1.1
(n = 1019, 2 mice) |
187 ± 53
(n = 643, 2 mice) |
| RyR1Y522S/wt (2–3 months) | 9.1*
(n = 1493, 4 mice) |
243 ± 72 *
(n = 1069, 4 mice) |
To assess the functional consequences of aging under conditions of chronic oxidative/nitrosative stress in muscle of RyR1Y522S/wt mice, we measured the ability of the soleus muscle to generate force and found that the muscles from older ( 8 month old) RyR1Y522S/wt mice display a significant decrease in maximal developed force and this decrease is prevented by chronic administration of NAC in their water supply (Figure 4H and I).
Our findings are consistent with mitochondrial damage resulting from prolonged oxidative/nitrosative stress contributing to contractile dysfunction in aged RyR1Y522S/wt. However, SR Ca2+ leak and store depletion could also directly impact muscle function. We find that maximal caffeine induced stress is reduced in the RyR1Y522S/wt mice and this is also prevented by chronic feeding of NAC to the mice in their drinking water (Figure 4J). The observed reduction in maximal caffeine-induced contracture could result from a decrease in releasable Ca2+ stores, reduced myofilament Ca2+ sensitivity, or both.
Discussion
Environmental heat stress triggers sudden death in RyR1Y522S/wt mice. We suggest that this is due to a cycle whereby elevated cytosolic Ca2+, combined with temperature dependent increases in RNS, produce nitrosative modifications of the mutant channel that enhance RyR1 channel activity at elevated temperatures. The net result is a destructive feed-forward cycle of increased myoplasmic Ca2+ and RNS with temperature (Figure 5), ultimately producing EHS in RyR1Y522S/wt mice. Over an extended period, this cycle appears to produce a myopathy characterized by decreased force generation and damaged mitochondria.
Figure 5. Proposed model of exertional/environmental heat stress and myopathy in RyR1Y522S/wt mice.

➀ SR Ca2+ release channels in RyR1Y522S/wt mice are more sensitive to voltage, ligand, and Ca2+ activation and open more readily, producing small, possibly local, increases in resting Ca2+. ➁ Increased cytosolic Ca2+ levels enhance ROS/RNS production. Increases in RNS are likely to be produced by nitric oxide synthases (NOS). Possible sources of ROS include mitochondria, xanthine oxidase (XO) and NAD(P)H oxidases (NOX). ➂ Although both S-glutathionylation and S-nitrosylation of RyR1Y522S/wt occurs, our data suggest that S-nitrosylation alone enhances the temperature sensitivity of RyR1. ➃ S-nitrosylation of RyR1Y522S/wt increases its sensitivity to temperature and decreases its sensitivity to Ca2+ inhibition further promoting SR Ca2+ leak. ➄ Ca2+ increases further enhance ROS/RNS production. ➅ In response to heat stress, Ca2+ release from the modified RyR1Y522S/wt is greatly and persistently augmented, leading to heat stroke. ➆ and ➇ Chronically-elevated levels of Ca2+ and ROS/RNS damage mitochondria and contribute to the development of myopathy.
RyR1 blockers, ROS/RNS scavengers and inhibitors of NOS abolish temperature dependent increases in cytosolic Ca2+ and RNS. Since NOS inhibition blocks temperature dependent increases in Ca2+ and RNS, but not ROS, these results suggest that the increase in Ca2+ results from the increase in RNS. We do not know which NOS isoform(s) is involved, but skeletal muscle is rich in nNOS and this isoform may be activated by increases in cytosolic Ca2+ since it colocalizes with ryanodine receptors in cardiac myocytes (Barouch, 2002; Hare, 2003). Although most nNOS localizes to the sarcolemma in skeletal muscle (Wells, 2003), close juxtapositioning of RyR1 and a subpopulation of nNOS in the triad junction might allow for a very local RyR1-mediated Ca2+ leak from RyR1 to stimulate NO production by nNOS.
