

Abstract
The enzymatic methylation of inorganic As (iAs) is catalyzed by As(+3 oxidation state)-methyltransferase (AS3MT). AS3MT is expressed in rat liver and in human hepatocytes. However, AS3MT is not expressed in UROtsa, human urothelial cells that do not methylate iAs. Thus, UROtsa cells are an ideal null background in which the role of iAs methylation in modulation of toxic and cancer-promoting effects of this metalloid can be examined. A retroviral gene delivery system was used in this study to create a clonal UROtsa cell line (UROtsa/F35) that expresses rat AS3MT. Here, we characterize the metabolism and cytotoxicity of arsenite (iAsIII) and methylated trivalent arsenicals in parental cells and clonal cells expressing AS3MT. In contrast to parental cells, UROtsa/F35 cells effectively methylated iAsIII, yielding methylarsenic (MAs) and dimethylarsenic (DMAs) containing either AsIII or AsV. When exposed to MAsIII, UROtsa/F35 cells produced DMAsIII and DMAsV. MAsIII and DMAsIII were more cytotoxic than iAsIII in UROtsa and UROtsa/F35 cells. The greater cytotoxicity of MAsIII or DMAsIII than of iAsIII was associated with greater cellular uptake and retention of each methylated trivalent arsenical. Notably, UROtsa/F35 cells were more sensitive than parental cells to the cytotoxic effects of iAsIII but were more resistant to cytotoxicity of MAsIII. The increased sensitivity of UROtsa/F35 cells to iAsIII was associated with inhibition of DMAs production and intracellular accumulation of MAs. The resistance of UROtsa/F35 cells to moderate concentrations of MAsIII was linked to its rapid conversion to DMAs and efflux of DMAs. However, concentrations of MAsIII that inhibited DMAs production by UROtsa/F35 cells were equally toxic for parental and clonal cell lines. Thus, the production and accumulation of MAsIII is a key factor contributing to the toxicity of acute iAs exposures in methylating cells.
Keywords: As, Methyltransferase, AS3MT, Metabolism, Toxicity, Urinary bladder, Human
Introduction
Biomethylation is the major pathway for the metabolism of inorganic As (iAs) in humans and other mammals. In humans, iAs is converted to methylarsenic (MAs) and dimethylarsenic (DMAs) that contain As (As) in +3 (AsIII) or +5 (AsV) oxidation state (Cullen et al., 1984). In rats and hamsters, DMAs is further methylated to yield trimethylarsine oxide (TMAsVO) (Yamauchi and Yamamura, 1985; Yoshida et al., 1998). Two classes of enzymes are involved in the methylation of iAs, including AsV-reductases (Gregus and Nemeti, 2002; Radabaugh et al., 2002; Zakharyan and Aposhian, 1999; Zakharyan et al., 2001) and AsIII-methyltransferases (Lin et al., 2002; Zakharyan et al., 1995). The metabolic conversion of iAs is generally recognized to be a determinant of the toxic and carcinogenic effects of this metalloid. However, the roles of specific AsIII- and AsV-containing metabolites in mediation of these adverse effects have not been characterized. Methylated pentavalent arsenicals, methylarsonic acid (MAsV), dimethylarsinic acid (DMAsV), and TMAsVO, which are less acutely toxic than either arsenate (iAsV) or arsenite (iAsIII) (Yamauchi and Fowler, 1994), are thought to be detoxification products. However, unlike iAs species, DMAsV and TMAsVO at high doses are complete carcinogens in adult rats (Shen et al., 2003; Wei et al., 1999). In contrast, methylated trivalent arsenicals, methylarsonous acid (MAsIII) and dimethylarsonous acid (DMAsIII), are more toxic than iAs in vivo (Petrick et al., 2001) and are more potent than iAs as enzyme inhibitors (Lin et al., 2001; Petrick et al., 2001; Styblo et al., 1997), cytotoxins (Petrick et al., 2000; Styblo et al., 2000, 2002), and as modulators of cell signaling pathways (Drobná et al., 2003; Walton et al., 2004). In addition, MAsIII and DMAsIII are more potent than iAs as DNA-damaging agents in vitro and in cultured cells (Kligerman et al., 2003; Mass et al., 2001). Thus, MAsIII and DMAsIII formed in the course of iAs methylation likely contribute to the toxicity and cancer-promoting effects associated with acute and chronic exposures to iAs. The pattern and magnitude of this contribution depend on rates of the production of MAsIII and DMAsIII from iAs and on the distribution and retention of these metabolites.
The kinetics of iAs methylation have been studied in vitro using recombinant rat As(+3 oxidation state)-methyltransferase (AS3MT; previously designated as cyt19) (Devesa et al., 2004; Waters et al., 2004a). Rat AS3MT (rAS3MT) is an S-adenosylmethionine-dependent enzyme (Mr 42 kDa) that is widely expressed in rat tissues, e.g., liver, heart, lung, or kidney (Lin et al., 2002). In the presence of a reductant, Tris(2-carboxylethyl)-phosphine, or a thioredoxin/thioredoxin reductase/NADPH coupled reaction system, rAS3MT methylates iAsIII, yielding all known mono-, di-, and trimethylated AsIII- and AsV-containing metabolites (Devesa et al., 2004). Thus, rAS3MT catalyzes both the oxidative methylation of trivalent arsenicals and the reduction of pentavalent intermediates to trivalency: iAsIII → MAsV → MAsIII → DMAsV → DMAsIII → TMAsVO. Addition of glutathione (GSH) to a rAS3MT-containing reaction mixture stimulates synthesis of MAs and DMAs from iAsIII, but higher GSH concentrations (≥0.5 mM) reduce TMAsVO formation (Waters et al., 2004a, 2004b). In the absence of GSH, TMAsVO is the predominant product of iAsIII methylation catalyzed by recombinant rAS3MT. In addition, a fraction of TMAsVO is reduced by rAS3MT to volatile trimethylarsine (TMAsIII) (Waters et al., 2004b). Human AS3MT is encoded by a gene orthologous to rat AS3MT (Lin et al., 2002). Human AS3MT is expressed in primary human hepatocytes (Drobná et al., 2004) that are efficient methylators of iAsIII (Drobná et al., 2004; Styblo et al., 1999). In contrast, AS3MT is not expressed in UROtsa cells, an SV-40-transformed normal human urinary bladder epithelial cell line that does not methylate iAsIII (Lin et al., 2002; Styblo et al., 2000). Despite this marked difference in capacity to methylate iAsIII, cultured human hepatocytes and UROtsa cells are about equally sensitive to the cytotoxic effects of this arsenical (Styblo et al., 2000, 2002). Thus, the relationship between the capacity to methylate iAs and the susceptibility to acute toxic effects of iAs exposure remains unclear.
To examine the role of the AS3MT-dependent metabolism in modulation of the toxic and cancer-promoting effects of iAs in cells, we created a clonal UROtsa cell line (UROtsa/F35) that expresses rAS3MT and, unlike the parent UROtsa cell line, methylates iAsIII and MAsIII. Studies with UROtsa and UROtsa/F35 cell lines show that AS3MT expression and capacity to methylate trivalent arsenicals are associated with decreased resistance to cytotoxicity of iAsIII and increased resistance to cytotoxicity of MAsIII. The differences in susceptibility to the cytotoxicities of iAsIII and MAsIII are examined in relation to the kinetics and dose–response patterns of iAsIII and MAsIII metabolism.
Materials and methods
Arsenicals
The following arsenicals were used for the treatment of cultured cells and/or as standards for instrumental analysis of As metabolites in treated cell cultures: Radiolabeled carrier-free [73As]iAsIII was prepared by reduction of [73As]iAsV (Oak Ridge National Laboratory, Oak Ridge, TN) with metabisulfite–thiosulfate reagent (Reay and Asher, 1977). iAsIII and iAsV, sodium salts (>98% pure), were obtained from Sigma (Saint Louis, MO). MAsV, disodium salt (98% pure), was obtained from Chem Service (West Chester, PA). DMAsV (dimethylarsinic acid, 98% pure) was purchased from Strem Chemicals Inc. (Newburyport, MA). Methylarsine oxide, iododimethylarsine, and TMAsVO were synthesized by Professor William R. Cullen (University of British Columbia, Vancouver, Canada) as previously described (Cullen et al., 1984; Styblo et al., 1997). In aqueous solutions, methylarsine oxide and iododimethylarsine form MAsIII and DMAsIII, respectively (Petrick et al., 2001). Identity and purity of the custom-synthesized arsenicals were confirmed by 1H-NMR, mass spectrometry, and hydride generation-atomic absorption spectrometry.
