

Abstract
The primary granule proteins (PGP) of myeloid cells are a source of multiple antigens with immunotherapeutic potential for myeloid leukemias. Therefore, we developed a method to induce T-cell responses to PGP protein sequences. We found that gene-transfected antigen-presenting cells efficiently expand functionally competent PGP-specific CD4 and CD8 T cells. The system was optimized using T-cell responses to autologous CD40-activated B cells (CD40-B) transfected with a cytomegalovirus pp65-encoding expression vector. To generate leukemia-specific T cells, expression vectors encoding the PGP proteinase 3 (PR3) human neutrophil elastase, and cathepsin-G were transfected into CD40-B cells to stimulate postallogeneic stem cell transplantation T cells from five patients with myeloid and three with lymphoid leukemias. T-cell responses to PGP proteinase 3 and human neutrophil elastase were observed in CD8+ and CD4+ T cells only in patients with myeloid leukemias. T-cell responses against cathepsin-G occurred in both myeloid and lymphoblastic leukemias. T cells from a patient with chronic myelogenous leukemia (CML) and from a posttransplant CML patient, expanded against PGP, produced IFN-γ or were cytotoxic to the patient's CML cells, demonstrating specific antileukemic efficacy. This study emphasizes the clinical potential of PGP for expansion and adoptive transfer of polyclonal leukemia antigen-specific T cells to treat leukemia.
Primary granule proteins (PGP) are aberrantly expressed in CD34-positive cells in myeloid leukemias. This abnormal expression may contribute to the leukemic phenotype. One of these proteins, human neutrophil elastase (HNE), may be responsible for the clonal dominance of chronic myeloid leukemia (CML) cells (1, 2). Due to their aberrant and high expression in myeloid leukemic progenitor cells, PGP are of great interest as a source of leukemia-restricted antigens for immunotherapy. Proteinase 3 (PR3), HNE, and peptides derived from PR3 (PR1, PR7) can induce T cells cytotoxic to myeloid leukemia progenitors but not normal cells (3–5). Furthermore, PR3- and HNE-specific CD4 and CD8 T cells can be generated from healthy donors, facilitating adoptive immunotherapy following allogeneic stem cell transplantation (6–8). PR1, an HLA-A2–binding epitope originally identified in PR3, was also found in HNE, and we previously found that HNE-pulsed antigen-presenting cells (APC) allowed the generation of PR1-specific CD8 T cells recognizing CML progenitor cells (8), indicating that one antigenic epitope has arisen from two closely related proteins. Cathepsin-G, another PGP, can also be processed and presented by CML progenitor cells (9). These data suggest that PGP can serve as important immunotherapeutic target antigens for myeloid leukemias.
Given the therapeutic potential of PGP-specific cytotoxic T lymphocytes, an important issue is how best to develop clinically applicable immunotherapeutic strategies with these proteins. Most translational research has focused on using small peptide sequences with defined binding to a specific HLA allele to elicit HLA-restricted tumor-specific T-cell responses. This strategy limits immunotherapy to individuals carrying a particular HLA allele (usually HLA-A*0201, represented in around 40% of the Caucasian population) and is primarily applicable CD8 T-cell responses (10, 11). Clinical experience with adoptively transferred cytomegalo-virus (CMV)-specific T cells shows the importance of CMV-specific CD4 T cells in maintaining a sustained and functional level of CD8 cytotoxic T-lymphocyte frequencies to the virus (12). The use of entire protein sequences as a source of antigen can overcome the limitations of peptide-based immunotherapy. Experience with T-cell responses to viruses indicates that antigenic viral proteins typically contain multiple epitopes that bind diverse HLA molecules and induce broadly directed antigen-specific T-cell responses (13). Furthermore, complete proteins contain peptide sequences available for presentation by MHC class II as well as class I molecules, thus making possible combined CD4 and CD8 T-cell responses to the same protein; this situation enables maintenance of the CD8 T-cell response through the provision of cognate CD4 T-cell help (14). Whereas advantageous, it is more difficult to produce protein antigens at purity levels comparable with those achievable for peptides; further, if recombinantly expressed in bacteria, such proteins must be free of endotoxin. As an alternative, investigators have used DNA coding for the desired protein antigen, either by transfecting APC so as to produce the protein of interest or by vaccinating with antigen-encoding DNA (15, 16). Most commonly, dendritic cells are used as APCs, because they reliably stimulate naïve T-cell responses (17, 18). Recently, CD40-activated B cells (CD40-B cells), which share many cell surface molecules with dendritic cells and can be readily expanded in vitro, have been investigated as alternatives to dendritic cells. They can be pulsed with peptide antigens (19, 20), loaded with purified proteins or tumor cell lysate (21, 22), retrovirally transduced (23) or transfected with tumor cell–derived or in vitro–transcribed RNA or with DNA vectors encoding the antigen of choice (24).
In this study, we show that CD40-B cells, transfected with PGP-encoding expression vectors, induced antigen-specific CD4 and CD8 T-cell responses against the three target antigens PR3, HNE, and cathepsin-G. Thus, CD40-activated B cells can express, process, and present endogenous leukemia-associated antigens in the context of both MHC class I and class II molecules. Furthermore, the induced CD4 and CD8 T-cell responses not only recognized naturally processed antigens on CD40B cells but also killed leukemia cells.
Materials and Methods
Cells
All subjects tested gave written permission for their blood and marrow cells to be used for research under Institutional Review Board–approved National Heart, Lung, and Blood Institute stem cell allotransplantation protocols (97-H-0099, 99-H-0046, 02-H-0111, 03-H-0192, and 04-H-0112). Apheresis collections from patients before and after transplant, and from their donors, were cryopreserved in liquid nitrogen in 20% of FCS and 10% DMSO until use. All patients and donors studied expressed HLA-A*0201. Peripheral blood mono-nuclear cells (PBMC) from nine leukemia patients (four CML chronic phase, one CML blastic phase, one acute myelogenous leukemia, and three acute lymphocytic leukemia cases) and their HLA-identical related donors were studied together with PBMC from eight CMV-seropositive and eight CMV-seronegative donors.
