

Abstract
Purpose
RDH11 and RDH12 are closely related retinol dehydrogenases expressed in the retina. RDH12 has been linked to the early-onset retinal dystrophy Leber congenital amaurosis, whereas RDH11 has not been associated with human disease. To understand their physiological roles, the authors investigated their expression during development and their regulation by light-induced oxidative stress in mouse retina.
Methods
Quantitative RT-PCR and immunoblot analysis were used for quantification of RDH11 and RDH12 during development and oxidative stress. Expression during development was measured between embryonic day (E) 12 and postnatal day (P) 210 (7 months) in C57BL/6 mouse eyes. Expression during light-induced oxidative stress was measured between 2 and 24 hours of exposure to light in BALB/c mouse retina.
Results
The RDH11 level was low and remarkably constant during development and oxidative stress. RDH12 expression started at P7 and increased until P30 to approximately sevenfold higher than RDH11. Oxidative stress induced by exposure to constant bright light led to a rapid and significant decrease of RDH12 protein.
Conclusions
The low and constant expression of RDH11 suggested a housekeeping function for this enzyme. The onset of RDH12 expression during the maturation of photoreceptor cells suggested a function related to the visual process. The light-induced rapid decrease of RDH12 protein, preceding the decrease of the mRNA, suggested a specific degradation of the protein rather than a regulation of gene expression.
Leber congenital amaurosis (LCA), a severe and early-onset form of retinal dystrophy, has been linked to mutations in several distinct genes, including RDH12.1–5 The RDH12 gene encodes a microsomal retinol dehydrogenase that is expressed in photoreceptor cells in retina.6–9 RDH11, the most closely related sequence, shares 73% identity with RDH12 in humans6 and 67% identity with RDH12 in mice. RDH11 is a microsomal retinol dehydrogenase expressed in many tissues in mice including liver, testis, and retina.10 RDH11 has not been linked to any human disease.
The physiological role of these enzymes has been investigated in vivo using knockout mice. As retinol dehydrogenases, these enzymes were expected to be important in the retinoid cycle for the recycling of the visual chromophore 11-cis retinal, allowing the regeneration of photosensitive rhodopsin pigment after bleaching.11 However, a decrease in the regeneration of 11-cis retinal has not been found in Rdh11 or Rdh12 knockout mice.8,9,12,13 These knockout mice, however, exhibit a significant delay in dark adaptation after bleaching.8,12,13 This phenotype cannot be explained by a delay of 11-cis retinal recycling, leaving the molecular mechanism for the delay in dark adaptation unknown. It was hypothesized that RDH12 catalyzes the reduction of all-trans retinal in photoreceptor inner segments, indirectly participating to the clearance of all-trans retinal in the outer segments. A delayed clearance in the knockout would then block the regeneration of photosensitive rhodopsin.8 Interestingly, clinical observations in patients with LCA caused by mutations of the RDH12 gene revealed that almost all patients had a defect in dark adaptation during the first years of life, followed by progressive rod-cone dystrophy.1,4,5,14
Although Rdh11 knockout mice have not been tested for sensitivity to light damage, Rdh12 knockout mice have a greater sensitivity to light-induced photoreceptor apoptosis.8 When exposed to constant bright light for 48 hours, photoreceptor cell loss was induced in the pigmented Rdh12 knockout mice but not in the wild-type mice.8 The molecular mechanism leading to this murine phenotype is unknown. It was hypothesized that the accumulation of all-trans retinal in photoreceptor inner segments of the knockout mice could be a sensitizer for light damage far beyond what is observed in the wild-type mice.8 LCA patients with mutated RDH12 have progressive photoreceptor degeneration affecting rods and cones. Onset of symptoms begins in early childhood (2–4 years) and progression to legal blindness begins in early adulthood, but it is unknown whether exposure to light or oxidative stress plays a role in this pathogenesis.1,2,4,5
The substrate specificity and catalytic activities of RDH11 and RDH12 have been characterized in vitro by using purified enzymes and microsomal fractions of transfected cells.6,7,10 These studies have shown that RDH11 and RDH12 are able to catalyze the reduction of two distinct groups of aldehydic substrates: the retinaldehydes and the short-chain (hydroxy)aldehydes.7,10 Because of their activity toward retinaldehydes, these enzymes were named retinol dehydrogenases and the idea that they could play a direct part in the visual cycle was proposed.3,6,7,15 Their activity toward the short-chain (hydroxy)aldehydes suggests that they may have additional or alternative roles.7,10 Short-chain (hydroxy)aldehydes are toxic end products of the lipid peroxidation of membrane polyunsaturated fatty acids. This nonenzymatic, auto-amplified degradation of lipids is induced by reactive oxygen species generated in excess during oxidative stress.16 Short-chain (hydroxy)aldehydes are thought to mediate, at least in part, the apoptotic response induced by oxidative stress in cells.17–19 Because RDH11 and RDH12 catalyze the reduction of these toxic aldehydes to less toxic alcohols, they may protect photoreceptor cells against the toxicity and apoptosis induced by oxidative stress.