Cell lines and cell culture
UROtsa cells were obtained from Dr. N. Unimye, Department of Urology, School of Medicine, West Virginia University. The clonal UROtsa/F35 cell line expressing rAS3MT was created from the parent UROtsa cell line, using a retroviral pLEGFP-N1 gene delivery system (Clontech, Palo Alto, CA). Briefly, rat liver cDNA was PCR amplified, using primers specific for the rAS3MT mRNA sequence (Lin et al., 2002). The PCR product (1.1 kb) was cloned into the pLEGFP-N1 retroviral vector. The pLEGFP-N1/rAS3MT vector was used to transform DH5α Escherichia coli strain (Stratagene, La Jolla, CA). Ampicillin-selected E. coli colonies were screened for the presence of the rAS3MT insert. To confirm its identity, the insert was sequenced, using CMV (forward) and EGFP-N (reverse) PCR primers: 5′-GGATAGCGGTTTGACTCACGC-3′ and 5′-CGTCGCCGTCCAGCTCGACCAG-3′, respectively. Positive E. coli colonies were multiplied overnight at 37 °C in Luria–Bertani broth (Invitrogen, Carlsbad, CA) in the presence of ampicillin (50 μg/ml). The pLEGFP-N1/rAS3MT plasmid was purified from the E. coli culture, using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA). AmphoPack-293 cells (Stratagene) were transfected with the pLEGFP-N1/rAS3MT plasmid in the presence of FuGENE 6 (Roche, Florence, SC). Stable AmphoPack-293 cell lines producing AS3MT-encoding retrovirus were established in medium containing 1 mg geneticin (G418, GIBCO)/ml. Retrovirus-containing media from these cell lines were used to transduce UROtsa cells. Transduced UROtsa clone (UROtsa/F35) was selected in culture medium containing G418 (250 μg/ml). Both UROtsa and UROtsa/F35 cells were cultured in MEM (GIBCO BRL, Grand Island, NY) medium supplemented with 10% FBS (GIBCO), 50 U penicillin (Sigma)/ml, and 50 μg streptomycin (Sigma)/ml. Cells were cultured in 96-, 24-, or 6-well plates (Costar Corning Incorporated, Corning, NY) and in 60-mm diameter culture dishes (Falcon, Becton Dickinson Lab-ware, Lincoln Park, NJ) at 37 °C in a humidified incubator in a 95% air/5% CO2 atmosphere. Confluent cell cultures were serum starved for 16 h before use in experiments. These or similar culture conditions, including serum starvation, have previously been used by this and other laboratories in studies that examined the metabolism and toxic effects of iAs in UROtsa cells (Drobná et al., 2003; Simeonova et al., 2000, 2002; Styblo et al., 2000, 2002). Notably, serum starvation for 16 h had no effect on the integrity or morphology of cultured cells.
Analysis of rAS3MT expression by RT-PCR
Total RNA from UROtsa or UROtsa/F35 cells was isolated using TRIzol reagent (Life Technology Inc., Gaithersburg, MD). RNA was treated with RQ1 DNase (Promega, Madison, WI) and purified with an RNeasy Minikit (Qiagen). cDNA was synthesized from 1 μg of total RNA by reverse transcription, using SuperScript II Reverse Transcriptase (Invitrogen) and random primers (Invitrogen). Residual RNA was digested by RNase H (Promega). Aliquots of cDNA corresponding to 100 ng of total RNA each were amplified by a “Touchdown” PCR. The AS3MT-specific PCR primers (forward, 5′-ATTTTGGATCTGGGCAGTGGGAGT-3′, and reverse, 5′-AGTGACCAAACGTGGAGGGCAGA-3′) were used, yielding a 477 bp product. β-Actin served as a housekeeping gene control for RT-PCR analyses. A pair of β-actin-specific PCR primers yielding a 288-bp PCR product was used: forward, 5′-TCATGAAGTGTGACGTTGACATCCGT-3′, and reverse, 5′-CCTAGAAGCATTTGCGGTGCACGATG-3′. The PCR reactions were carried out in a GeneAmp 9700 thermo-cycler (PE Applied Biosystems, Foster City, CA). The PCR products were analyzed by electrophoreses in 1% agarose gel. The pLEGFP-N1/rAS3MT plasmid was used as a positive control for the PCR step of the RT-PCR analysis.
Analysis of rAS3MT expression by immunoblot
The immunoblot analysis of rAS3MT in cultured cells used polyclonal rabbit antiserum raised against the recombinant rAS3MT (Alpha Diagnostic International, San Antonio, TX). For this analysis, cells were lysed in RIPA buffer (50 mM Tris–HCl pH 7.4, 1% NP-40, 0.25% sodium deoxy-cholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and 1 μg/ml of each leupeptin, aprotinin, and pepstatin). Cellular proteins (85 μg) were separated by 10% SDS–PAGE and electroblotted to PVDF membrane. The membrane was blocked for 1 h with 3% non-fat milk in PBS–Tween 20 and probed overnight at 4 °C with the rabbit anti-rAS3MT antiserum (1:1000). The antigen–antibody complex was then visualized, using an anti-rabbit HRP-conjugated antibody (sc-2004; Santa Cruz Biotechnology, Santa Cruz, CA) and the enhanced chemiluminescence Super Signal West Pico reagent (Pierce, Rockford, IL). Optical densities of antigen–antibody complexes were quantified by a computerized GeneGnome imaging system (Syngene, Frederick, MD). A purified recombinant rAS3MT containing a histidine tag (Mr~45 kDa) was used as a positive control for this analysis. The recombinant protein was prepared as previously described (Walton et al., 2003; Waters et al., 2004a).
Analysis of As metabolites by TLC
To evaluate the capacity of UROtsa and UROtsa/F35 cells to methylate iAs, confluent cultures were incubated with carrier-free [73As]iAsIII in the presence or absence of iAsIII carrier. After incubation, radiolabeled metabolites were extracted from cells and media by CuCl (Styblo et al., 1996) and oxidized with hydrogen peroxide. The oxidized (AsV-containing) metabolites were analyzed by TLC, using an isopropanol–acetic acid–water (10:1:2.5) solvent system (Styblo et al., 1995). This solvent system separates iAsV, MAsV, DMAsV, and TMAsVO (Waters et al., 2004b). The distribution of radioactivity on TLC plates was evaluated with a computerized Fuji FLA-2000 imaging system (Fujifilm, Stamford, CT). Because arsenicals in CuCl extracts are oxidized by hydrogen peroxide, this method cannot determine the original oxidation state of As in metabolites in cell lysates and culture media before extraction.
Analysis of AsIII- and AsV-containing metabolites
A pH-specific hydride generation atomic absorption spectrometry (HG-AAS) (Del Razo et al., 2001) was used to analyze arsenicals in cell cultures exposed to iAsIII, MAsIII, or DMAsIII. To permit selective determination of AsIII- and AsV-containing metabolites, the HG-AAS analysis was carried out in freshly harvested cells and media without prior digestion. Here, arsines from iAsIII, MAsIII, DMAsIII, and TMAsVO were generated at pH 6; HG at pH 1 was used to generate arsines from both tri- and pentavalent arsenicals (Devesa et al., 2004). Arsines generated in these reactions were cold-trapped, separated by their boiling points, and analyzed using a Perkin-Elmer model 5100 PC atomic absorption spectrometer (Perkin-Elmer, Norwalk, CT, USA) equipped with a Perkin-Elmer Model EDL II electrodeless discharge lamp operated at 197.3 nm. Differences between atomic absorption spectra detected at pH 1 and 6 were used to calculate concentrations of AsIII- and AsV-containing metabolites. For analysis of iAs, MAs, and DMAs without regard to the oxidation state of As, culture media and cells were digested overnight at 90 °C in 2 M phosphoric acid (ULTREX grade, J.T. Baker, Inc., Phillipsburg, NJ). The HG reaction was carried out at pH 1. The identities of arsenicals in spectral peaks were confirmed using aliquots of samples spiked with standards. Calibration curves for each of the arsenicals (0.5, 2.5, 10, 20, 80 ng As) were generated to quantify results of the analyses at both pH 1 and 6. The detection limits of the pH-specific HG-AAS (ng As) are as follows: iAsIII, 0.27 ng; MAsIII, 0.22 ng; DMAsIII, 0.38 ng; TMAsVO, 0.14 ng; iAsV, 0.40 ng; MAsV, 0.24 ng; and DMAsV, 0.31 ng (Devesa et al., 2004).
Cell viability assay
Cytotoxicities of trivalent arsenicals in cultures of UROtsa and UROtsa/F35 cells were evaluated using the MTT (thiazolyl blue) assay that determines the activity of mitochondrial dehydrogenases in viable cells (Carmichael et al., 1987). For this assay, cells were cultured in 96-well plates to confluence and then exposed to iAsIII, MAsIII, or DMAsIII for 24 h. The MTT assay was performed as previously described (Drobná et al., 2003).
Statistical analysis
Results of the As speciation analyses and the MTT assay were statistically evaluated using Graph-Pad Instat software package (GraphPad Software Inc., San Diego, CA). The one-way analysis of variance (ANOVA) followed by Tukey or Student–Newman–Keuls multiple comparison tests was used to evaluate differences between experimental groups. The differences at the P < 0.05 level were considered statistically significant.