CD3+ T cells (>98%) were negatively selected from PBMC using magnetic beads (Resting T-Cell Isolation kit; Miltenyi, Auburn, CA). For reverse transcription-PCR detection of PGP mRNA from CML and normal progenitor cells, CD34+ cells were selected using anti-CD34 beads (Dynal Biotech, Brown Deer, WI) from two separate donor granulocyte colony-stimulating factor–mobilized PBMC samples and two PBMC samples from patients with CML.
Reverse transcription-PCR
Expression of the PGP HNE, PR3, and cathepsin-G by CD34 cells from two CML patients and by G-CSF–mobilized CD34 cells from two healthy subjects was determined using reverse transcription-PCR. HL60 cells were used as the positive control. The genes were amplified using the primers listed in Table 1. PCR conditions were 94°C for 5 minutes, then 30 cycles of 94°C for 30 seconds, 65°C for 30 seconds, and 72°C for 30 seconds. Finally, PCR products were extended at 72°C for 7 minutes.
Table 1.
PCR primers used
| Sense (5′-3′) | Antisense (5′-3′) | |
|---|---|---|
| HNE | CCACCATGACCCCCGCCGACTCGCTGTCTTTTCC | GCTCTAGAGAGAGTGGTGTGGGCAGCTGAGGTGACCCG |
| PR3 | CTGCCACCATGGCTCACCGCCCCCAGCCCTGCCCTGGCGTC | CGTTCAGACAAAGATCCGCCTCGAGGTTTGGGCCAGGCTC |
| CG | CTGCCACCATGCAGCCACTCCTGCTTCTGCTGGCCTTTCTCC | CGTCTAGAGGAGCTGGCCTGTGTCCCCGAGAAGAAGAGTCAG |
| Cloning primers | ||
| HNE, outer | GCACGGAGGGGCAGAGACCC | GTGCCAGATGCTGGAGAGTGTG |
| HNE, inner | CTGCCACCATGACCCTCGGCCGCCGACTCGCGTGTCTTTTCC | CGTCTAGAGAGAGTGTGGGTGTGGGCAGCTGAGGTGACCCG |
| PR3, outer | GAGGAGCTTGATCGTGGGTGC | GAGCTGCTTCTGTCCAAAGATC |
| PR3, inner | CTGCCACCATGGCTCACCGGCCCCCCAGCCCTGCCCTGGCGTC | CGTCTAGACAAAGATCCGCCTCGAGGGTTTGGAGCCAGGCTC |
| CG, outer | GCACAGCAGCAACTGACTGGG | GCAACACTGTGGAGCTGGCCTG |
| CG, inner | CTGCCACCATGCAGCCACTCCTGCTTCTGCTGGCCTTTCTCC | CGTCTAGAGGAGCTGGCCTGTGTCCCCGAGAAGAAGAGTCAG |
Constructs
Reverse-transcribed mRNA isolated from the myeloid leukemia cell line HL60 served as a template for the PCR amplification of the PGP open reading frames. The indicated open reading frames were amplified in a nested PCR reaction. First, the open reading frames were enriched from the cDNA using the outer primer set. Next, this outer PCR product served as a template for the inner PCR primers. The primers are listed in Table 1.
The inner primer sets contained a Kozak sequence at the 5′ end and an XbaI linker at the 3′ end. Conditions for both the outer and nested (inner) PCR were as follows: 1 minute at 95°C, followed by 35 cycles, each consisting of 30 seconds at 95°C, 50 seconds at 52°C, and 1 minute at 72°C, followed by a final extension of unfinished products for 5 minutes at 72°C.
The PCR products were ligated into the TA cloning vector system (TOPO TA cloning system, Invitrogen, Carlsbad, CA). The identity and integrity of the inserts was confirmed by sequence analysis (ABI Prism System, Applied Biosystems, Foster City, CA). The inserts were excised from the TA cloning vector with EcoRI and XbaI and cloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen). All cloned gene sequences were again confirmed using the ABI Prism system. Sequence comparison revealed that HNE and cathepsin-G genes were identical to the original genes. PR3 had three nucleotide alterations causing amino acid substitutions; C256 to G (P70 to A), A451 to G (T135 to A), T454 to A (S136 to T). These three amino acid alterations have been reported as variations (irrelevant to enzymatic activity) by BLAST search. Therefore, we considered these three cloned genes suitable for further experiments. In addition, the PGP open reading frames were cloned 5′ of the internal ribosome entry site of the expression plasmid pIRES2-eGFP (Clontech, Palo Alto, CA), facilitating expression analysis of the cloned genes due to coexpression of eGFP detectable by flow cytometry from the same RNA transcript. For the CMV experiments, pcDNA3.1 containing the IE1-pp65 fusion gene (a kind gift from Dr. Christian Davrinche, Institut National de la Sante et de la Recherche Medicale; Toulouse, France; ref. 13) was used as a stimulatory antigen. peGFP was also used as control. All plasmids were amplified and purified for transfection using the HiSpeed plasmid purification kit (Qiagen, Valencia, CA).