We previously identified 7 specific cysteines in one subunit of RyR1 (out of 100) that can be S-nitrosylated (Aracena-Parks, 2006). Of these, four (C315, C811, C906, C3635) are endogenously nitrosylated. Stamler and coworkers (Eu, 2000; Sun, 2001a, 2003a, 2001b) found that C3635 is the primary RyR1 cysteine that is S-nitrosylated at low pO2. At high pO2, some cysteines (not C3635) are oxidized, preventing RyR1 S-nitrosylation (Sun, 2003). Thus, C3635 is the best candidate for S-nitrosylation of RyR1Y522S/wt in our mice and studies are currently ongoing to determine if nitrosylation of this residue in necessary and sufficient for enhancing RyR1 temperature sensitivity.
The Y522S mutation in humans is associated with a myopathy and central cores in muscle fibers. Although we did not detect central cores in our mice, young mice displayed evidence of a myopathy in terms of mitochondrial alterations, while muscle function is not greatly compromised. In contrast, older RyR1Y522S/wt solei display marked mitochondrial structural damage and a decreased ability to generate force that is prevented by treating the mice with NAC. Mitochondrial lipid peroxidation is greatly increased in the muscle of older RyR1Y522S/wt mice and this is also prevented by NAC. Thus, chronic exposure to elevated Ca2+ and ROS/RNS leads to progressive mitochondrial damage and decreased ability to generate force, suggesting that these pathways contribute to the myopathy observed in older mice.
The temperature and exercise sensitivity of RyR1Y522S/wt mice provide new mechanistic insight into environmental and/or exertional heat illness, disorders that have previously been linked in humans to MH mutations in RyR1 (Davis, 2002; Wappler, 2001). Consistent with this linkage, the probability of an MH response to exercise or volatile anesthetics in RyR1Y522S/wt mice is decreased by cooling (unpublished observation). Recent evidence indicates that intensive exercise also promotes RyR1 S-nitrosylation (Bellinger, 2008). Our data are the first to demonstrate that exercise, EHS, and heat-induced sudden death can result from a disease mutation in RyR1 that promotes Ca2+ leak, enhances nitrosative stress and promotes subsequent S-nitrosylation of the mutant RyR1. It remains to be determined if EHS in humans is caused by RyR1 mutations that promote a similar feed-forward mechanism of uncontrolled Ca2+ leak and nitrosative stress.
In summary, our data demonstrate that Ca2+ release channels in RyR1Y522S/wt mice are leaky, producing elevations in resting Ca2+, ROS, RNS and basal stress at physiologically relevant temperatures. Our data support the involvement of a destructive feed-forward cycle whereby Ca2+ leak enhances RNS production, and subsequent S-nitrosylation of RyR1 further increases Ca2+ leak, resulting in regenerative Ca2+ release that underlies uncontrolled contractions during heat stress. We also suggest that increased Ca2+, RNS,. and/or ROS ultimately contribute to the progressive development of a myopathy characterized by decreased muscle performance and mitochondrial damage. An intriguing aspect of this study is the possibility that RyR1 mutations together with nitrosative stress represent a “double-hit mechanism” that underlies a subset of human cases of enhanced susceptibility to heat stroke, exertional/environmental illness, and/or sudden death.
Experimental Procedures
Mice
All procedures were approved by the IACUC at Baylor College of Medicine and UCAR at the University of Rochester. For in vivo antioxidant treatment, mice were provided ad libitum access to drinking water containing NAC (1% w/v), L-NAME (1% w/v), or NAC plus L-NAME (both at 1% w/v).
In vivo temperature sensitivity
Mice were anesthetized with etomidate, which does not trigger MH episodes in either MH susceptible humans (Robertson, 1992) or RyR1Y522S/wt mice. Two minutes following etomidate injection, mice were placed in an environmental chamber at 41°C. The initial core body temperature, as well as the temperature every minute thereafter, was monitored over the next 15 minutes of exposure.
Contractile studies
Muscle collection and contractile studies were performed as previously described (Chelu, 2006). For determination of maximal developed stress, the data were fitted to a sigmoidal curve with the peak value of this curve reported as the maximal stress.
Isolation of sub-cellular fractions from mouse skeletal muscle
Muscle from each mouse was quickly collected, snap frozen in liquid nitrogen and stored at −80°C for up to 2 weeks. Microsomes or mitochondrial-enriched samples were obtained from thawed muscle by differential centrifugation (see Supplemental Information).