The similarities between RDH11 and RDH12 (sequence, activity, substrate specificity, and the common phenotype of delayed dark adaptation in knockout mice) suggest that the two enzymes may have overlapping physiological roles. In this study, we describe the expression levels and regulation of RDH11 and RDH12 enzymes during development and during exposure to oxidative stress. We found a significantly higher expression level of RDH12 than RDH11 in the mouse retina at all times after postnatal day (P) 7. We also found that exposure to oxidative stress greatly affects RDH12 levels but not RDH11 levels. We concluded that the two enzymes differ in their expression during development and in their regulation by light-induced oxidative stress.
MATERIALS AND METHODS
Materials
Rabbit polyclonal antibodies against mouse RDH11 and RDH12 were produced and affinity purified by Pacific Immunology Corp. (Ramona, CA). Synthetic peptides (amino acids [aa] 301–316 of RDH11 and aa 289–304 of RDH12) were coupled with keyhole limpet hemocyanin and were used for immunization. Chemicals and mouse monoclonal anti-Flag antibody were from Sigma (St. Louis, MO). Mouse monoclonal anti-β actin was from Abcam (Cambridge, MA). The protein oxidation detection kit was from Chemicon (OxyBlot; Temecula, CA).
Animals
The animals were maintained in a 12-hour dim light-dark cycle. BALB/c and C57BL/6 mice were purchased from Charles River Laboratory (Raleigh, NC), and Rdh11 and Rdh12 knockout lines were maintained at the Dean A McGee Eye Institute. BALB/c mice were exposed to damaging light, as described.20 Briefly, unanesthetized mice were exposed to 3000-lux diffuse, cool, white fluorescent light for indicated times in clear plastic cages with wire tops. Drinking water was supplied by a bottle attached to the side of each cage and food was placed inside the cage such that there was no obstruction between the light and each animal. Each cage contained one mouse. All exposures to light were stopped at 6 PM. The animals either were killed immediately for tissue collection or were returned to the dim light-dark cycle for 7 days before the eyeballs were collected for histologic examination. This 7-day period after light treatment allowed the retina to clear all dead cells and to return to a well-organized morphology. Mice were killed by CO2 inhalation, a method approved by the Panel of Euthanatization of the American Veterinary Medical Association. All procedures were performed according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the University of Oklahoma Health Sciences Center (OUHSC) Guidelines for Animals in Research. All protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the OUHSC and the Dean A. McGee Eye Institute.
Tissue Dissection and Processing
Eyeballs were collected from 10-month-old C57BL/6 mice in RNase-free phosphate-buffered saline after 12 hours of dark adaptation. Clean, uncontaminated retina, iris-ciliary body, and posterior segment containing RPE/choroid/scleral tissues were collected for RNA preparation, as described.21 For expression during development, whole eye globes without lenses from C57BL/6 mice were individually frozen for all time points after birth. Before birth, at embryonic day (E) 12, a part of the head containing both eyes was frozen, and at E15 and E18, both eye globes from each embryo were pooled and frozen. For light-induced oxidative stress, dissected retinas from 10-week-old BALB/c mice were individually frozen immediately after light exposure. From each mouse, one retina was used for RNA preparation and one retina was used for protein preparation. For histology of these retinas, eyeballs were oriented by labeling the superior half with a permanent dye. Oriented eyes were embedded in paraffin, and sections were cut along the vertical meridian, through the optic nerve head, and stained with hematoxylin and eosin for viewing.
RNA Preparation and Quantitative RT-PCR
Total RNA was prepared from individual frozen retinas using reagent (Trizol; Invitrogen, Carlsbad, CA). cDNA was prepared using the reverse transcription system from Promega (Madison, WI) with oligo primers. Gene-specific primers for quantitative RT-PCR were (dT)15 chosen close to the 3′ end of the cDNA and were designed to span at least an intron to avoid amplification of any genomic contamination. Sequences of primers are available on request. Each quantitative RT-PCR reaction was set up in a final volume of 25 μL containing 12.5 μL SYBR Green from Bio-Rad (Hercules, CA), cDNA made from 65 ng total RNA, and 0.4 μM each primer. Reactions were set up in triplicate on 96-well plates and quantified (iQ Real-Time PCR Detection System; Bio-Rad). Expression data were calculated from four independent samples, each performed in triplicate, and were normalized with the expression of the housekeeping gene Rpl19, as indicated.22
Protein Preparation and Immunoblot Analysis
Four retinas from four different mice were pooled and homogenized in reagent (T-PER; Pierce, Rockford, IL) in the presence of protease inhibitors (2 μg/mL aprotinin, 5 μg/mL pepstatin A, 10 μg/mL leupeptin, and 0.5 mM phenylmethylsulfonyl fluoride; final concentrations). After 15-minute incubation at 4°C, the samples were centrifuged at 10,000g for 5 minutes, and the supernatants were collected. Protein concentrations were measured using the Coomassie reagent (Pierce). Immunoblot analysis was carried out as described using a luminol substrate (SuperSignal West Pico Chemiluminescent Substrate; Pierce).13 Signals were quantified using the (Image Station 4000R; Eastman Kodak; Rochester, NY). As loading controls, membranes were stained with Ponceau red from Sigma, and each immunoblot was probed with anti-actin antibody. To perform the immunoblot detection of carbonyl groups introduced into proteins by oxidative reactions, 20 μg each protein extract was derivatized with 2,4-dinitrophenylhy-drazine and immunoblotted (OxyBlot Protein Oxidation Detection Kit; Chemicon) according to the manufacturer’s protocol.