Results
Expression of rAs3MT and methylation of iAs in UROtsa/F35 cells
The mRNA levels for rAS3MT were examined by RT-PCR in the total RNA extracts from UROtsa cells and newly established clonal UROtsa/F35 cells after selection with geneticin (Fig. 1a). The pLEGFP-N1/rAS3MT plasmid was used as a positive control for the PCR reaction step. A single DNA band corresponding to a 500-kb amplicon was detected in the PCR reaction mixture containing either pLEGFP-N1/rAS3MT plasmid or cDNA from UROtsa/F35 cells. No PCR product was found in the PCR reaction mixture containing cDNA from parental UROtsa cells. Immunoblot analysis of protein extracts from either UROtsa or UROtsa/F35 cells detected a strongly immunoreactive protein with an estimated molecular weight of about 48 kDa (Fig. 1b). An immunoreactive 41- to 42-kDa protein band consistent with rAS3MT was detected in UROtsa/F35 cells, but not in parental cells. Immunoblot analysis of a recombinant His-tagged rAS3MT that served as a positive control yielded a protein band of about 44 kDa. The capacity to methylate iAs was examined in cultures of UROtsa and UROtsa/F35 cells incubated for 63 h with carrier-free [73As]iAsIII. Radiolabeled methylated arsenicals, MAsV and DMAsV, but not TMAsVO, were detected by TLC in oxidized extracts from UROtsa/F35 cells and from media (Fig. 1c). In contrast, TLC analysis of UROtsa cultures found only iAsV. Over the 63-h incubation period, UROtsa/F35 cells methylated 75% of carrier-free [73As]iAsIII, yielding mainly DMAs. Metabolic profiles of iAs were also examined in UROtsa and UROtsa/F35 cells incubated for 24-h with [73As]iAsIII at final concentrations from 1 to 100 μM iAsIII (Fig. 2). At low concentrations of iAsIII (1–5 μM), UROtsa/F35 cells retained significantly greater amounts of total As than did UROtsa cells. In contrast, at higher iAsIII concentrations (50 and 100 μM), UROtsa cells retained more total As than did clonal cells. In UROtsa/F35 cultures, the yield of DMAs increased as the concentration of iAsIII increased from 1 to 10 μM. Over this concentration range, DMAs was the major methylated metabolite in both cells and media. At higher iAsIII concentrations, DMAs production decreased. In contrast, the yield of MAs increased in proportion to the initial concentration of iAsIII in culture. The rAS3MT expression patterns and the capacity to methylate iAsIII remained stable and reproducible in UROtsa/F35 cells kept in culture for more than 4 months (i.e., about 35 passages) and did not change after cryopreservation in liquid nitrogen for 4 weeks (data not shown).
Fig. 1.
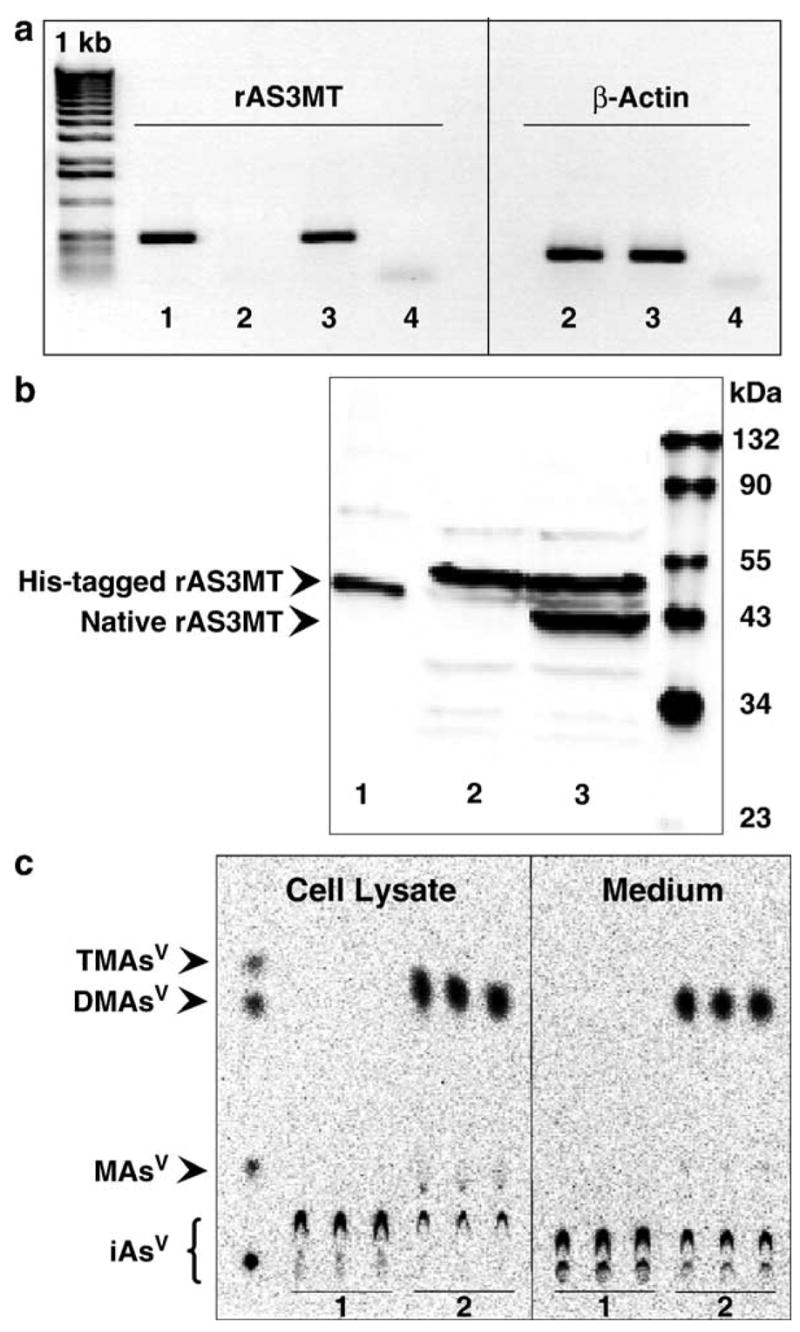
Expression and activity of rAS3MT in UROtsa/F35 cells. (a) RT-PCR analysis: (1) positive control (pLEGFP-N1/rAS3MT plasmid); (2) UROtsa cells; (3) UROtsa/F35 cells; (4) negative control. (b) Immunoblot analysis: (1) recombinant His-tagged rAS3MT (Mr~45 kDa); (2) UROtsa cells; (3) UROtsa/F35 cells expressing native rAS3MT (Mr~41 kDa). (c) TLC analysis of 73As-metabolites in cell cultures exposed to carrier-free [73As]iAsIII for 63 h: (1) UROtsa culture; (2) UROtsa/F35 culture.
Fig. 2.
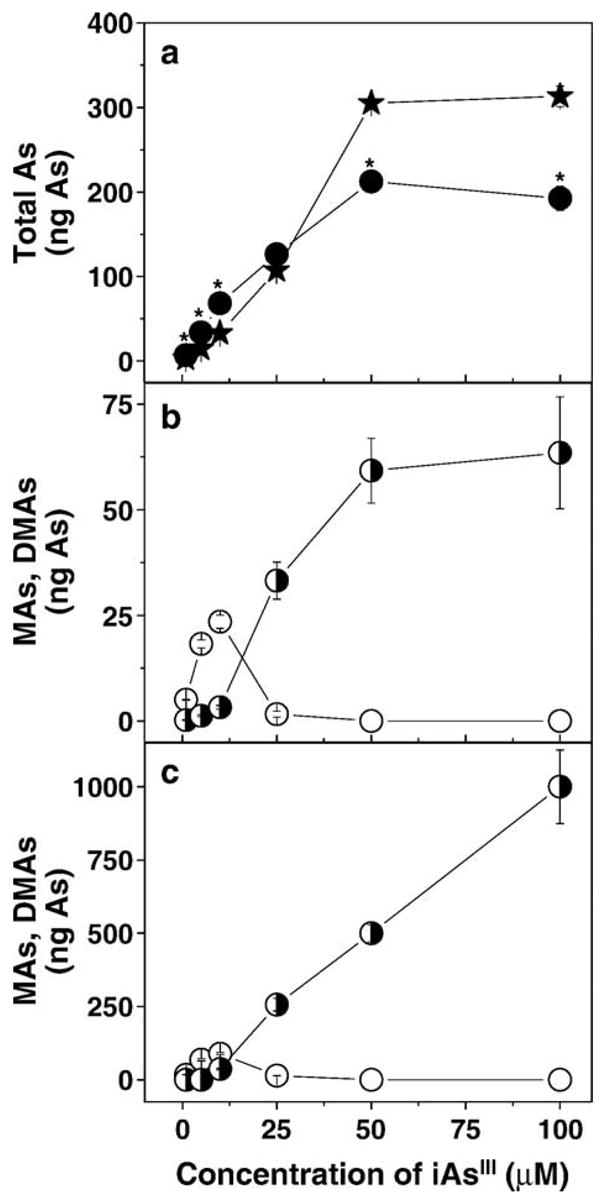
Production and distribution of metabolites in UROtsa and UROtsa/F35 cultures exposed to 1, 5, 10, 25, 50, or 100 μM iAsIII for 24 h. (a) The total intracellular As in UROtsa (★) and UROtsa/F35 (●) cultures; (b) intracellular MAs (◑) and DMAs (○) in UROtsa/F35 cultures; (c) MAs (◑) and DMAs (○) in media from UROtsa/F35 cultures. Values are expressed as ng of As in cells or medium from one well of a 12-well culture plate (mean ± SD, n = 3).