Ca2+ flux studies
Microsomal vesicles (1mg/ml) from RyR1wt/wt or RyR1Y522S/wt skeletal muscle were passively loaded with Ca2+ and Ca2+ release kinetics was analyzed at 25°C, as described previously (Donoso et al., 2000), using a KinTek SF-2002 thermoregulated stopped-flow spectrometer (KinTek Corp.). Studies at 37°C were performed after 5-minute equilibration of samples in the equipment. Release rate constants were obtained from peak differential analysis of raw fluorescence data.
Lipoperoxidation assay
Basal lipoperoxidation levels were measured using thiobarbituric acid reactive substances (TBARS) as described elsewhere (Letelier, 2005).
Glutathione assays
Total GSH and GSSG were assayed using deproteinized muscle homogenates in a 96-well format according to Tietze (Tietze, 1969), as modified by Griffith (Griffith, 1980).
Primary cultures and fluorescent microscopy
Primary myotube cultures were grown on Matrigel (BD Biosciences) coated glass coverslips from 1–4 day old mice as previously described (Pollard, 1948). ROS and RNS imaging were performed using the probes DCF or DAF, as detailed in Supplemental Experimental Procedures. Ca2+ imaging was performed as previously described in Long et al (Long, 2007). Perforated patch clamp recordings of L-type Ca2+ currents (L-currents) and intracellular Ca2+ transients were recorded as described in Chelu et al (Chelu, 2006). Details are described in Supplemental Experimental Procedures.
Western blotting
Western blotting with mouse anti-glutathione 1:10,000 (Virogen) and rabbit anti-S-nitrosocysteine 1:10,000 (Sigma) were performed and analyzed as previously described (Aracena-Parks, 2006). Stripping of membranes was performed with Li-COR Stripping Solution, following the manufacturer’s directions. Stripped membranes were re-probed with a mouse anti-RyR1 1:10,000 (Affinity Bioreagents) only to confirm the identity of the analyzed band. This antibody displays different affinities for RyR1 electrophoresed under reducing or non-reducing conditions. Thus, fluorescent data was normalized to the Coomasie stain of the blots.
[3H]ryanodine binding
Equilibrium binding was performed with microsomes as detailed previously (Aracena-Parks, 2006). Ca2+ titration of binding was performed with 5 nM [3H]ryanodine as described by Rodney et al. (Rodney, 2000). Kinetic assays with 5 nM [3H]ryanodine at 23°C or 37°C were measured as detailed in Hawkes et al. (Hawkes, 1992). Details of buffers used can be found in Supplemental Experimental Procedures.
Preparation and analysis of samples for electron microscopy (EM)
EM was performed in FDB muscles from 2–3 month and 1 year-old mice as detailed in Paolini et al (Paolini, 2007). Details are described in Supplemental Experimental Procedures.
Statistical analyses
All analyses were performed in Sigma Plot (Systat Software, Inc).