RNA In Situ Hybridization
For in situ hybridization, paraffin sections were deparaffinized using all RNase-free solvents and buffers. The mouse Rdh11 antisense probe (5′-GACCAAGTACAGGATGAAGGGAAGAGAGAGCAGAAGC-AGGAATCCGAA-3′) was synthesized and biotin labeled by GeneDetect (Bradenton, FL). In situ hybridizations were performed according to the protocol provided at http://www.genedetect.com/Merchant2/InsituParaffinBIOTIN.pdf, with pepsin for permeabilization and 1:50 dilution of the probe. The probe was detected (TSA Biotin System; Perkin Elmer, Waltham, MA) and visualized with horseradish peroxidase chromogenic substrate diaminobenzidine.
RESULTS
Transcription of Rdh11 and Rdh12 Genes in Mouse Retina
RDH12 localization in the retina has been previously investigated by using in situ hybridization6 and immunohistochemistry.8,9 These studies showed that both the mRNA and the protein are located in rod inner segments and cell bodies. Localization of RDH11 in the retina was also investigated.6,10,13 Immunohistochemistry results were contradictory. One group showed expression in the retinal pigment epithelium (RPE),6 and our group showed expression in various layers of the retina, including photoreceptor inner segments.10 In another study, we further investigated the localization of Rdh11 gene transcription by taking advantage of the knockout mice in which the Rdh11 gene was replaced by LacZ, leaving the promoter of Rdh11 intact.13 In these mice, β-galactosidase activity was found exclusively in photoreceptor inner segments, demonstrating an active transcription in photoreceptor cells.13 However, it could be argued that Rdh11 expression in some tissues, such as RPE, was strictly dependent on intronic sequences of Rdh11 removed in the knockout.
We dissected mouse eyes and measured by quantitative RT-PCR the levels of Rdh11 and Rdh12 mRNA in the retina, iris/ciliary body (Iris/CB), and RPE/choroid/sclera (RPE/Ch/Sc) fractions (Fig. 1A). Rdh11 mRNA was found primarily in the retina, but it was also expressed in the RPE/Ch/Sc at a lower level (25% of the signal is located in this fraction). We quantified rhodopsin (Rho) and Rpe65 levels in the retina and RPE/Ch/Sc as a control for the purity of the dissected fractions (Fig. 1B). We found less than 3% cross-contamination between the two fractions. Therefore, the expression of Rdh11 in the RPE/Ch/Sc cannot be explained by a contamination of retina. Rdh12 mRNA was found in the retina and was undetectable in the RPE/Ch/Sc, confirming the previously published results.6,8,9
Figure 1.

Localization and quantification of Rdh11 and Rdh12 mRNAs in adult C57BL/6 mouse eye. (A, B) Quantification of indicated mRNAs in dissected mouse retina, iris/ciliary body, and RPE/choroid/sclera (RPE/CH/Sc). Total RNA from three 10-month-old adult mice were prepared individually and subjected to real-time PCR quantification. Each value represents the average amount of indicated mRNA relative to that of Rpl19, which was arbitrarily defined as 1. Error bars represent the SD of the three samples. (C) Low-power view of bright-field–illuminated eye sections from wild-type (WT) and Rdh11 knockout (KO) albino mice hybridized with an antisense Rdh11 probe. Note the absence of signal in photoreceptor inner segments (IS, arrow) in the knockout mouse, indicating that this signal is specific for Rdh11 mRNA. OS, rod outer segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
We further investigated the localization of Rdh11 mRNA by in situ hybridization (Fig. 1C). As a negative control, we used a section from Rdh11 knockout mouse. The Rdh11-specific signal (present in wild-type but not in knockout mice) was located in photoreceptor inner segments (IS; arrow), confirming our previous β-galactosidase results.13 No specific signal was detected in the RPE/Ch/Sc, suggesting that Rdh11 expression level is lower in these cells, probably below the minimum level of detection for in situ hybridization.