Analysis of AsIII- and AsV-containing metabolites in UROtsa/F35 cells exposed to iAsIII and MAsIII
The pH-specific HG-AAS was used to determine oxidation states of arsenicals in cultures of UROtsa/F35 cells incubated with 1 μM iAsIII or 3 μM MAsIII for 24 h. To minimize oxidation of trivalent arsenicals, freshly harvested cells and media were snap frozen in dry ice and submitted immediately for HG-AAS analysis. HG-AAS analysis at pH 6 (i.e., analysis of trivalent arsenicals) was completed within 4 h of sample collection. Analysis at pH 1 was completed within 8 h. DMAsIII was the major methylated metabolite in cells and media from cultures exposed to 1 μM iAsIII (Table 1a). DMAsV and traces of MAsIII were found in cells. Traces of DMAsV were also detected in media. About 27% of iAsIII initially added into the culture were methylated over the 24-h incubation period. Unmetabolized iAsIII was present mainly in culture media. DMAsIII was also the major metabolite in cultures exposed to 3 μM MAsIII (Table 1b). Most DMAsIII were found in the media; all DMAsV were retained in cells. About 91% of MAsIII initially added into the culture were converted to DMAsIII or DMAsV. The remaining MAsIII were equally distributed between cells and media. A small amount of MAsV was in media at the end of the incubation. TMAsVO was not detected by HG-AAS in UROtsa/F35 cultures exposed to either iAsIII or MAsIII. Overall recoveries of As from cultures incubated with iAsIII or MAsIII ranged from 91% to 110%.
Table 1.
As species in UROtsa/F35 cultures exposed to (a) 1 μM iAsIII and (b) 3 μM MAsIII
| Sample | As species
|
Sum of species | |||||
|---|---|---|---|---|---|---|---|
| AsV | AsIII | MAsV | MAsIII | DMAsV | DMAsIII | ||
| a | |||||||
| Cell lysate | 0 | 1.78 ± 0.24 | 0 | 0.13 ± 0.09 | 6.59 ± 0.46 | 12.40 ± 1.33 | 20.90 ± 1.82 |
| Medium | 0 | 119.59 ± 4.81 | 0 | 0 | 0.15 ± 0.31 | 25.28 ± 1.23 | 145.02 ± 4.11 |
| b | |||||||
| Cell lysate | 0 | 0 | 0 | 16.98 ± 1.41 | 64.45 ± 9.59 | 12.29 ± 3.29 | 93.71 ± 11.51 |
| Medium | 0 | 0 | 4.07 ± 3.78 | 17.26 ± 4.76 | 0 | 294.33 ± 24.82 | 315.66 ± 24.32 |
Confluent cells in 6-well culture plates were exposed for 24 h to 1 μM iAsIII or 3 μM MAsIII. Speciation analysis of As was carried out in freshly isolated cell lysates and media by HG-AAS at pH 6 and pH 1. Results are expressed as ng As per well (mean ± SD, n = 4).
Uptake of trivalent arsenicals by UROtsa and UROtsa/F35 cells
Uptake of trivalent arsenicals was examined in confluent cultures of UROtsa and UROtsa/F35 cells incubated with 1 μM iAsIII, 1 μM MAsIII, or 1 μM DMAsIII for up to 2 h. Total iAs (iAsIII + iAsV), total MAs (MAsIII + MAsV), and total DMAs (DMAsIII + DMAsV) were determined by HG-AAS in cells and media after digestion with phosphoric acid. Fig. 3 shows intracellular concentrations of arsenicals normalized for cellular protein content. The uptake of iAsIII and MAsIII was characterized by biphasic kinetics. The first fast phase occurred over the first 15 min and was followed by a slower phase. In UROtsa/F35 cells exposed to iAsIII, the end of the fast phase coincided with the onset of methylation, as indicated by DMAs formation. MAs was detected in cells only after 2 h. No methylated metabolites were found in culture media (data not shown). Between 30 and 90 min, total concentrations of arsenicals were significantly higher in UROtsa cells than in UROtsa/F35 cells. However, at the end of 2-h incubation period, UROtsa and UROtsa/F35 cells contained similar amounts of arsenicals, representing 3.4% and 3.0% of total As in the culture, respectively. In UROtsa/F35 cultures exposed to MAsIII, DMAs was detected in cells as early as at 2.5 min. The concentration of DMAs in cells increased steadily over the 2-h incubation period. In the medium, DMAs was first detected only after 30 min when the intracellular concentration of DMAs reached 20 ng As/mg of protein. The accumulation of DMAs in cells was responsible for significantly higher levels of total As in UROtsa/F35 cells as compared to UROtsa cells between 1 and 2 h of exposure. After 2-h incubation, UROtsa/F35 and UROtsa cells contained 21.0% and 15.2% of total As in the culture, respectively. Notably, 37.3% of MAsIII was converted to DMAs during the 2-h incubation period. In UROtsa and UROtsa/F35 cultures incubated with DMAsIII, the intracellular concentration of DMAs increased rapidly during the first 15 min. At the later time points, the intracellular concentrations of DMAs remained practically unchanged. At the end of the 2-h incubation period, UROtsa/F35 cells contained a slightly larger fraction of DMAs (39.0%) than UROtsa cells (30.3%). No other metabolites were detected in either cells or culture medium.
Fig. 3.

Uptake of trivalent arsenicals by UROtsa and UROtsa/F35 cells during 120-min exposures to iAsIII, MAsIII, or DMAsIII. The total intracellular As in UROtsa (★) and UROtsa/F35 (●) cultures exposed to 1 μM iAsIII(a), 1 μM MAsIII(b), or 1 μM DMAsIII(c). Intracellular iAs (⊕), MAs (◑), and DMAs (○) in UROtsa/F35 cells exposed to 1 μM iAsIII (d) or 1 μM MAsIII (e). Inset in panel e shows DMAs in media of UROtsa/F35 cells exposed to 1 μM MAsIII. Values are expressed as ng of As per mg of cellular protein (mean ± SD, n = 3).
Production and distribution of metabolites in UROtsa and UROtsa/F35 cells exposed to trivalent arsenicals
Following the 2-h uptake experiments, the cellular retention and distribution of metabolites were examined in UROtsa and UROtsa/F35 cultures exposed to 1 μM iAsIII, 1 μM MAsIII, or 1 μM DMAsIII for 6, 24, 48, and 72 h. Total iAs, total MAs, and total DMAs were determined in digested cell lysates and media by HG-AAS. Fig. 4 shows distribution of metabolites in UROtsa and UROtsa/F35 cultures exposed to 1 μM iAsIII. In UROtsa cultures incubated with iAsIII for 6 h, only 4.8% of total iAs was found in cells. Later, the amount of iAs associated with cells decreased. After 72 h, cells retained 3.7% of total iAs in culture. UROtsa/F35 cells exposed to 1 μM iAsIII for 6 h contained almost equal amounts of iAs, MAs, and DMAs that in sum accounted for 12.4% of total As in culture. After 24 h, UROtsa/F35 cells retained more than 23% of total As and DMAs was the major intracellular metabolite. Only traces of iAs and MAs were detected in cells after 48 and 72 h. The amount of DMAs retained in cells decreased between 24 and 72 h. The amount of DMAs found in medium steadily increased. A total of 70.2% of iAsIII was methylated by UROtsa/F35 cells over the 72-h incubation period, yielding almost exclusively DMAs. UROtsa cells exposed to 1 μM MAsIII for 6 h retained 15.5% of MAs in the culture (Fig. 5). The amount of intracellular MAs increased to 57.7% between 6 and 24 h and remained practically unchanged for the remainder of the 72-h incubation interval. DMAs was the major metabolite and MAs a minor metabolite in UROtsa/F35 cells after 6-h incubation with 1 μM MAsIII. The intracellular metabolites accounted for 43.8% of total As in culture. The conversion of MAsIII to DMAs was largely completed during the first 24 h. Only traces of MAs were detected in cells or culture media in later time points. The fraction of DMAs retained in cells decreased from 42.8% to 22% of total As between 24 and 72 h with a concurrent increase in the levels of DMAs in medium. Similar retention and distribution patterns for DMAs were found in UROtsa and UROtsa/F35 cells exposed to 1 μM DMAsIII (Fig. 6). In both cell types, the amount of DMAs retained in cells peaked after 6 or 24 h, representing 23–36% of total DMAs, and then steadily decreased to 11–12% by the end of the 72-h incubation period. TMAsVO was not detected in URotsa/F35 cultures over this time period.
Fig. 4.