Generation of B-cell lines
CD40-B cells were generated as previously described (25). In brief, irradiated (75 Gy) human CD40L-transfected murine fibroblasts (LTK-CD40L), kindly provided by Dr. Van Kooten (Department of Nephrology, Leiden University Medical Centre, Leiden, Netherlands), were plated in six-well plates (BD Bioscience, Franklin Lakes, NJ) at a concentration of 0.1 × 106 cells/well, in RPMI complete medium [25 mmol/L HEPES buffer (pH 7.55), 2 mmol/L l-glutamine, 100 units/mL penicillin, and 100 μ/mL streptomycin; Life Technologies, Inc., Gaithersburg, MD] supplemented with 10% FCS and cultured overnight at 37°C, 5% CO2. After washing twice with PBS, 2 × 106 cells/mL PBMC were cocultured with LTK-CD40L cells in the presence of recombinant human interleukin-4 (rhIL-4; 4 ng/mL; Peprotech, Rocky Hill, NJ) and 0.7 μg/mL cyclosporin A in Iscove's modified Dulbecco's medium (Invitrogen), supplemented with 10% human AB serum (Gemini Bio-Product, Woodland, CA), 50 μg/mL of transferrin (Boehringer Mannheim, Indianapolis, IN), 5 μg/mL insulin (Sigma Chemical, Co., St. Louis, MO), and l-glutamine/penicillin/streptomycin as above at 37°C, 5% CO2. Cultured cells were transferred to new plates with freshly prepared, irradiated LTK-CD40L cells every 3 to 5 days. Before use, dead cells were removed from the CD40-B cells by Ficoll density centrifugation, followed by washing twice with PBS. The viability of this fraction was >99%, and >95% of the cells expressed CD19 and CD20 after 2 weeks of culture.
Transfection of CD40-B
CD40-B cells were transfected using the Amaxa (Gaithersburg, MD) B-cell Nucleofection kit according to manufacturer's instruction. CD40-B cells were transferred to B-cell medium and cultured with irradiated LTK-CD40L stimulator cells as described above until needed. Dead cells were removed from the transfected CD40-B cells before each T-cell stimulation using Ficoll density centrifugation, which typically resulted in a viability >98%.
Intracellular cytokine assay and cell surface staining
The procedure was modified from Waldrop et al. (26), and in a previous study we defined the reproducibility of this assay by replicate testing cytokine production in 24 CMV-seropositive subjects stimulated with a CMV-infected fibroblast lysate (27). In brief, 1 × 106 purified CD3 T-cells were incubated with transfected CD40-B cells, or CD40-B cells loaded separately with 10 μmol/L HLA-A*0201–restricted PR1 peptide (VLQELNVTV; amino acids 169−177 in PR3 and amino acids 168−176 in HNE; Biosynthesis, Inc., Lewisville, TX), 10 μmol/L HLA-A*0201–restricted CMV pp65 peptide (NLVPMVATV; pp65 amino acids 495−503; Biosynthesis), 10 μmol/L HLA DRB1*0301–restricted IE1 peptide (VRVDMVRHRIKEHMLKKYTQ; Biosynthesis; ref. 13), 10 μg IE1-pp65 chimeric protein, 10 μg CMV lysate (AD169, Advanced Biotechnologies, Inc., Columbia, MD); unloaded and mock-transfected CD40-B cells were used as negative controls, respectively. Anti-CD28 (Biosource International, Camarillo, CA) and anti-CD49d (Becton Dickinson, San Jose, CA) were included in all assays at 1 μg/mL each in RPMI 1640 complete medium with 10% human AB serum to provide costimulation. CD40-B cells (1 × 106) were pulsed with peptides, IE1-pp65 chimeric protein, or CMV cell lysate overnight in RPMI 1640 without serum at 37°C, 5% CO2. The cells were irradiated (75 Gy), washed once, and used as stimulators. After 2-hour culture with stimulators, Brefeldin A (Sigma) was added at 10 μg/mL for an additional 4 hours. After harvesting, cells were labeled with the following conjugated monoclonal antibodies as previously described: anti-CD3 (phycoerythrin), anti-CD4 (PerCP), anti-CD8 (APC) for cell surface antigens, and anti-human IFN-γ FITC (Becton Dickinson) for intracellular antigens (27). Staphylococcus enterotoxin B (Sigma) was used as a positive control at a concentration of 1 μg/mL. Data were collected using a FACSCalibur flow cytometer (Becton Dickinson) and analyzed with CellQuest software (Becton Dickinson).
Cytokine array
CD40-B cells cultured with LTK-CD40L cells for >2 to 3 weeks (>99% CD19-positive B cells) were irradiated (75 Gy) and cultured overnight in RPMI 1640 without serum. Supernatants were harvested and examined for their cytokine profile using an array kit (RayBiotech, Inc., Norcross, GA) according to the manufacturer's instructions.
Generation of T-cell lines
CD40-B cells were transfected, cultured for several days, and harvested. Dead cells were removed by Ficoll density centrifugation, and the remaining viable cells were irradiated (75 Gy) and used to stimulate autologous CD3+ T cells. Responder CD3+ T cells were stimulated weekly for up to 4 weeks with transfected CD40-B cells at a 1:1 stimulator/responder ratio for the first stimulation and at a lower stimulator/responder ratio (1:5−1:10) for subsequent stimulations, in 10% human AB serum/RPMI complete medium containing 10 ng/mL of recombinant human rhIL-7 and rhIL-12 (Biosource). rhIL-2 (20 IU/mL; Tecin, Roche Pharmaceutical, Indianapolis, IN) was added at 20 IU/mL the following day. After four stimulations, cultured cells were nonspecifically expanded with 50 IU/mL IL-2 and 30 ng/mL anti-CD3 antibody (Biosource).