Relative Expression Levels of RDH11 and RDH12
As shown in Figure 1A, Rdh12 mRNA is 40-fold more abundant than Rdh11 mRNA (Rdh12 mRNA level, 2; Rdh11 mRNA level, 0.05) in the 10-month-old retina. To confirm the higher expression of RDH12 at the protein level, we used affinity-purified polyclonal antibodies raised against a specific sequence of 16 amino acids located in the C-terminal portion of each protein. We first estimated the relative affinities of each antibody against its antigen (Fig. 2A). We transfected HEK-293 cells with expression constructs encoding flag-tagged versions of RDH11 and RDH12. After immunoprecipitation with anti-flag antibody, we compared the amounts of RDH11-Flag and RDH12-Flag proteins (1.0 and 1.9, respectively) using the anti-flag antibody. We then stripped the blot, cut it in half, and hybridized the half containing RDH11-Flag with anti-RDH11 antibody and the half containing RDH12-Flag with anti-RDH12 antibody. Dilution of antibodies, incubation times, and exposure times were the same for the two halves. Signal intensities for RDH11-Flag and RDH12-Flag were 1.0 and 2.3, respectively. We concluded that if the affinity of anti-RDH11 for RDH11 is arbitrarily defined as 1.0, the affinity of anti-RDH12 for RDH12 is 1.2 (2.3/1.9).
Figure 2.
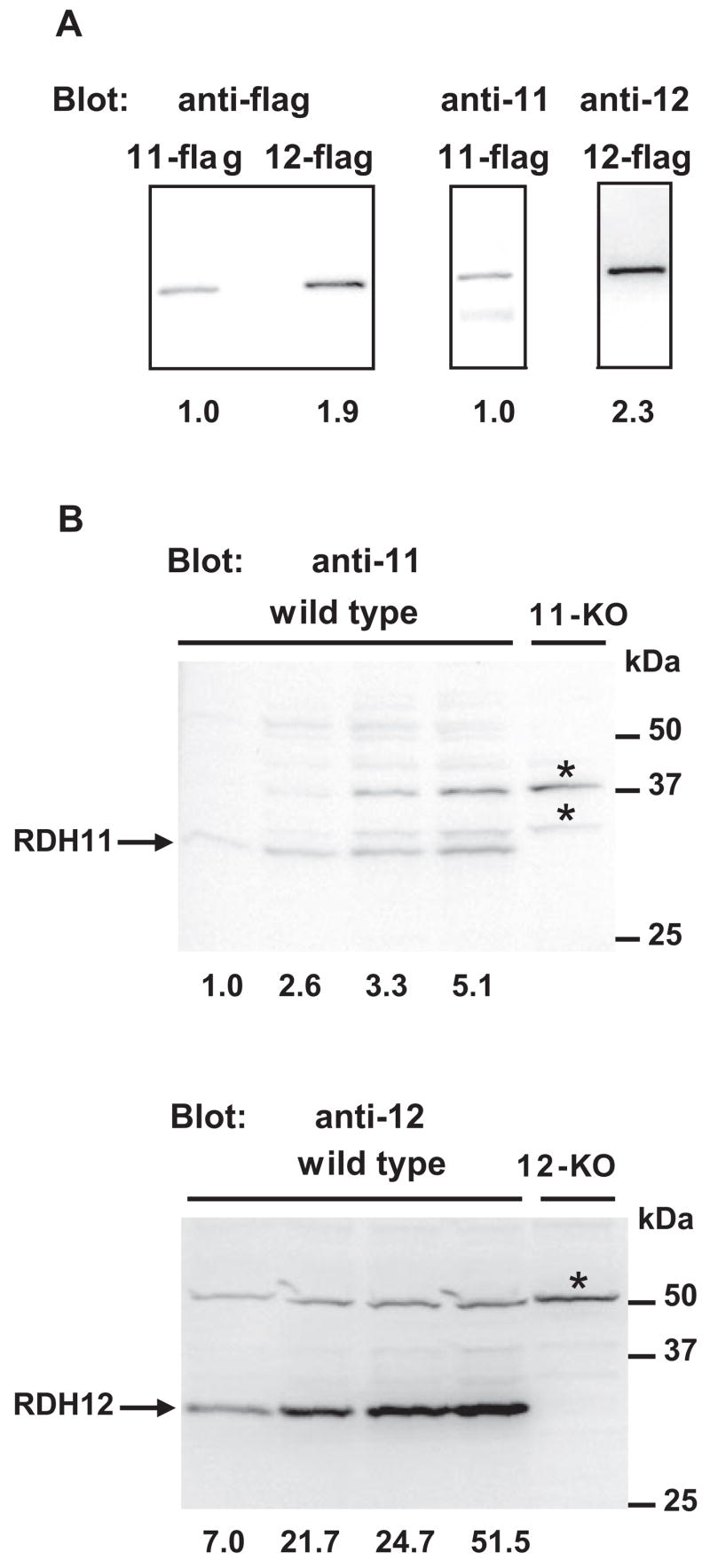
Quantification of the relative expression levels of the RDH11 and RDH12 proteins in C57BL/6 mouse retina. (A) Measure of the relative affinities of anti-RDH11 and anti-RDH12 antibodies for their respective antigens. On day 0, HEK293 cells were transfected with flag-tagged RDH11 and RDH12 (11-flag and 12-flag, respectively) using transfection reagent (FuGene 6; Roche, Indianapolis, IN), according to the manufacturer’s protocols. On day 3, cells were harvested and proteins were immunoprecipitated with anti-flag antibody coupled to agarose beads (Sigma) according to the manufacturer’s instructions. After elution in Laemmli buffer, samples were subjected to SDS-PAGE, then immunoblotted with an anti-flag antibody coupled with HRP. The blot was then stripped and reprobed with a 1:500 dilution of anti-RDH11 or anti-RDH12 antibody, as indicated. Signals were quantified using the (Image Station 4000R; Kodak) and expressed relative to 11-flag arbitrarily defined as 1. (B) RDH11 and RDH12 relative expression in C57BL/6 mouse retina. Four retinas from 2-month-old wild-type, Rdh11 knockout (11-KO), or Rdh12 knockout (12-KO) mice were pooled before homogenization and protein extraction using reagent (T-PER; Pierce). Aliquots (5, 10, 20, 40 μg) of wild-type protein extract and 40 μg indicated knockout extract were subjected to SDS-PAGE, then immunoblotted with a 1:500 dilution of anti-RDH11 or anti-RDH12 antibody. Signals were quantified and expressed relative to the first line containing 5 μg protein and blotted with anti-RDH11, which was arbitrarily defined as 1. *Nonspecific bands.