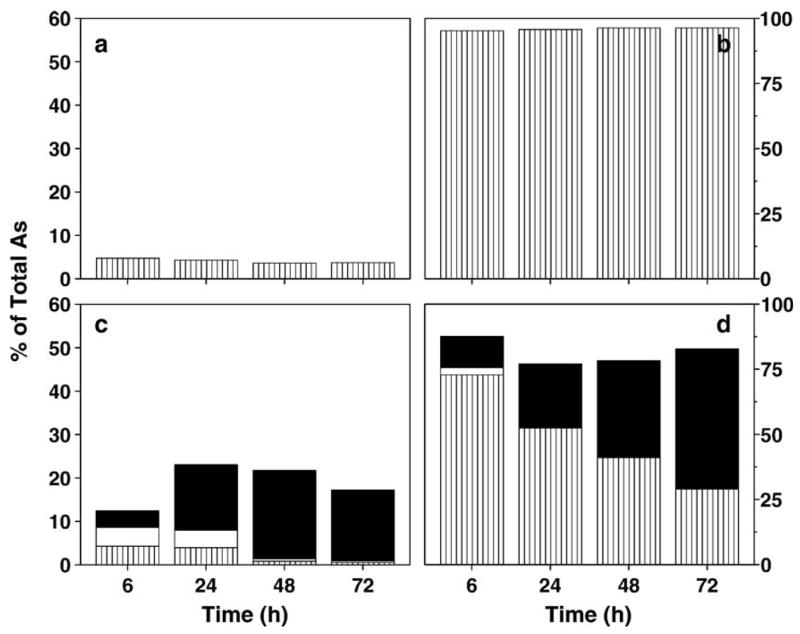
Production and distribution of metabolites, including iAs (▥), MAs (□), and DMAs (■), in UROtsa and UROtsa/F35 cultures exposed to 1 μM iAsIII for 6, 24, 48, and 72 h. (a) Cell lysates and (b) media from UROtsa cultures; (c) cell lysates and (d) media from UROtsa/F35 cultures. Values are expressed as percentage of total As in culture (mean, n = 3).
Fig. 5.
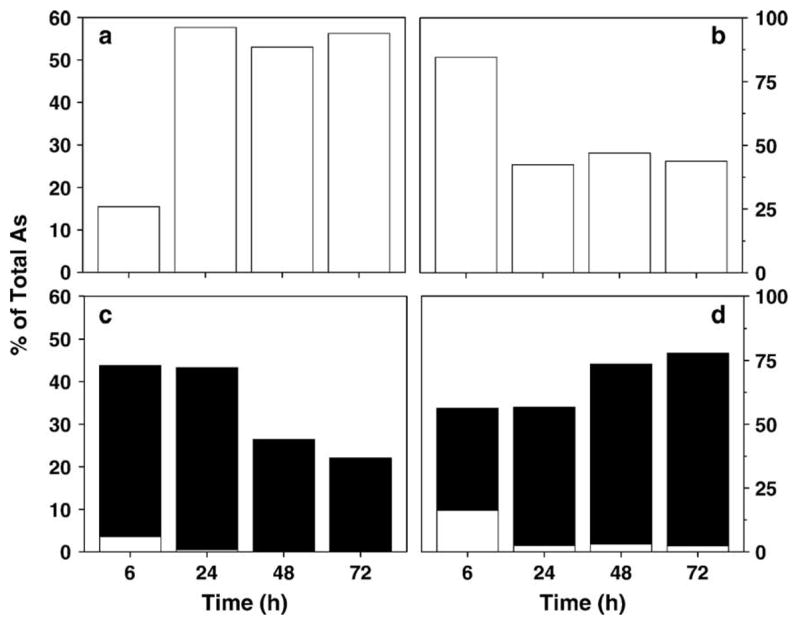
Production and distribution of metabolites, including MAs (□) and DMAs (■), in UROtsa and UROtsa/F35 cultures exposed to 1 μM MAsIII for 6, 24, 48, and 72 h. (a) Cell lysates and (b) media from UROtsa cultures; (c) cell lysates and (d) media from UROtsa/F35 cultures. Values are expressed as percentage of total As in culture (mean, n = 3).
Fig. 6.

Distribution of DMAs (■) in UROtsa and UROtsa/F35 cultures exposed to 1 μM DMAsIII for 6, 24, 48, and 72 h. (a) Cell lysates and (b) media from UROtsa cultures; (c) cell lysates and (d) media from UROtsa/F35 cultures. Values are expressed as percentage of total As in culture (mean, n = 3).
Cytotoxicities of trivalent arsenicals in UROtsa and UROtsa/F35 cells
Cytotoxicities of trivalent arsenicals were examined in confluent UROtsa and UROtsa/F35 cultures exposed to iAsIII (0.5–100 μM), MAsIIIO (0.1–5 μM), or DMAsIIII (0.1–25 μM). Cell viability was measured after 24-h exposures using MTT assay (Fig. 7). For both cell lines, methylated trivalent arsenicals were more cytotoxic than iAsIII. MAsIII was a more potent cytotoxin than DMAsIII. Statistically significant differences were found between UROtsa and UROtsa/F35 cells in the susceptibility to iAsIII and MAsIII. In cultures exposed to 50–75 μM iAsIII, the loss of viability was more pronounced in UROtsa/F35 cells as compared to UROtsa cells. In contrast, UROtsa cells were more susceptible than UROtsa/F35 cells to cytotoxicity of 2,3, and 4 μM MAsIII. Small, but statistically significant differences were found between the two cell lines in susceptibility to 1 and 5 μM DMAsIII with UROtsa/F35 cells more resistant than UROtsa cells. Additional experiments were carried out to characterize relationship between the susceptibility of UROtsa and UROtsa/F35 to cytotoxicity of MAsIII and the intracellular retention and metabolic conversion of this arsenical. Here, confluent UROtsa and UROtsa/F35 cultures were exposed to 1–5 μM MAsIII for 24 h. After exposure, cell viability was determined by the MTT assay and As species in cells and media were determined by HG-AAS after acid digestion. Cell viability in UROtsa cells was significantly lower than in UROtsa/F35 cells over this range of MAsIII concentrations. LC25 and LC50 values calculated from the cell viability versus MAsIII concentration charts (Fig. 8) were used to characterize the cytotoxicity of MAsIII in UROtsa and UROtsa/F35 cells. The LC25 and LC50 values were defined as concentrations of MAsIII that decreased cell viability by 25% and 50%, respectively. The LC25 and LC50 values were higher for UROtsa/F35 cells (3.4 and 4.1 μM) than for UROtsa cells (1.6 and 3.1 μM). In UROtsa cells, the intracellular content of MAs increased proportionally to the concentration of MAsIII in culture, reaching a plateau at 3 μM MAsIII. UROtsa/F35 cells effectively methylated MAsIII at concentrations up to 3–4 μM. DMAs was the major or the only metabolite found in cells under these exposure conditions. At higher concentrations of MAsIII, DMAs production declined (data not shown) and MAs was the major metabolite detected in cells. After exposure to 5 μM MAsIII, the average intracellular concentration of MAs in UROtsa/F35 culture (252 ng As/well) resembled that found in UROtsa culture (224 ng As/well).
Fig. 7.
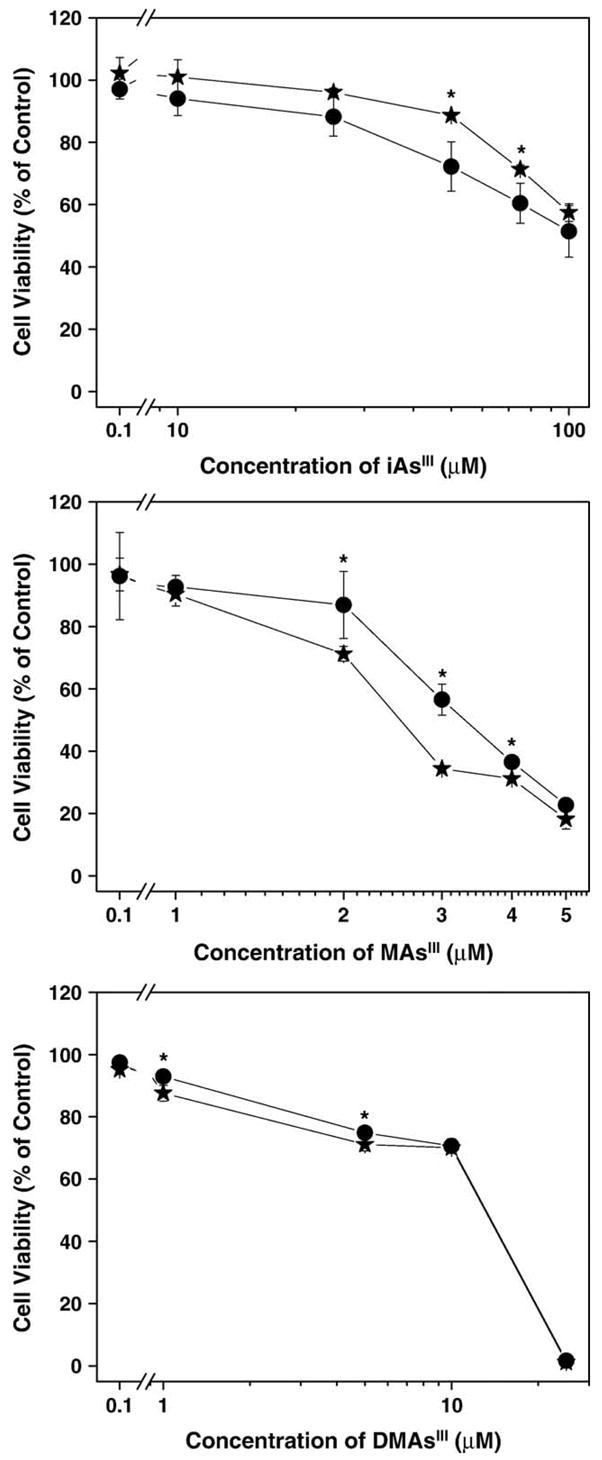
Cell viability in UROtsa (★) and UROtsa/F35 (●) cultures exposed to (a) iAsIII, (b) MAsIII, and (c) DMAsIII for 24 h (mean ± SD; n = 4).