Tetramer staining
At baseline, and after four stimulations, responder cells were stained for 30 minutes at 37°C with 1 μg (monomer weight) phycoerythrin-conjugated HLA-A*0201 PR1 tetramer-phycoerythrin (PR1 peptide: VLQELNVTV) or with APC- or phycoerythrin-conjugated HLA-A*0201 CMV pp65 tetramer (CMV pp65 peptide: NLVPMVATV) in a final volume of 50 μL. After washing with 0.5% bovine serum albumin/PBS, cells were stained for cell surface markers (CD3, CD8, and CD4) for 15 minutes at room temperature and then washed once. The cells were resuspended in 200 μL 1% paraformaldelyde/PBS and analyzed by flow cytometry on the same day.
Cytotoxicity assay
Cytotoxicity assays with Calcein-AM (Molecular Probes, Eugene, OR) were done as published elsewhere (28). In brief, bone marrow cells from HLA-A*0201–positive CML patients were labeled with Calcein-AM and used as target cells at various effector/target ratios. Results are expressed as the mean value of six replicates.
Immunocytochemical staining
Expression of PR3 was determined in transfected CD40-B cells 24, 48, and 72 hours posttransfection. Cytospin preparations of the transfectants were prepared and fixed with 4% paraformaldehyde/PBS for 10 minutes at 4°C, and permeabilized with 0.1% Triton X-100/PBS for 15 minutes at room temperature. After rinsing, nonspecific sites were blocked with 1% bovine serum albumin/PBS for 15 minutes at room temperature, and the cells stained with anti-PR3 monoclonal antibody (clone CLB12.8, Accurate Chemical & Scientific Corp., Westbury, NY) as the first antibody and goat antimouse (Fab2) IgG-FITC as the second antibody (Beckman Coulter, Miami, FL). IE1-pp65 chimeric protein expression in transfected CD40-B cells was assessed similarly using the CMV pp65 antigenemia antibody kit (Chemicon International, Inc., Temecula, CA) according to manufacturer's instructions. Specimens were examined by fluorescence microscopy.
Results
Immunologic characteristics of CD40-B cells
After 3 to 4 weeks of culture of PBMC on CD40 ligand–expressing fibroblasts, B cells dominated the cultures. Over 95% of these activated B cells were shown to express high levels of HLA class I and II, the costimulatory molecules CD80 and CD86, and the adhesion molecule CD54 (intercellular adhesion molecule-I), and moderate levels of CD83 (23 ± 4%; data not shown). The transfected CD40-B cells were irradiated (75 Gy) before use. The supernatants of these cells were examined for the presence of proinflammatory or anti-inflammatory cytokines using a cytokine array. No significant cytokine production was observed in the supernatant of serum-free culture medium (data not shown).
Optimization of transfection and T-cell stimulation procedure using IE1-pp65
CD40-B cells were harvested after 2 weeks of culture, when the majority of cells expressed B-cell markers, and transfected with constructs encoding eGFP or the fusion gene of IE1 and pp65, both considered immunodominant antigens of CMV. The transfected cells were examined daily by flow cytometry (for eGFP expression) and immunocytochemistry (IE1-pp65). The highest expression levels were achieved 48 hours posttransfection for both genes (IE1-pp65, 38.7 ± 1.0%; eGFP, 61.3 ± 6.4%). PGP expression levels by transfected CD40-B were similarly examined in four independent experiments using an eGFP-PGP transfected system, and also showed maximum expression at 48 hours after transfection as determined by flow cytometry (63.2% for PR-3, 47.8% for HNE, and 58.9% for cathepsin-G). In addition, cytoplasmic expression of PR-3 by transfected CD40-B was confirmed by immunocytochemistry (data not shown). Recognition by memory T cells was next determined using the intracellular cytokine (ICC) assay following stimulation with CD40B transfectants derived from PBMC from four CMV-seropositive donors. The highest percentage of CD4 and CD8 responder cells were detected using IE1-pp65–transfected CD40-B cells 48 hours posttransfection (Fig. 1), indicating that optimal antigen expression levels and presentation of antigenic peptide through the MHC class I and II pathways coincided. This time point was chosen for all further stimulations.
Fig. 1.
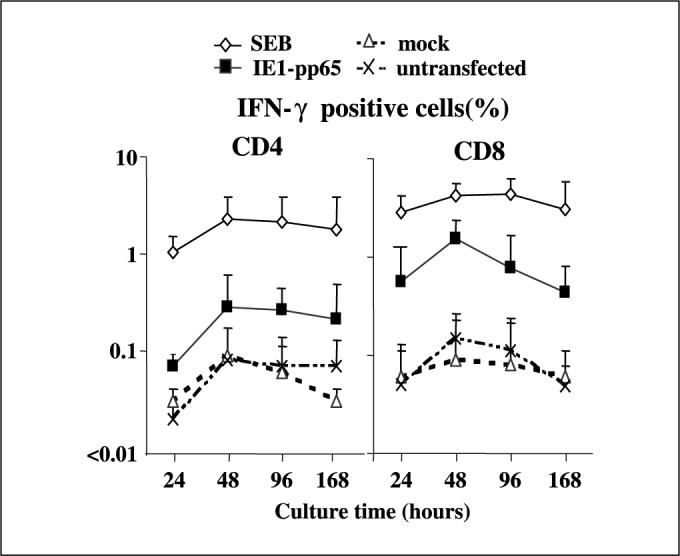
Maximum presentation of antigenic peptides processed from transfected protein by CD40B for both MHC class I and II occurs 48 hours after transfection. CD4 and CD8 T cells from four CMV-seropositive, HLA-A*0201–positive healthy donor PBMC were examined for IFN-γ production by ICC assay in response to IE1-pp65 fusion gene or mock transfected autologous CD40B. Both CD4 and CD8 Tcells showed maximum IFN-γ production after 48 hours culture with IE1-pp65 fusion gene transfectants at the 48-hour time point (0.3% and 1.6%, respectively). CD8 responses against transfectants were greater than those of CD4 Tcells but the transfected protein was processed with similar kinetics through both MHC class I and II pathways. SEB, Staphylococcus enterotoxin B (positive control); mock, CD40B cells transfected with vector alone; untransfected, CB40B cells alone (negative control). Points, mean; bars, SD.