Figure 2B shows the specificity of each antibody for its antigen. Increasing amounts of protein (5–40 μg) from wild-type retina were run with 40 μg protein from Rdh11 knockout retina (Fig. 2B, top). As expected, one band corresponding to RDH11 was found around 31 kDa; it was the only band that disappeared in the Rdh11 knockout. Two unknown proteins cross-reacted with the anti-RDH11 antibody, but they did not correspond to a modified version of RDH11 because they were still present in the knockout. A similar experiment was conducted using the Rdh12 knockout retinal extract as a negative control (Fig. 2B, bottom). The specific signal corresponding to RDH12 was found around 31 kDa and was absent in the knockout. Another unknown protein cross-reacted with the anti-RDH12 antibody, but no cross-reactivity occurred between the RDH12 antibody and the anti-RDH11 antibody. We quantified the RDH11 and RDH12 signals in these blots. Protein amounts, antibody dilutions, incubation times, and exposure times were the same for the two blots. As shown in Figure 2B, the amount of RDH12 was 7- to 10-fold higher than the amount of RDH11 (average result was 8.2-fold). Taking into account the slightly higher affinity of RDH12 antibody, we concluded that we have sevenfold (8.2/1.2 = 6.8) approximately more RDH12 protein than RDH11 protein in 2-month-old dissected mouse retina. mRNA and protein quantifications showed that RDH12 was significantly more abundant than RDH11 in the mouse retina, but the differences were less pronounced at the protein than at the RNA level.
Expression of RDH11 and RDH12 during Development
We then quantified the expression levels of Rdh11 and Rdh12 in mouse eye globes without lens from E12 to 7 months using real time RT-PCR. As shown in Figure 3A, Rdh11 mRNA levels were low but relatively constant at all times. Rdh12 mRNA levels were lower than Rdh11 and almost undetectable until P3. Then Rdh12 increased rapidly between P3 and P15. After P15, Rdh12 expression continued to increase at least to 7 months of age but at a lower rate.
Figure 3.

Expression levels of RDH11 and RDH12 during development in C57BL/6 mouse eye. (A) Quantification of Rdh11 and Rdh12 mRNA. For each time point, four samples were prepared from eye globes minus the lens and subjected to real-time PCR quantification. Each value represents the average amount of mRNA relative to that of Rpl19, which was arbitrarily defined as 1. Error bars represent the SD for the four samples. (B) Immunoblots showing RDH11 and RDH12 during development. For each time point, four individual samples (eye globes minus the lens) were pooled before homogenization and protein extraction using T-PER reagent. Aliquots (40 μg) of protein extracts were subjected to SDS-PAGE, then immunoblotted with a 1:500 dilution of anti-RDH11 or anti-RDH12 antibody. (C) Protein levels during development. Signals for RDH11 and RDH12 were quantified from the immunoblots shown in (B) and plotted.
We performed a similar analysis at the protein level (Figs. 3B, 3C). The results were essentially similar to those obtained with mRNAs; RDH11 levels were low but detectable and relatively constant at all times. RDH12 expression started at P7 and increased until P30. The RDH12 protein level then remained stable even though mRNA levels were still increasing.