Fig. 8.
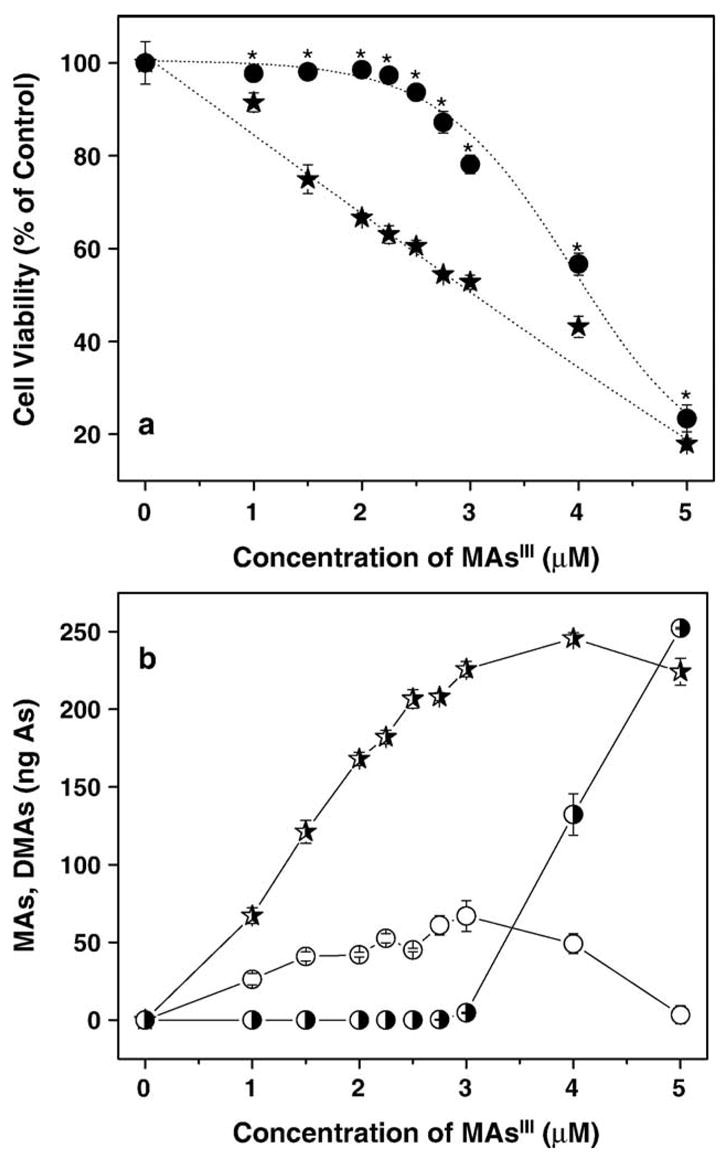
Relationship between the cytotoxicity and metabolism of MAsIII in UROtsa and UROtsa/F35 cells. (a) Cell viability in UROtsa (★) and UROtsa/F35 (●) cultures exposed to MAsIII for 24 h; (b) intracellular metabolites: MAs in UROtsa cells (
 ), MAs in UROtsa/F35 cells (◑), and DMAs in UROtsa/F35 cells (○) after 24-h exposure to MAsIII (mean ± SD; n = 4).
), MAs in UROtsa/F35 cells (◑), and DMAs in UROtsa/F35 cells (○) after 24-h exposure to MAsIII (mean ± SD; n = 4).
Discussion
A variety of studies indicate that the enzymatic methylation of iAs plays a significant role in modulation of toxic and cancer-promoting effects of iAs exposure. However, previous work comparing the acute toxicities of iAsIII in a variety of mammalian cell types with and without capacity to methylate iAsIII have not clarified the relationship between methylation and toxicity (Styblo et al., 2000, 2002). In this study, we developed a novel model in which a parental and a transduced cell line, UROtsa and UROtsa/F35, differ only in capacity to methylate iAs. RT-PCR, immunoblot, and As speciation analyses show that UROtsa/F35 cells express rAS3MT and methylate iAsIII, yielding mono- and dimethylated AsIII- and AsV-containing metabolites. In addition, UROtsa/F35 cells methylate MAsIII, yielding DMAsIII and DMAsV. The patterns of methylated metabolites in UROtsa/F35 cells exposed to various concentrations of iAsIII resembled those found in cultured primary rat and human hepatocytes (Drobná et al., 2004; Styblo et al., 1999). For example, in UROtsa/F35 cells, high concentrations of iAsIII inhibited formation of DMAs from MAs with little effect on the formation of MAs from iAsIII. Similarly, exposures of UROtsa/F35 cells to high concentrations of MAsIII inhibited its conversion to DMAs. For both cultured primary hepatocytes and UROtsa/F35 cells, inhibition of DMAs production resulted in accumulation of MAs in cells. Previous work found TMAsVO and volatile TMAsIII to be final products of the in vitro methylation of iAsIII, MAsIII, or DMAsIII by recombinant rAS3MT (Waters et al., 2004b). The rAS3MT-catalyzed in vitro production of TMAsVO and TMAsIII was inhibited by millimolar GSH (Waters et al., 2004b). Notably trimethylated arsenicals were not detected in this study in UROtsa/F35 cells treated with iAsIII, MAsIII, or DMAsIII. Relatively high cellular concentrations of GSH (2–4 mM) may prevent formation of TMAsVO and TMAsIII from their metabolic precursors. The mechanism by which GSH inhibits the conversion of DMAsIII to trimethylated arsenicals has not been determined.
It should be noted that a recent study of Bredfeldt et al. (2004) found small amounts of MAsIII and MAsV in UROtsa cells cultured in the presence of iAsIII, thus contradicting our data that show these cells lacking the capacity to methylate iAs. These authors indicate that HPLC-ICP-MS was used for As speciation in low molecular weight (<10 kDa) fractions of cell lysates. However, no details regarding the speciation technique or data validation are provided. Our studies on the metabolism and toxic effects of As in UROtsa cells (Drobná et al., 2003; Styblo et al., 2000, 2002) have consistently shown that these cells do not methylate iAs. Notably, T24 cells, another cell line derived from human bladder epithelium, also lack the capacity to methylate iAs (Styblo et al., 2000). These finding have been validated using two independent speciation techniques, including metabolic radiolabeling with 73As and HG-AAS. We have also shown that UROtsa cells do not express AS3MT (Lin et al., 2002). Importantly, comparison of our previous work (Drobná et al., 2003; Lin et al., 2002;) with results of the present study suggests that serum starvation for 16 h has no effect on AS3MT expression or on the metabolic pattern of iAs in UROtsa cells.