Next, CD4 and CD8 T-cell responses of eight HLA-A*0201–positive, CMV-seropositive and three HLA-A*0201–positive CMV-seronegative subjects were examined using autologous transfected CD40-B cells. Seven of eight seropositive subjects showed cytokine production in CD4 and CD8 T cells in response to IE1-pp65 –transfected CD40-B cells but not to the mock-transfected control (Fig. 2A); none of the seronegative controls produced IFN-γ in response to these autologous CD40-B antigen transfectants (Fig. 2B). As a further control, CD40-B cells were loaded with CMV lysate or with HLAA*0201–binding peptide. With one exception, the responses to these antigens paralleled the data from transfectant stimulations, suggesting that all seven (87.5% of total) positive responders to the IE1-pp65 fusion gene transfectant had a memory CD8 T-cell response against pp65. The frequency of responder cells to transfected CD40-B cells was higher than to peptide-loaded CD40-B cells (2.1 ± 0.8% versus 0.57 ± 0.26% in transfected and peptide-pulsed CD40-B cells, respectively; P < 0.001), suggesting that multiple epitopes were recognized by the CD8 T cells. Both CD4 and CD8 T cells responded specifically to CMV lysate-loaded CD40 B cells (Fig. 2).
Fig. 2.

IFN-γ production by HLA-A*0201–positive, CMV-seropositive (A) and CMV-seronegative (B)donor PBMC stimulated with E1-pp65 fusion gene–transfected CD40-B cells. Seven of eight HLA-A*0201+/CMV+ donor CD8 T lymphocytes responded to IE1-pp65 fusion gene transfectants (IE1-pp65 fusion gene versus mock: 2.09% versus 0.06%), with smaller responses in CD4 Tcells (0.11% versus 0.05%, respectively). Seronegative donors did not show significant responses to CMV stimulation. The numbers and bars represent mean frequency (%) of IFN-γ–positive CD8 and CD4 Tcells determined by ICC assay.
To assess whether the transfectants were capable of supporting the expansion of type I cytokine (IFN-γ)-producing CD8 and CD4 T cells, T cells from three CMV-seropositive/HLAA*0201–positive healthy controls were stimulated weekly with IE1-pp65 or mock DNA–transfected autologous CD40-B up to three times, and then tested for IFN-γ expression. IE1-pp65 DNA-transfected CD40-B preferentially expanded CD8 T cells (mean CD8; 37 ± 24% before versus 87 ± 7% after expansion; mean CD4: 53 ± 11% before versus 11 ± 10% after expansion; Table 2). Using pp65[495−503] peptide-pulsed CD40-B cells and tetramer staining, it was found that the CD8 responders included an expansion of pp65[495−503]–specific cells. In Fig. 3, the relative increase in cell numbers was 2 log (165−790–fold increase) in IE1-pp65 DNA–responsive CD8 T cells and 2 to 3 log (140−3345–fold increase) in CMV-pp65/HLA-A*0201 peptide –responsive CD8 T cells.
Table 2.
Characteristics of expanded T cells with repetitive stimulations by IE1-pp65 fusion gene — transfected autologous CD40-B cells in CMV-seropositive/HLA-A*0201 — positive healthy donors D1, D3, and D8
|
CD4/8 phenotype (%) |
IE1-pp65 fusion gene |
IE1-pp65 protein |
pp65[495−503] peptide |
CMV lysate, 10 μg |
Mock transfected |
CD40B alone |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post | |
| D1 | ||||||||||||||
| CD4 | 58.5 | 9 | 0.04 | 0.3 | 0.1 | 2.44 | 0.02 | 0.08 | 0.07 | ND | 0.01 | 0.33 | 0.01 | 0.18 |
| CD8 | 39.5 | 87 | 5.4 | 18.5 | 0.07 | 0.48 | 0.38 | 3.93 | 0.03 | ND | 0.03 | 0.04 | 0.03 | 0.04 |
| D3 | ||||||||||||||
| CD4 | 59.5 | 18 | 0.07 | 2.9 | 0.3 | 5.0 | 0.04 | 0.27 | 0.38 | 1.2 | 0.04 | 0.38 | 0.03 | 0.15 |
| CD8 | 39 | 80 | 0.84 | 5.1 | 0.4 | 1.0 | 0.38 | 1.95 | 0.23 | 0.32 | 0.37 | 0.37 | 0.23 | 0.16 |
| D8 | ||||||||||||||
| CD4 | 40 | 6 | 0.1 | 1.71 | 0.06 | 1.24 | 0.03 | 0.14 | 0.14 | 1.32 | 0.02 | 0.35 | 0.03 | 0.14 |
| CD8 | 60 | 94 | 2.22 | 34.4 | 0.04 | 0.98 | 1.04 | 14.3 | 0.02 | 0.05 | 0.04 | 0.19 | 0.03 | 0.05 |
NOTE: Results show percentage of IFN-γ production after 3 weekly stimulations, determined by ICC assay for each stimulator cell type; phenotypic data is shown as percentage of cells expressing either CD4 or CD8. Significant differences are in bold.
Abbreviation: ND, not done.
Fig. 3.

IE1-pp65 ^ transfected autologous CD40-B cells expand CD4 and CD8 responses to the fusion protein and CD8 responses to pp65[495−503] peptide. CD3 Tcells from three CMV-seropositive/HLA-A*0201 ^ positive individuals (D1, D3,and D8) were stimulated weekly 3with IE1-pp65 fusion gene ^ transfected autologous CD40-B
× cells. Pre, at start of culture; post, after expansion in cultureT-cell responses were measured by IFN-γ ICC assay. Absolute numbers of responding CD4 and CD8 Tcells are shown before and after stimulation. All individuals showed 2 to 3 log-fold increases in target-specific CD4 and CD8 Tcells.