Regulation of Rdh11 and Rdh12 Expression during Light Exposure
Because of the possibility that RDH11 and RDH12 could be protective against the toxicity of (hydroxy)aldehydes produced during lipid peroxidation, we quantified Rdh11 and Rdh12 mRNA levels during light-induced oxidative stress. An induction of these genes by oxidative stress would argue in favor of such function. Exposure to bright constant light has been shown to induce oxidative stress in the rodent retina.23 We used BALB/c mice because their sensitivity to light damage has been well characterized.24,25 All the mice were humanely killed at the same time, after being dark adapted, light adapted, or subjected to light damage (constant bright light set at 3000 lux) from 2 to 24 hours. Histologic examination of these retinas performed 7 days after light exposure (Fig. 4A) showed shortening of rod outer segments after 2 hours of light exposure and a progressive loss of photoreceptor cell nuclei starting after 2 hours of light exposure and reaching its maximum after 12 hours of light exposure. Oxidative modification of retinal proteins was detected by immunoblotting immediately after light exposure (Fig. 4B). Protein carbonylation started to increase at 2 hours of light exposure and reached its maximum at 12 hours of light exposure. These results showed the extent of retinal damage caused by exposure of BALB/c mice to bright light in this experiment. We measured the mRNA and protein levels of RDH11 and RDH12 immediately after light exposure (Figs. 5, 6). As shown in Figure 5, Rdh11 mRNA remained remarkably stable until 24 hours of light damage, whereas Rdh12 mRNA decreased rapidly after 8 hours of light damage to reach 25% of its original level at 24 hours. As a control for the induction of specific genes during light-induced oxidative stress, we used heme oxygenase 1 (HO-1).26 As shown in Figure 5, the level of HO-1 mRNA increased after 6 hours of light damage until 12 hours of light damage. We concluded that there is no such induction of Rdh11 and Rdh12 genes during acute light-induced oxidative stress.
Figure 4.

Retinal damage induced by light exposure. (A) Histology showing morphologic changes in retina after light exposure. Ten-week-old BALB/c mice were light adapted (LA) or exposed to bright light (3000 lux) for the indicated times and were killed 7 days after light exposure. Hematoxylin and eosin staining was performed on paraffin sections of eyeballs. Three independent experiments were conducted with reproducible results, and representative data are shown. Shortening of the rod outer segments and loss of photoreceptor cell nuclei in the ONL were observed after light exposure. RPE/Ch/Sc, RPE/choroid/sclera; OS, rod outer segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. All panels are 0.5 to 1 mm superior to the optic nerve head. (B) Immunoblot showing oxidative modification of proteins during light exposure. BALB/c mice were light adapted (LA) or exposed to light damage (LD) for indicated times and were killed immediately after light exposure. For each condition, retinas from three BALB/c mice were pooled before homogenization and protein extraction using reagent (T-PER; Pierce). Aliquots (20 μg) of fresh protein extracts were derivatized and immunoblotted using a protein oxidation detection kit (OxyBlot; Chemicon) according to the manufacturer’s protocol.
Figure 5.

Quantification of Rdh11 and Rdh12 mRNA during light exposure. Four 10-week-old BALB/c mice were used for each condition—dark-adapted (DA), light-adapted (LA), and light-damaged (LD)—for the indicated times. Total RNAs were prepared from one retina from each mouse and subjected to real-time PCR quantification. Each value represents the average amount of indicated mRNA relative to that of Rpl19, which was arbitrarily defined as 1. Error bars represent the SD for the four samples.
Figure 6.

Quantification of RDH11 and RDH12. (A) Immunoblots showing RDH11 and RDH12 during light exposure. For each condition, four retinas from four BALB/c mice were pooled before homogenization and protein extraction using reagent (T-PER; Pierce). Aliquots (40 μg) of protein extracts were subjected to SDS-PAGE, then immunoblotted with a 1:500 dilution of anti-RDH11 or anti-RDH12 antibody. As a loading control, immunoblots were probed with anti-actin antibody at a 1:1000 dilution. (B) Protein levels during light exposure. Signals for RDH11 and RDH12 were quantified from the immunoblots shown in (A) and from two additional sets of immunoblots. Averages were plotted relative to RDH12 DA, arbitrarily defined as 1. Error bars represent the SD from the three immunoblots.
We confirmed the stable amount of RDH11 and the decrease of RDH12 at the protein level (Figs. 6A, 6B). However, as shown in Figure 6B, the decrease of RDH12 starts earlier than 8 hours. After a slight increase of RDH12 protein level at 2 hours of light damage, the protein level decreased until 8 hours of light damage, even though the mRNA level was still stable during this time. Then the protein level remained stable until 24 hours of light damage, at approximately 50% of its original level. In this experiment, we used one retina for RNA and one retina for protein from the same mouse so that we could relate the mRNA and protein changes in the same set of mice.
DISCUSSION
To gain some insight into the physiological functions of RDH11 and RDH12, we investigated their gene expression in the developing and aging (7-month-old) mouse retina and under conditions of acute oxidative stress.
Retinal development is a complex process including the generation of seven distinct retinal cell types, neuronal differentiation, vascularization, and the onset of vision.27,28 Retinal expression profiles of thousands of genes have been analyzed during mouse retinal development and grouped by clusters of distinct expression profiles.29,30 Correlation between gene expression and specific postnatal developmental events can be used as an indication for a gene function. According to Dorrell et al.,29 50% of the genes expressed in the retina have minimal expression changes and vary less than threefold during development. Many common housekeeping genes have expression that remains constant even during extreme physiological changes, such as those occurring during development. According to our study, Rdh11 falls into this category; it remains constant during development and during light-induced acute oxidative stress, suggesting a housekeeping function for this gene.