The present work shows that expression and activity of rAS3MT in UROtsa/F35 cells have striking effects on the uptake and retention patterns for iAsIII and MAsIII. However, these effects depended strongly on the exposure time and the concentrations of arsenicals in the culture. UROtsa/F35 cells exposed to low concentrations of iAsIII (1–10 μM) for 6–72 h retained significantly greater amounts of total As than did UROtsa cells (Figs. 2 and 4). Greater cellular retention of total As in UROtsa/F35 cells was associated with the production and accumulation of methylated metabolites, MAs and DMAs, that were present in cells mainly as MAsIII or DMAsIII (Table 1). In contrast, at high iAsIII concentrations (50–100 μM) UROtsa/F35 cells retained less total As than did UROtsa cells (Fig. 2). Under these conditions, the formation of DMAs from MAs was inhibited and significant amounts of MAs were retained in cells. MAs represented about 30% of intracellular As. Despite of the lower total As load, UROtsa/F35 cells were more susceptible than UROtsa cells to cytotoxicity of 50–100 μM iAsIII (Fig. 7). These data suggest intracellular MAs, probably present as MAsIII, to be the cytotoxic species. Interestingly, UROtsa/F35 cells were more resistant than UROtsa cells to the cytotoxic effects of 1–5 μM MAsIII (Figs. 7 and 8). UROtsa/F35 cells effectively methylated MAsIII over the concentration range of 1–3 μM (Figs. 5 and 8). At these exposure levels, UROtsa/F35 cells retained less total As than did UROtsa cells. In addition, As retained by UROtsa/F35 cells was present mostly as DMAs. Higher concentrations of MAsIII (4–5 μM) that inhibited DMAs production in UROtsa/F35 cells increased the intracellular levels of MAs such that it was the major intracellular metabolite. As a result of the inhibition of DMAs production, MAs concentration in UROtsa/F35 cells exposed to 5 μM MAsIII was almost identical as MAs concentration in UROtsa cells under the same exposure conditions. Exposure to 5 μM MAsIII had similar effects on viability of both UROtsa/F35 and UROtsa cells. Thus, increased resistance of UROtsa/F35 cells to moderate concentrations of MAsIII was associated with fast conversion of MAsIII to DMAs and with efflux of DMAs from cells. The cytotoxicity of high MAsIII concentrations is linked to the inhibition of DMAs production and accumulation of MAs in cells. Taken together, these results suggest that DMAs (possibly DMAsIII) is the major excretory product of the methylation of iAsIII and MAsIII by UROtsa/F35 cells. MAs (probably MAsIII) is avidly retained by cells and cannot be effectively released without conversion to DMAs. We have previously shown that accumulation of MAs in rat hepatocytes treated with subtoxic concentrations of iAsIII is associated with inhibition of thioredoxin reductase (Lin et al., 2001), a sensitive marker of the exposure to MAsIII. The current study definitively links the production and/or accumulation of MAs to cytotoxicity that has been associated with acute exposure to iAsIII.
The present work also shows that the uptakes of MAsIII and DMAsIII by UROtsa and UROtsa/F35 cells exceed the uptake of iAsIII by an order of magnitude. The greater uptakes combined with accumulation of MAs in cells and with the slow efflux of DMAs contribute to significantly higher retention of total As in cells exposed to MAsIII or DMAsIII as compared to cells exposed to iAsIII. Similar differences between the uptake and/or retention of iAsIII and methylated trivalent arsenicals have been found in primary rat hepatocytes (Styblo et al., 2000) and differentiated 3T3-L1 adipocytes (Devesa et al., unpublished results). Notably, MAsIII and DMAsIII are significantly more cytotoxic than iAsIII for all these and other cell types (Styblo et al., 2000, 2002; Walton et al., 2004). Thus, greater cellular uptakes and retention of MAsIII and DMAsIII are likely to contribute to the greater toxicities of methylated trivalent arsenicals in cell culture systems. Greater cellular uptakes and retention may at least in part explain why during subtoxic exposures MAsIII or DMAsIII affect major cellular functions, e.g., activator protein-1 or insulin signaling (Drobná et al., 2003; Walton et al., 2004), to a greater extent than does iAsIII. Two transport systems have been shown to contribute to the uptake of iAsIII by yeast and mammalian cells, including aquaglyceroporins (Carbrey et al., 2003; Liu et al., 2002) and hexose transporters (Liu et al., 2004). The roles of these transport systems in the uptake of methylated arsenicals, including MAsIII and DMAsIII, by yeast, UROtsa, and UROtsa/F35 cells are currently under investigation.
We have shown that MAsIII and DMAsIII produced in the course of in vitro methylation of iAs in rat liver cytosol bind to specific proteins (Styblo and Thomas, 1997). Binding of arsenicals to cellular proteins may contribute the accumulation of MAs and DMAs in UROtsa/F35 cells exposed to iAsIII or MAsIII. The relatively slow efflux of methylated metabolites may also account for accumulation in cells. Glutathione transferase Pi (GST-Pi) and the multidrug resistance-associated protein-1 and -2 (MRP-1/2) facilitate the transport of arsenicals from cells exposed to iAsIII (Kala et al., 2000; Leslie et al., 2004; Vernhet et al., 2001; Wang and Lee, 1993). This process may involve a GST-Pi-catalyzed formation of arsenothiol complexes with GSH and efflux of these complexes via an MRP-mediated mechanism. The presence of GSH was essential for the optimal function of this mechanism (Leslie et al., 2004). Arsenicals, including iAs, MAs, and DMAs, have been shown to form complexes with GSH even in the absence of GST-Pi (Delnomdedieu et al., 1994; Scott et al., 1993). Arsenic in GSH complexes, including iAs(GS)3, MAs(GS)2, and DMAsGS, were found in the 3 + oxidation state. These findings are consistent with results of our experiments that showed DMAs in media of UROtsa/F35 cultures exposed to either iAsIII or MAsIII to be almost exclusively in the form of DMAsIII. Thus, the cellular GSH levels, as well as GST-Pi and MRP-1/2 activities, may play important roles in the cellular retention of arsenicals. These factors may modulate the adverse effects associated with the formation and retention of methylated trivalent arsenicals. The role of GST-Pi, MRP-1/2, and of the intracellular GSH levels in the regulation of DMAs efflux from UROtsa/F35 cells and in the modulation of cytotoxicities of trivalent arsenicals in this cell line will be examined in future studies.
The establishment of UROtsa and UROtsa/F35 cells that differ genetically in AS3MT expression allows study of the role of phenotype for As methylation as a factor in the uptake, retention, and toxicity of various arsenicals. Using this model, we showed that the enzymatic methylation of iAs has a profound effect on the kinetics of cellular metabolism and cytotoxicity of this metalloid. The data presented here indicate that the formation and efflux of DMAs contribute to detoxification of iAs in the cell. However, inhibition of DMAs formation that occurs in cells exposed to high iAs concentrations results in increased production and accumulation of MAs. The accumulation of MAs, probably as MAsIII, in cells contributes significantly to the overall cytotoxic effects associated with this exposure. These data provide additional evidence that the formation of MAsIII is a process that potentiates acute toxicity of iAs.
Acknowledgments
The authors thank Professor William R. Cullen, Department of Chemistry, University of British Columbia, who provided methylated trivalent arsenicals for this study. Z.D. is supported by a grant from CEHS, UNC Chapel Hill. S.B.W. was a postdoctoral fellow in the Curriculum in Toxicology, University of North Carolina at Chapel Hill, and was supported by Training Grant T901915 of the U.S. Environmental Protection Agency-University of North Carolina Toxicology Research Program. V.D. is a recipient of an MECD/Fulbright Grant for Postdoctoral training administrated by the Ministry of Education, Culture and Sports, Spain. M.S. is supported by NIH grant ES010845 and a Clinical Nutrition Research Center Grant DK 56350. The manuscript has been reviewed in accordance with the policy of the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- Bredfeldt TG, Kopplin MJ, Gandolfi AJ. Effects of arsenite on UROtsa cells: low-level arsenite causes accumulation of ubiquitinated proteins that is enhanced by reduction in cellular glutathione levels. Toxicol Appl Pharmacol. 2004;198:412–418. doi: 10.1016/j.taap.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Carbrey JM, Gorelick-Feldman DA, Kozono D, Praetorius J, Nielsen S, Agre P. Aquaglyceroporin AQP9: solute permeation and metabolic control of expression in liver. Proc Natl Acad Sci USA. 2003;100:2945–2950. doi: 10.1073/pnas.0437994100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- Cullen WR, McBride BC, Reglinski J. The reaction of methylarsenicals with thiols: some biological implications. J Inorg Biochem. 1984;21:179–194. [Google Scholar]
- Del Razo LM, Styblo M, Cullen WR, Thomas DJ. Determination of trivalent methylated arsenicals in biological matrices. Toxicol Appl Pharmacol. 2001;174:282–293. doi: 10.1006/taap.2001.9226. [DOI] [PubMed] [Google Scholar]