The IE1-pp65 specificity of the CD4 responders was confirmed using recombinant IE1-pp65 chimeric protein-loaded autologous CD40-B cells (Table 2; Fig. 3). There was an ∼2 log (60−117×) increase in CD4 T-cell responses to the chimeric protein. The epitope-specific expansion of CD4 T cells with IE1-pp65 DNA-transfected CD40-B cells was also shown for an HLA-DRB1*0301-binding, IE1-derived epitope in donor D8. The frequency increased from 0.1% before expansion to 1.23% after in vitro expansion. In the same cultures, the frequency of pp65[495−503]–responsive CD8 cells rose from 1.04% to 14.3%, indicating that the DNA-transfected CD40-B cells were capable of specifically expanding both CD8 and CD4 T cell subsets. Overall, the data indicate that the antigenic peptides derived from endogenously synthesized protein translated from transfected DNA can be simultaneously processed to both MHC class I and II pathways in CD40-B.
Expansion of primary granule protein–specific T cells
Having established the optimal conditions for the generation and transfection of CD40-B cells, and for generating antigen-specific CD4 and CD8 T cells, we next focused on PGP as candidates for immunotherapy of myeloid malignancies using transfected CD40-B to expand PGP-specific T cells. PBMC from five HLA-A*0201+ patients transplanted for myeloid leukemias (four CML and one acute myelogenous leukemia patients), at the time of complete remission and full-donor T-cell chimerism, were examined for T-cell responses against PGP-transfected CD40-B cells generated from their respective donors. All five patients had been selected because they had low frequencies of circulating CD8 T cells recognizing PR3, as shown by PR1 tetramer staining. CD4 and CD8 T-cell responses were examined in vitro using PGP-encoding DNA-transfected CD40-B cells (Fig. 4A and B). After one stimulation, most of the patients had both CD4 and CD8 responses against PR-3 (three of five CD4 and five of five CD8 cells), HNE (four of five CD4 and four of five CD8 cells), and cathepsin-G (five of five CD4 and four of five CD8 cells). Notably, in responders, CD8 T-cell frequencies achieved antigen-specific stimulation frequencies around 1%. In contrast, posttransplant PBMC from three HLAA*0201+ patients with acute lymphocytic leukemia showed weak responses in CD8 cells mainly to cathepsin-G (Fig. 4C). These observations suggests that circulating PGP-responsive T cells exist in a majority of patients with myeloid leukemia who received allogeneic bone marrow transplantation, but that cathepsin-G may be more widely recognized.
Fig. 4.
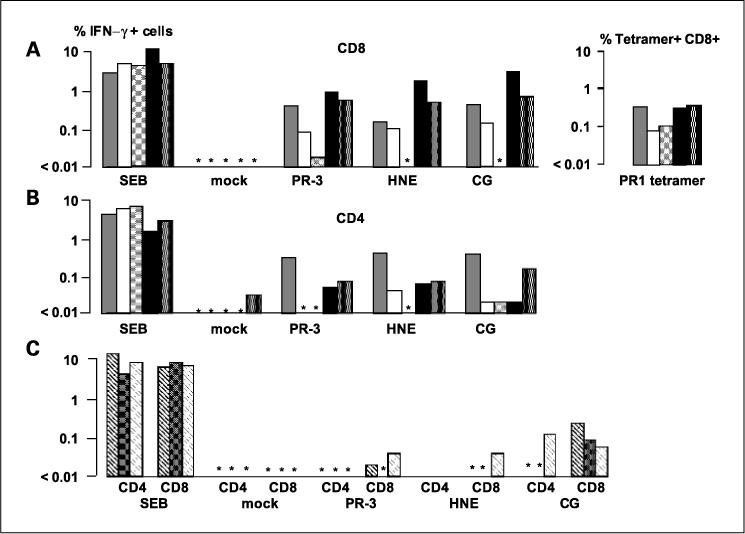
CD8 and CD4 PGP transfectant-responsive T cells are detected more readily in day 100 posttransplantation samples from patients with myeloid leukemias than in patients with lymphoid malignancies. PGP-specific IFN-γ responses measured by ICC in five HLA-A*0201–positive patients with myeloid leukemia (three CML chronic phase, one CML in blastic phase, and one acute myelogenous leukemia) 100 days following stem cell transplant from an HLA identical sibling donor. Asterisks, undetectable IFN-γ. A and B, CD8 and CD4 responses, respectively. All patients showed detectable frequencies of PR1 tetramer-positive cells. C, absent or weak IFN-γ responses to PR3 and HNE but stronger responses to cathepsin-G (CG)in three patients with acute lymphocytic leukemia 100 days posttransplant. SEB, Staphylococcus enterotoxin B–positive control; mock, mock transfected CD40-B cells negative control.
Repeated stimulations with primary granule protein–transfected CD40-B can induce T-cell responses against chronic myelogenous leukemia blasts and chronic myelogenous leukemia progenitor cells
A sixth patient (HLA-A*0201+) with CML blast crisis pretransplant had a CD8 T-cell response to PR1 peptide before antigen-specific expansion as determined by tetramer staining and IFN-γ production by quantitative PCR (29). CD3 cells obtained from PBMC of this patient were stimulated weekly with mock-transfected, PR3-, HNE-, or cathepsin-G–encoding DNA-transfected autologous CD40-B up to four times and tested for recognition of CML cells. All three T-cell lines stimulated with PGP-encoding constructs showed IFN-γ production in CD4 and CD8 T-cell subsets upon stimulation with autologous leukemic cells, whereas the mock-transfected control cultures were negative (Table 3).