In contrast, Rdh12 has a low expression level at birth, with expression increasing after P3 and throughout postnatal development. The onset of RDH12 protein expression coincides with the elongation of the rod outer segments (the first disks appear at P631), and RDH12 increases rapidly until P30. During this period the onset of vision (at P12, when the pups open their eyes) and the maturation of the visual process take place.31 The elongation of the rod outer segments takes place during the third trimester in human retinal development,31,32 suggesting that in humans, RDH12 may already be expressed at birth with the onset of vision.
Many of the genes in this category of expression profile are directly or indirectly related to the visual process.29 It has been shown that a functional visual cycle and efficient recycling of photosensitive rhodopsin are required to mediate light-induced oxidative stress,23 establishing the concomitant onset of vision and light-induced oxidative stress. Therefore, an enzyme that detoxifies byproducts of oxidative stress would be needed at the onset of vision and at all times thereafter.
In the second part of this study, we explored the possibility that Rdh11 and Rdh12 can have a detoxification function during oxidative stress. We reasoned that if Rdh11 and Rdh12 do have such function, then these genes would be induced during oxidative stress. We found that Rdh11 mRNA and protein were not induced during light-induced oxidative stress. Similarly, we did not find any upregulation of the Rdh12 gene during oxidative stress, as tested in our conditions. However, we found a rapid decrease of Rdh12 mRNA after 8 hours of light damage. cDNA microarray analysis of retinal gene transcription during light damage has shown that during the early phase of light damage, up to 7 hours in BALB/c mice, gene transcription does not decrease in photoreceptors. In fact, some genes are even upregulated.33 After 8 hours of light damage, however, we observed a general shutdown of gene transcription in photoreceptor cells. We quantified the expression of two other photoreceptor-specific genes, Elovl4 and Rom-1, and found that their mRNA levels were stable until 8 hours of light damage and then decreased between 8 and 24 hours of light damage, as in Rdh12 (not shown). Therefore, the decrease in Rdh12 mRNA probably was not caused by a specific downregulation of this gene but rather by a nonspecific shutdown of gene transcription.
Interestingly, we found a moderate and transient increase of RDH12 protein at 2 hours of light damage that was followed by a decrease in RDH12 protein level starting after only 2 hours of light damage This was 6 hours before the mRNA decrease started in the same set of mice, and it suggested that the regulation of RDH12 takes place at the protein level. Our results suggested that an induction in RDH12 protein degradation takes place early during light damage. We can speculate that this is induced by oxidative modification of the protein, leading to its degradation by the proteasome system. Oxidation of proteins can be induced directly by reactive oxygen species and by indirect modification by the secondary byproducts of oxidative stress such as 4-hydroxynonenal (4-HNE).34 Because of its microsomal localization and its catalytic activity on 4-HNE,7 RDH12 may be particularly exposed to 4-HNE modifications during oxidative stress. In the hypothesis that RDH12 does have a detoxification role, the rapid decrease of the protein level may precipitate the damage caused by light-induced oxidative stress. The mechanism for this decrease will have to be investigated further.
Acknowledgments
The authors thank Krzysztof Palczewski for the Rdh12 knockout mice, Robert E. Anderson and John D. Ash for helpful discussions, and Linda Boone and Louisa Williams for technical assistance with histology.
Supported by National Center for Research Resources Grant P20RR017703 and National Eye Institute Grants P30EY12190 and R01EY14052. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, the National Eye Institute, or the National Institutes of Health.
Footnotes
Disclosure: Y. Kanan, None; L.D. Wicker, None; M.R. Al-Ubaidi, None; M.N.A. Mandal, None; A. Kasus-Jacobi, None
References
- 1.Perrault I, Hanein S, Gerber S, et al. Retinal dehydrogenase 12 (RDH12) mutations in Leber congenital amaurosis. Am J Hum Genet. 2004;75(4):639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janecke AR, Thompson DA, Utermann G, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36(8):850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 3.Thompson DA, Janecke AR, Lange J, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum Mol Genet. 2005;14(24):3865–3875. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- 4.Schuster A, Janecke AR, Wilke R, et al. The phenotype of early-onset retinal degeneration in persons with RDH12 mutations. Invest Ophthalmol Vis Sci. 2007;48(4):1824–1831. doi: 10.1167/iovs.06-0628. [DOI] [PubMed] [Google Scholar]