Supplementary Material
Acknowledgments
This work was supported by grants from NIH (AR 050503 and AR053349 to S.L.H., AR44657 to R.T.D., and 5P01AR052354 to S.L.H. and R.T.D.), the Muscular Dystrophy Association to S.L.H, Research Grant GGP030289 from the Italian Telethon Foundation to F.P, and a NIH Dental and Craniofacial training grant T32-DE07202 to AER.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aghdasi B, Reid MB, Hamilton SL. Nitric Oxide Protects the Skeletal Muscle Ca2+ Release Channel from Oxidation Induced Activation. The Journal of biological chemistry. 1997;272:25462–25467. doi: 10.1074/jbc.272.41.25462. [DOI] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of Cysteines Involved in S-Nitrosylation, S-Glutathionylation, and Oxidation to Disulfides in Ryanodine Receptor Type 1. The Journal of biological chemistry. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric Oxide Regulates the Heart by Spatial Confinement of Nitric Oxide Synthase Isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, Lacampagne A, Marks AR. Remodeling of Ryanodine Receptor Complex Causes “Leaky” Channels: A Molecular Mechanism for Decreased Exercise Capacity. Proceedings of the National Academy of Sciences of the United States of America; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahan D, Kozak-Ribbens G, Confort-Gouny S, Ghattas B, Figarella-Branger D, Aubert M, Cozzone PJ. A Noninvasive Investigation of Muscle Energetics Supports Similarities Between Exertional Heat Stroke and Malignant Hyperthermia. Anesth Analg. 2001;93:683–689. doi: 10.1097/00000539-200109000-00030. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Knochel JP. Heat Stroke. N Engl J Med. 2002;346:1978–1988. doi: 10.1056/NEJMra011089. [DOI] [PubMed] [Google Scholar]
- Carroll S, Skarmeta JG, Yu X, Collins KD, Inesi G. Interdependence of Ryanodine Binding, Oligomeric Receptor Interactions, and Ca2+ Release Regulation in Junctional Sarcoplasmic Reticulum. Arch Biochem Biophys. 1991;290:239–247. doi: 10.1016/0003-9861(91)90615-p. [DOI] [PubMed] [Google Scholar]
- Chelu MG, Goonasekera SA, Durham WJ, Tang W, Lueck JD, Riehl J, Pessah IN, Zhang P, Bhattacharjee MB, Dirksen RT, Hamilton SL. Heat- and Anesthesia-Induced Malignant Hyperthermia in an RyR1 Knock-In Mouse. FASEB J. 2006;20:329–330. doi: 10.1096/fj.05-4497fje. [DOI] [PubMed] [Google Scholar]
- Chu A, Diaz-Munoz M, Hawkes MJ, Brush K, Hamilton SL. Ryanodine as a Probe For the Functional State of the Skeletal Muscle Sarcoplasmic Reticulum Ca2+Release Channel. Mol Pharmocol. 1990;37:735–741. [PubMed] [Google Scholar]
- Clark CB, Zhang Y, Martin SM, Davies LR, Xu L, Kregel KC, Miller FJ, Buettner GR, Kerber RE. The Nitric Oxide Synthase Inhibitor N(omega)-Nitro-L-Arginine Decreases Defibrillation-Induced Free Radical Generation. Resuscitation. 2004;60:351–357. doi: 10.1016/j.resuscitation.2004.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Brown R, Dickson A, Horton H, James D, Laing N, Marston R, Norgate M, Perlman D, Pollock N, Stonwell K. Malignant Hyperthermia Associated with Exercise-Induced Rhabdomyolysis or Congenital Abnormalities and a Novel RYR1 Mutation in New Zealand and Australian Pedigrees. Br J Anaesth. 2002;88:508–515. doi: 10.1093/bja/88.4.508. [DOI] [PubMed] [Google Scholar]
- Donoso P, Aracena P, Hidalgo C. Sulfhydryl Oxidation Overrides Mg(2+) Inhibition of Calcium-Induced Calcium Release in Skeletal Muscle Triads. Biophysical journal. 2000;79:279–286. doi: 10.1016/S0006-3495(00)76290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducreux S, Zorzato F, Muller C, Sewry C, Muntoni F, Quinlivan R, Restagno G, Girard T, Treves S. Effect of Ryanodine Receptor Mutations on Interleukin-6 Release and Intracellular Calcium Homeostasis in Human Myotubes from Malignant Hyperthermia-Susceptible Individuals and Patients Affected by Central Core Disease. The Journal of biological chemistry. 2004;279:43838–43846. doi: 10.1074/jbc.M403612200. [DOI] [PubMed] [Google Scholar]