Table 3.
Lymphocytes from a patient with CML in blast crisis were pulsed with PR3, HNE, and cathepsin-G — transfected CD40B cells or untransfected or mock-transfected controls
| Stimulators | CD4 (%) | CD8 (%) | CD4+/IFN-γ+ (%) | CD8+/IFN-γ+ (%) |
|---|---|---|---|---|
| Untransfected | ||||
| None | 54 | 46 | — | — |
| CD40B | 72 | 28 | 0.59 | 0.13 |
| CD40B+ PR1 | 77 | 23 | 0.1 | 0.19 |
| Transfected | ||||
| Mock | 67 | 33 | 0.12 | 0.16 |
| PR3 | 55 | 45 | 1.07 | 0.47 |
| HNE | 75 | 25 | 0.72 | 1.34 |
| Cathepsin-G | 70 | 30 | 0.87 | 0.51 |
NOTE: Only PGP-transfected CD40B cells elicited significant increases in IFN-γ upon stimulation with autologous leukemic blasts. Significant differences are in bold.
PBMC from another HLA-A*0201–positive CML patient taken posttransplant during complete molecular remission and with full-donor chimerism were similarly expanded with the PGP-encoding DNA-transfected HLA-identical donor CD40-B cells. The resulting lines were tested for cytotoxicity against pretransplant leukemic cells, and, as a control, bone marrow cells from the donor. Although the leukemia cells were generally more susceptible to killing, all three lines (but especially PR3-stimulated cells) specifically recognized the leukemic cells and not the donor-derived cells (Fig. 5). The leukemia specificity correlated with mRNA expression levels as assessed in CD34 cells from two CML patients and two healthy controls: only CML cells showed PR3, HNE, and cathepsin-G expression (Fig. 6).
Fig. 5.

Repeated stimulations with PGP-transfected CD40B cells induces a T-cell response with specificity for CML cells. CD3+ T cells obtained from PBMC of patient 5 with CML at day 100 post–bone marrow transplantation were stimulated weekly with PGP-transfected CD40B cells from the HLA-identical sibling donor, and tested for cytotoxicity against autologous CML cells and against normal bone marrow cells from the donor after the fourth stimulation (week 4). The PR3, HNE, and cathepsin-G–induced Tcell lines selectively recognized CML cells but not normal cells. E:Tratio, effector-to-target ratio.
Fig. 6.
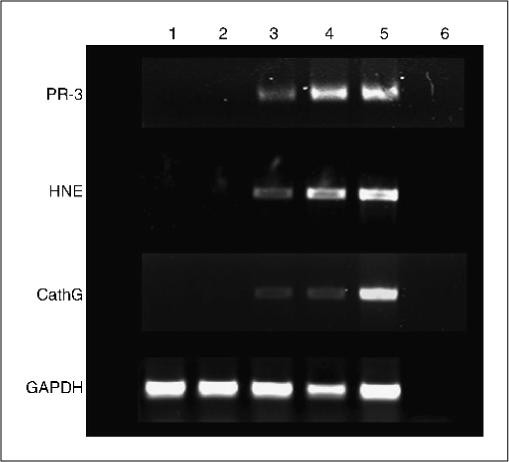
PGP mRNA is expressed in CML but not in normal CD34 cells. CD34 cells from two CML patients and from two healthy donors were examined for the expression of PGP transcripts using reverse transcription-PCRas described in Materials and Methods. Lanes 1and 2, CD34 cells from healthy donors; lanes 3 and 4, CML CD34 cells; lane 5, HL-60 cells; lane 6, water as a negative control. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a RNA quality control in the reverse transcription-PCR. CathG, cathepsin-G.
Discussion
These data expand our previous findings that PR3- and HNE-specific T-cell responses can be amplified from post–bone marrow transplantation samples (8, 29). In addition, we identified cathepsin-G as an immunotherapeutic candidate leukemia-associated antigen of myeloid, and possibly of lymphoid, leukemias. As with PR3 and HNE, cathepsin-G is overexpressed in myeloid leukemias but the function of cathepsin-G in leukemic cells is not known. Low levels of memory T cells recognizing PGP in normal individuals suggest that tolerance to these self-antigens is normally maintained (29). The potential of PGP to induce T-cell responses may be related to their aberrant expression in CD34 cells of patients with myeloid malignancies coupled with this immunologic memory for PGP. Our results suggest that the entire family of PGP represent an important immunotherapeutic target in myeloid malignancies. At present, only the PR3 peptide PR1 has been evaluated in clinical trials (30). However, the dramatic clinical responses to PR1 peptide vaccine in HLA-A*0201 individuals with leukemia supports the likelihood that T-cell responses to the entire protein sequence of PR3, as well as to other PGP, would be more powerful because of their polyclonality, and would be better sustained by CD4 help through MHC class II–presented antigens. The use of entire protein sequences as the antigen source would more be generally applicable because leukemias other than those in patients expressing HLA-A*0201 could be targeted. Furthermore, interindividual variability of immune responses, e.g., to PGP vaccines, would be minimized because of the diversity of potential antigens available to induce an immunodominant response to PGP.
Here, we describe CD4 and CD8 T-cell responses against the leukemia-associated antigens PR3, HNE, and cathepsin-G. The resultant T-cell lines displayed full effector activity against leukemic cells, showing specific killing of leukemic cells and synthesizing IFN-γ upon stimulation with the leukemic cells. These results suggest that our approach to induction and expansion of leukemia-associated antigen-specific T cells resulted in effective graft-versus-leukemia responses.