- 5.Sun W, Gerth C, Maeda A, et al. Novel RDH12 mutations associated with Leber congenital amaurosis and cone-rod dystrophy: biochemical and clinical evaluations. Vis Res. 2007;47(15):2055–2066. doi: 10.1016/j.visres.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haeseleer F, Jang GF, Imanishi Y, et al. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem. 2002;277(47):45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry. 2005;44(18):7035–7047. doi: 10.1021/bi050226k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda A, Maeda T, Imanishi Y, et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem. 2006;281(49):37697–37704. doi: 10.1074/jbc.M608375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurth I, Thompson DA, Rèuther K, et al. Targeted disruption of the murine retinal dehydrogenase gene Rdh12 does not limit visual cycle function. Mol Cell Biol. 2007;27(4):1370–1379. doi: 10.1128/MCB.01486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasus-Jacobi A, Ou J, Bashmakov YK, et al. Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2003;278(34):32380–32389. doi: 10.1074/jbc.M304969200. [DOI] [PubMed] [Google Scholar]
- 11.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Ann Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim TS, Maeda A, Maeda T, et al. Delayed dark adaptation in 11-cis-retinol dehydrogenase deficient mice: a role of RDH11 in visual processes in vivo. J Biol Chem. 2005;280(10):8694–8704. doi: 10.1074/jbc.M413172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasus-Jacobi A, Ou J, Birch DG, et al. Functional characterization of mouse RDH11 as a retinol dehydrogenase involved in dark adaptation in vivo. J Biol Chem. 2005;280(21):20413–20420. doi: 10.1074/jbc.M413789200. [DOI] [PubMed] [Google Scholar]
- 14.Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23(4):306–317. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 15.Kedishvili NY, Chumakova OV, Chetyrkin SV, et al. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1) J Biol Chem. 2002;277(32):28909–28915. doi: 10.1074/jbc.M202588200. [DOI] [PubMed] [Google Scholar]
- 16.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30(4):277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 17.Awasthi YC, Sharma R, Cheng JZ, et al. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol Aspects Med. 2003;24(4–5):219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Bioch Polonica. 2003;50(2):319–336. [PubMed] [Google Scholar]
- 19.Tanito M, Elliott MH, Kotake Y, Anderson RE. Protein modifications by 4-hydroxynonenal and 4-hydroxyhexenal in light-exposed rat retina. Invest Ophthalmol Vis Sci. 2005;46(10):3859–3868. doi: 10.1167/iovs.05-0672. [DOI] [PubMed] [Google Scholar]
- 20.Tanito M, Anderson RE. Bright cyclic light rearing-mediated retinal protection against damaging light exposure in adrenalectomized mice. Exp Eye Res. 2006;83(3):697–701. doi: 10.1016/j.exer.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 21.Mandal MN, Ayyagari R. Complement factor H: spatial and temporal expression and localization in the eye. Invest Ophthalmol Vis Sci. 2006;47(9):4091–4097. doi: 10.1167/iovs.05-1655. [DOI] [PubMed] [Google Scholar]
- 22.Mandal MN, Vasireddy V, Reddy GB, et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci. 2006;47(12):5505–5513. doi: 10.1167/iovs.06-0312. [DOI] [PubMed] [Google Scholar]
- 23.Wenzel A, Grimm C, Samardzija M, Remâe CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retinal Eye Res. 2005;24(2):275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 24.LaVail MM, Gorrin GM, Repaci MA, Yasumura D. Light-induced retinal degeneration in albino mice and rats: strain and species differences. Prog Clin Biol Res. 1987;247:439–454. [PubMed] [Google Scholar]
- 25.Kaldi I, Martin RE, Huang Brush RS, Morrison KA, Anderson RE. Bright cyclic rearing protects albino mouse retina against acute light-induced apoptosis. Mol Vis. 2003;9:337–344. [PubMed] [Google Scholar]
- 26.Kutty RK, Kutty G, Wiggert B, Chader GJ, Darrow RM, Organisciak DT. Induction of heme oxygenase 1 in the retina by intense visible light: suppression by the antioxidant dimethylthiourea. Proc Natl Acad Sci USA. 1995;92(4):1177–1181. doi: 10.1073/pnas.92.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith RS, Kao WWY, John SWM. Ocular development. In: Smith RS, John SWM, Nishina PM, Sundberg JP, editors. Systematic Evaluation of the Mouse Eye: Anatomy, Pathology, and Biomethods. New York: CRC Press; 2002. pp. 45–63. [Google Scholar]
- 28.Kaufman MH, Bard JBL. The Anatomical Basis of Mouse Development. San Diego: Academic Press; 1999. [Google Scholar]
- 29.Dorrell MI, Aguilar E, Weber C, Friedlander M. Global gene expression analysis of the developing postnatal mouse retina. Invest Ophthalmol Vis Sci. 2004;45(3):1009–1019. doi: 10.1167/iovs.03-0806. [DOI] [PubMed] [Google Scholar]
- 30.Blackshaw S, Harpavat S, Trimarchi J, et al. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2(9):E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grün G. The development of the vertebrate retina: a comparative survey. Adv Anat Embryo Cell Biol. 1982;78:1–85. [PubMed] [Google Scholar]
- 32.Nag TC, Wadhwa S. Morphological and neurochemical development of the human neural retina. Neuroembryol Aging. 2006–07;4:19–30. [Google Scholar]
- 33.Chen L, Wu W, Dentchev T, et al. Light damage induced changes in mouse retinal gene expression. Exp Eye Res. 2004;79(2):239–247. doi: 10.1016/j.exer.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Chakravarti B, Chakravarti DN. Oxidative modification of proteins: age-related changes. Gerontology. 2007;53(3):128–139. doi: 10.1159/000097865. [DOI] [PubMed] [Google Scholar]