- Delnomdedieu M, Basti MM, Otvos JD, Thomas DJ. Reduction and binding of arsenate and dimethylarsenate by glutathione: a multinuclear magnetic resonance study. Chem-Biol Interact. 1994;90:139–155. doi: 10.1016/0009-2797(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Devesa V, Del Razo LM, Adair B, Drobná Z, Waters SB, Hughes MF, Styblo M, Thomas DJ. Comprehensive analysis of As metabolites by pH-specific hydride generation atomic absorption spectrometry. J Anal At Spectrom. 2004;19:1460–1467. [Google Scholar]
- Drobná Z, Jaspers I, Thomas DJ, Styblo M. Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals. FASEB J. 2003;17:67–69. doi: 10.1096/fj.02-0287fje. [DOI] [PubMed] [Google Scholar]
- Drobná Z, Waters SB, Walton FS, LeCluyse EL, Thomas DJ, Styblo M. Interindividual variation in the metabolism of As in cultured primary human hepatocytes. Toxicol Appl Pharmacol. 2004;201:166–177. doi: 10.1016/j.taap.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Gregus Z, Nemeti B. Purine nucleoside phosphorylase as a cytosolic arsenate reductase. Toxicol Sci. 2002;70:13–19. doi: 10.1093/toxsci/70.1.13. [DOI] [PubMed] [Google Scholar]
- Kala SV, Neely MW, Kala G, Prater CI, Atwood DW, Rice JS, Lieberman MW. The MRP2/cMOAT transporter and As-glutathione complex formation are required for biliary excretion of As. J Biol Chem. 2000;275:33404–33408. doi: 10.1074/jbc.M007030200. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, Mass MJ, DeMarini DM. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of As: induction of chromosomal mutations but not gene mutations. Environ Mol Mutagen. 2003;42:192–205. doi: 10.1002/em.10192. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Haimeur A, Waalkes MP. As transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J Biol Chem. 2004;279:32700–32708. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem Res Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- Lin S, Shi Q, Nix FB, Styblo M, Beck MA, Herbin-Davis KM, Simeonsson JB, Thomas DJ. A novel S-adenosyl-L-methionine:As(III) methyltransferase from rat liver cytosol. J Biol Chem. 2002;277:10795–10803. doi: 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Shen J, Carbrey JM, Mukhopadhyay R, Agre P, Rosen BP. Arsenite transport by mammalian aquaglyceroporins AQP7 and AQP9. Proc Natl Acad Sci USA. 2002;99:6053–6058. doi: 10.1073/pnas.092131899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Boles E, Rosen BP. As trioxide uptake by hexose permeases in Saccharomyces cerevisiae. J Biol Chem. 2004;279:17312–17318. doi: 10.1074/jbc.M314006200. [DOI] [PubMed] [Google Scholar]
- Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, Kligerman AD. Methylated trivalent As species are genotoxic. Chem Res Toxicol. 2001;14:355–361. doi: 10.1021/tx000251l. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Aposhian HV. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol. 2000;163:203–207. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Jagadish B, Mash EA, Aposhian HV. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem Res Toxicol. 2001;14:651–656. doi: 10.1021/tx000264z. [DOI] [PubMed] [Google Scholar]
- Radabaugh TR, Sampayo-Reyes A, Zakharyan RA, Aposhian HV. Arsenate reductase II. Purine nucleoside phosphorylase in the presence of dihydrolipoic acid is a route for reduction of arsenate to arsenite in mammalian systems. Chem Res Toxicol. 2002;15:692–698. doi: 10.1021/tx0101853. [DOI] [PubMed] [Google Scholar]
- Reay PF, Asher CJ. Preparation and purification of 74As-labeled arsenate and arsenite for use in biological experiments. Anal Biochem. 1977;78:557–560. doi: 10.1016/0003-2697(77)90117-8. [DOI] [PubMed] [Google Scholar]
- Scott N, Hatlelid KM, MacKenzie NE, Carter DE. Reaction of As(III) and As(V) species with glutathione. Chem Res Toxicol. 1993;6:102–106. doi: 10.1021/tx00031a016. [DOI] [PubMed] [Google Scholar]
- Shen J, Wanibuchi H, Wei EI, Kinoshita A, Yoshida K, Endo G, Fukushima S. Liver tumorigenicity of trimethylarsine oxide in male Fisher 344 rats-association with oxidative DNA damage and enhanced cell proliferation. Carcinogenesis. 2003;24:1827–1835. doi: 10.1093/carcin/bgg143. [DOI] [PubMed] [Google Scholar]
- Simeonova P, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, Luster MI. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transcription. Cancer Res. 2000;60:3445–3453. [PubMed] [Google Scholar]
- Simeonova P, Wang S, Hulderman T, Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. J Biol Chem. 2002;277:2945–2950. doi: 10.1074/jbc.M109136200. [DOI] [PubMed] [Google Scholar]
- Styblo M, Thomas DJ. Binding of arsenicals to proteins in an in vitro methylation system. Toxicol Appl Pharmacol. 1997;147:1–8. doi: 10.1006/taap.1997.8256. [DOI] [PubMed] [Google Scholar]
- Styblo M, Yamauchi H, Thomas DJ. Comparative methylation of trivalent and pentavalent arsenicals. Toxicol Appl Pharmacol. 1995;135:172–178. doi: 10.1006/taap.1995.1220. [DOI] [PubMed] [Google Scholar]
- Styblo M, Hughes MF, Thomas DJ. Liberation and analysis of protein-bound arsenicals. J Chromatogr B. 1996;677:161–166. doi: 10.1016/0378-4347(95)00490-4. [DOI] [PubMed] [Google Scholar]
- Styblo M, Serves SV, Cullen WR, Thomas DJ. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem Res Toxicol. 1997;10:27–33. doi: 10.1021/tx960139g. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, LeCluyse EL, Hamilton GA, Wang C, Cullen WR, Thomas DJ. Metabolism of As in primary cultures of human and rat hepatocytes. Chem Res Toxicol. 1999;12:560–565. doi: 10.1021/tx990050l. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000;74:289–299. doi: 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- Styblo M, Drobná Z, Jaspers I, Lin S, Thomas DJ. The role of biomethylation in toxicity and carcinogenicity of As. A research update. Environ Health Perspect. 2002;110(Suppl 5):767–771. doi: 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernhet L, Allain N, Payen L, Anger JP, Guillouzo A, Fardel O. Resistance of human multidrug resistance-associated protein 1-overexpressing lung tumor cells to the anticancer drug As trioxide. Biochem Pharmacol. 2001;61:1387–1391. doi: 10.1016/s0006-2952(01)00606-2. [DOI] [PubMed] [Google Scholar]
- Walton FS, Waters SB, Jolley SL, LeCluyse EL, Thomas DJ, Styblo M. Selenium compounds modulate the activity of recombinant rat AsIII-methyltransferase and the methylation of arsenite by rat and human hepatocytes. Chem Res Toxicol. 2003;16:261–265. doi: 10.1021/tx025649r. [DOI] [PubMed] [Google Scholar]
- Walton FS, Harmon AW, Paul DS, Drobná Z, Patel YM, Styblo M. Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: possible mechanism of As-induced diabetes. Toxicol Appl Pharmacol. 2004;198:424–433. doi: 10.1016/j.taap.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Wang HF, Lee TC. Glutathione S-transferase pi facilitates the excretion of As from As-resistant Chinese hamster ovary cells. Biochem Biophys Res Commun. 1993;192:1093–1099. doi: 10.1006/bbrc.1993.1529. [DOI] [PubMed] [Google Scholar]
- Waters SB, Devesa-Perez V, Del Razo LM, Styblo M, Thomas DJ. Endogenous reductants support catalytic function of recombinant rat cyt19, an As methyltransferase. Chem Res Toxicol. 2004a;17:404–409. doi: 10.1021/tx0342161. [DOI] [PubMed] [Google Scholar]
- Waters SB, Devesa V, Fricke MW, Creed JT, Styblo M, Thomas DJ. Glutathione modulates recombinant rat As (+3 oxidation state) methyltransferase-catalyzed formation of trimethylarsine oxide and trimethylarsine. Chem Res Toxicol. 2004b;17:1621–1629. doi: 10.1021/tx0497853. [DOI] [PubMed] [Google Scholar]
- Wei M, Wanibuchi H, Yamamoto S, Li W, Fukushima S. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis. 1999;20:1873–1876. doi: 10.1093/carcin/20.9.1873. [DOI] [PubMed] [Google Scholar]
- Yamauchi H, Fowler BA. Toxicity and metabolism of inorganic and methylated arsenicals. In: Nriagu JO, editor. As in the Environment, Part II: Human Health and Ecosystem Effects. Wiley; New York: 1994. pp. 35–43. [Google Scholar]
- Yamauchi H, Yamamura Y. Metabolism and excretion of orally administered As trioxide in the hamster. Toxicology. 1985;34:113–121. doi: 10.1016/0300-483x(85)90161-1. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Inoue Y, Kuroda K, Chen H, Wanibuchi H, Fukushima S, Endo G. Urinary excretion of As metabolites after long-term oral administration of various As compounds to rats. J Toxicol Environ Health, A. 1998;54:179–192. doi: 10.1080/009841098158890. [DOI] [PubMed] [Google Scholar]
- Zakharyan R, Wu Y, Bogdan GM, Aposhian HV. Enzymatic methylation of As compounds: assay, partial purification, and properties of arsenite methyltransferase and monomethylarsonic acid methyltransferase from rabbit liver. Chem Res Toxicol. 1995;8:1029–1038. doi: 10.1021/tx00050a006. [DOI] [PubMed] [Google Scholar]
- Zakharyan RA, Aposhian HV. Enzymatic reduction of As compounds in mammalian systems: the rate-limiting enzyme of rabbit liver As biotransformation is MMA(V) reductase. Chem Res Toxicol. 1999;12:1278–1283. doi: 10.1021/tx9901231. [DOI] [PubMed] [Google Scholar]
- Zakharyan RA, Sampayo-Reyes A, Healy SM, Tsaprailis G, Board PG, Liebler DC, Aposhian HV. Human monomethylarsonic acid (MMA(V)) reductase is a member of the glutathione-S-transferase superfamily. Chem Res Toxicol. 2001;14:1051–1057. doi: 10.1021/tx010052h. [DOI] [PubMed] [Google Scholar]