- Ellis FR, Halsall PJ, Harriman DG. Malignant Hyperpyrexia and Sudden Infant Death Syndrome. Br J Anaesth. 1988;60:28–30. doi: 10.1093/bja/60.1.28. [DOI] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The Skeletal Muscle Calcium Release Channel: Coupled O2 Sensor and NO Signaling Functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Griffith OW. Determination of Glutathione and Glutathione Disulfide Using Glutathione Reductase and 2-Vinylpyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- Hackl W, Winkler M, Mauritz W, Sporn P, Steinbereithner K. Muscle Biopsy for Diagnosis of Malignant Hyperthermia Susceptibility in Two Patients with Severe Exercise-Induced Myolysis. Br J Anaesth. 1991;66:138–140. doi: 10.1093/bja/66.1.138. [DOI] [PubMed] [Google Scholar]
- Hare JM. Nitric Oxide and Excitation-Contraction Coupling. J Mol Cell Cardiol. 2003;35:719–729. doi: 10.1016/s0022-2828(03)00143-3. [DOI] [PubMed] [Google Scholar]
- Hawkes MJ, Nelson TE, Hamilton SL. [3H] Ryanodine as a Probe of Changes in the Functional State of the Ca2+ -Release Channel in Malignant Hyperthermia. The Journal of biological chemistry. 1992;267:6702–6709. [PubMed] [Google Scholar]
- Hopkins PM. Malignant Hyperthermia: Advances in Clinical Management and Diagnosis. Br J Anaesth. 2000;85:118–128. [PubMed] [Google Scholar]
- Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and Pathogenesis of Malignant Hyperthermia. Muscle Nerve. 2000;23:4–17. doi: 10.1002/(sici)1097-4598(200001)23:1<4::aid-mus3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Letelier ME, Lepe AM, Faundez M, Salazar J, Marin R, Aracena P, Speisky H. Possible Mechanisms Underlying Copper-Induced Damage in Biological Membranes Leading to Cellular Toxicity. Chemicobiological interactions. 2005;151:71–82. doi: 10.1016/j.cbi.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Lichtman AD, Oribabor C. Malignant Hyperthermia Following Systemic Rewarming after Hypothermic Cardiopulmonary Bypass. Anesth Analg. 2006;102:372–375. doi: 10.1213/01.ane.0000189596.70694.36. [DOI] [PubMed] [Google Scholar]
- Long C, Cook LG, Hamilton SL, Wu GY, Mitchell BM. FK506 Binding Protein 12/12.6 Depletion Increases Endothelial Nitric Oxide Synthase Threonine 495 Phosphorylation and Blood Pressure. Hypertension. 2007;49:569–576. doi: 10.1161/01.HYP.0000257914.80918.72. [DOI] [PubMed] [Google Scholar]
- Marengo JJ, Hidalgo C, Bull R. Sulfhydryl Oxidation Modifies the Calcium Dependence of Ryanodine-Sensitive Calcium Channels of Excitable Cells. Biophysical journal. 1998;74:1263–1277. doi: 10.1016/S0006-3495(98)77840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba T, Murayama T, Ogawa Y. Redox States of Type 1 Ryanodine Receptor Alter Ca(2+) Release Channel Response to Modulators. Am J Physiol Cell Physiol. 2002;282:C684–692. doi: 10.1152/ajpcell.01273.2000. [DOI] [PubMed] [Google Scholar]
- Pamukcoglu T. Sudden Death Due to Malignant Hyperthermia. Am J Forensic Med Pathol. 1988;9:161–162. doi: 10.1097/00000433-198806000-00015. [DOI] [PubMed] [Google Scholar]
- Paolini C, Quarta M, Nori A, Boncompagni S, Canato M, Volpe P, Allen PD, Reggiani C, Protasi F. Reorganized Stores and Impaired Calcium Handling in Skeletal Muscle of Mice Lacking Calsequestrin-1. J Physiol. 2007;583:767–784. doi: 10.1113/jphysiol.2007.138024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Stambuk RA, Casida JE. Ca2+-Activated Ryanodine Binding: Mechanisms of Sensitivity and Intensity Modulation by Mg2+, Caffeine, and Adenine Nucleotides. Mol Pharmacol. 1987;31:232–238. [PubMed] [Google Scholar]
- Pollard JW, Walker JM. Methods in Molecular Biology. Totowa, NJ: Humana Press; 1948. Basic Cell Culture Protocol; p. 1997. [Google Scholar]
- Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of Superoxide Generation by Neuronal Nitric-Oxide Synthase. The Journal of biological chemistry. 1999;274:9573–9580. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- Quane KA, Keating KE, Healy JM, Manning BM, Krivosic-Horber R, Krivosic I, Monnier N, Lunardi J, McCarthy TV. Mutation Screening of the RYR1 Gene in Malignant Hypertherima: Detection of a Novel Tyr to Ser Mutation in a Pedigree with Associated Central Core. Genomics. 1994;23:236–239. doi: 10.1006/geno.1994.1483. [DOI] [PubMed] [Google Scholar]
- Robertson S. Advantages of Etomidate Use As An Anesthetic Agent. Vet Clinic North Am Small Anim Pract. 1992;22:277–280. doi: 10.1016/s0195-5616(92)50609-x. [DOI] [PubMed] [Google Scholar]
- Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL. Regulation of RYR1 Activity by Ca(2+) and Calmodulin. Biochemistry. 2000;39:7807–7812. doi: 10.1021/bi0005660. [DOI] [PubMed] [Google Scholar]
- Rossi AE, Boncompagni S, Protasi F, Dirksen RT. Developmental Regulation of Mitochondrial Triad Targeting in Skeletal Muscle. Biophysical journal. 2006;90:A269. [Google Scholar]
- Ryan JF, Tedeschi LG. Sudden Unexplained Death in a Patient with a Family History of Malignant Hyperthermia. J Clin Anesth. 1997;9:66–68. doi: 10.1016/S0952-8180(96)00207-3. [DOI] [PubMed] [Google Scholar]
- Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is Responsible for Skeletal Muscle Ryanodine Receptor Modulation by NO. Proceedings of the National Academy of Sciences of the United States of America. 2001a;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Xu L, Eu JP, Stamler JS, Meissner G. Nitric Oxide, NOC-12, and S-Nitrosoglutathione Modulate the Skeletal Muscle Calcium Release Channel/Ryanodine Receptor by Different Mechanisms. An Allosteric Function for O2 in S-Nitrosylation of the Channel. The Journal of biological chemistry. 2003;278 doi: 10.1074/jbc.M211940200. [DOI] [PubMed] [Google Scholar]
- Sun J, Xu L, Eu JP, Stamler JS, Meissner G. Classes of Thiols that Influence the Activity of the Skeletal Muscle Calcium Release Channel. The Journal of biological chemistry. 2001b;276:15625–15630. doi: 10.1074/jbc.M100083200. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Franklin BA, Balady GJ, Blair SN, Corrado D, Estes NA, 3rd, Fulton JE, Gordon NF, Haskell WL, Link MS, Maron BJ, Mittleman MA, Pelliccia A, Wenger NK, Willich SN, Costa F. Exercise and Acute Cardiovascular Events Placing the Risks into Perspective: A Scientific Statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism and the Council on Clinical Cardiology. Circulation. 2007;115:2358–2368. doi: 10.1161/CIRCULATIONAHA.107.181485. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymic Method for Quantitative Determination of Nanogram Amounts of Total and Oxidized Glutathione: Applications to Mammalian Blood and Other Tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Treves S, Anderson AA, Ducreux S, Divet A, Bleunven C, Grasso C, Paesante S, Zorzato F. Ryanodine Receptor 1 Mutations, Dysregulation of Calcium Homeostasis and Neuromuscular Disorders. Neuromuscul Disord. 2005;15:577–587. doi: 10.1016/j.nmd.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Wappler F, Fiege M, Steinfath M, Agarwal K, Scholz J, Singh S, Matschke J, Schulte Am Esch J. Evidence for Susceptibility to Malignant Hyperthermia in Patients with Exercise-Induced Rhabdomyolysis. Anesthesiology. 2001;94:95–100. doi: 10.1097/00000542-200101000-00019. [DOI] [PubMed] [Google Scholar]
- Wells KE, Torelli S, Lu Q, Brown SC, Partridge T, Muntoni F, Wells DJ. Relocalization of Neuronal Nitric Oxide Synthase (nNOS) as a Marker for Complete Restoration of the Dystrophin Associated Protein Complex in Skeletal Muscle. Neuromuscul Disord. 2003;13:21–31. doi: 10.1016/s0960-8966(02)00191-8. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang JZ, Danila CI, Hamilton SL. A Noncontiguous, Inter-Subunit Binding Site for Calmodulin on the Skeletal Muscle Ca2+ Release Channel. The Journal of biological chemistry. 2003;278:8348–8355. doi: 10.1074/jbc.M209565200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.