To advance immunotherapeutic approaches, investigators have focused on several ways to modify APC to induce antigen-specific T cells. These include CD40B cells (15), APC retrovirally transduced with CMV-DNA, or tumor antigen–encoding RNA (16–18), and loaded with tumor cell lysates (14). We confirmed that in vitro–activated B cells expressed high levels of MHC class I and II molecules, the costimulatory molecules CD80, CD83, CD86, and the adhesion molecule CD54, as previously reported (19). We then optimized our culture system using a chimeric molecule of the CMV immunodominant antigens IE1 and pp65. Optimal viral protein expression levels and T-cell stimulation capacity was reached 48 hours posttransfection. Because there is a tendency with CD40B cells to elicit the less cytotoxic Th2/Tc2 T-cell phenotypes (31), we added the Th1/Tc1-promoting dendritic cell–derived cytokine, IL-12. Using these 48-hour cultures, we showed that CMV-specific CD4 and CD8 T cells proliferated in response to transfected CD40-B cells, and that these cells retained the capacity to express IFN-γ in response to antigen stimulation, indicating that functional antigen-specific T cells were selectively amplified. Using CD40B cells as APC, Schultze et al. (19) were able to amplify tyrosinase peptide–specific CD8 T cells, which recognized melanoma cells. Whether the MHC class II–presented antigens are derived from endogenously processed antigens, or from uptake and processing of antigen-expressing, dying B cells is not known. The latter mechanism is supported by our observations that CD40-B cells could also present antigens from CMV-infected fibroblast lysate.
In this study, we used CD40-B cells as the source of the APC, because they are readily available in the laboratory. An alternative strategy would be to use gene-modified dendritic cell to present peptides from PGP protein to CD4 and CD8 T cells. Dendritic cells have the advantage of being the most reliable APC to generate cytotoxic T lymphocytes, and both dendritic-cell and B-cell APC can present peptides from transduced protein genes (32, 33). Translational research to make one or other of these approaches applicable for the induction and adoptive transfer of leukemia-specific T cells in clinical trials is now required.
Acknowledgments
Grant support: MEDIC Foundation.
Footnotes
Note: D.A. Price is a Medical Research Council (United Kingdom) Clinician Scientist.
References
- 1.El Ouriaghli F, Fujiwara H, Melenhorst JJ, Sconocchia G, Hensel N, Barrett AJ. Neutrophil elastase enzymatically antagonizes the in vitro action of G-CSF: implications for the regulation of granulopoiesis. Blood. 2003;101:1752–8. doi: 10.1182/blood-2002-06-1734. [DOI] [PubMed] [Google Scholar]
- 2.El Ouriaghli F, Sloand E, Mainwaring L, et al. Clonal dominance of chronic myelogenous leukemia is associated with diminished sensitivity to the antiproliferative effect of neutrophil elastase. Blood. 2003;102:3786–92. doi: 10.1182/blood-2003-03-0861. [DOI] [PubMed] [Google Scholar]
- 3.Molldrem J, Dermime S, Parker K, et al. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from protein-ase 3 preferentially lyse human myeloid leukemia cells. Blood. 1996;88:2450–7. [PubMed] [Google Scholar]
- 4.Molldrem JJ, Clave E, Jiang YZ, et al. Cytotoxic T lymphocytes specific for a nonpolymorphic proteinase 3 peptide preferentially inhibit chronic myeloid leukemia colony-forming units. Blood. 1997;90:2529–34. [PubMed] [Google Scholar]
- 5.Clave E, Molldrem J, Hensel N, Raptis A, Barrett AJ. Donor-recipient polymorphism of the proteinase 3 gene in bone marrow transplantation. J Immunother. 1999;22:1–6. doi: 10.1097/00002371-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Molldrem JJ, Lee PP, Wang C, Champlin RE, Davis MM. A PR1-human leukocyte antigen-A2 tetramer can be used to isolate low-frequency cytotoxicT lymphocytes from healthy donors that selectively lyse chronic myelogenous leukemia. Cancer Res. 1999;59:2675–81. [PubMed] [Google Scholar]
- 7.Molldrem JJ, Lee PP, Wang C, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6:1018–23. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- 8.Fujiwara H, El Ouriaghli F, Grube M, et al. Identification and in vitro expansion of CD4+ and CD8+ Tcells specific for human neutrophil elastase. Blood. 2004;103:3076–83. doi: 10.1182/blood-2003-07-2424. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos KP, Suciu-Foca N, Hesdorffer CS, Tugulea S, Maffei A, Harris PE. Naturally processed tissue- and differentiation stage-specific autologous peptides bound by HLA class I and II molecules of chronic myeloid leukemia blasts. Blood. 1997;90:4938–46. [PubMed] [Google Scholar]
- 10.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–7. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machiels JP, van Baren N, Marchand M. Peptide-based cancer vaccines. Semin Oncol. 2002;29:494–502. doi: 10.1053/sonc.2002.35244. [DOI] [PubMed] [Google Scholar]
- 12.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 13.Vaz-Santiago J, Lule J, Rohrlich P, et al. Ex vivo stimulation and expansion of both CD4(+) and CD8(+) T cells from peripheral blood mononuclear cells of human cytomegalovirus-seropositive blood donors by using a soluble recombinant chimeric protein, IE1-pp65. J Virol. 2001;75:7840–7. doi: 10.1128/JVI.75.17.7840-7847.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kierstead LS, Ranieri E, Olson W, et al. gp100/ pmel17 and tyrosinase encode multiple epitopes recognized by Th1-type CD4+ T cells. Br J Cancer. 2001;85:1738–45. doi: 10.1054/bjoc.2001.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630–41. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 16.Berzofsky JA, Terabe M, Oh S, et al. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest. 2004;113:1515–25. doi: 10.1172/JCI21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–47. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y, Bosch ML, Salgaller ML. Current methods for loading dendritic cells with tumor antigen for the induction of antitumor immunity. J Immunother. 2002;25:289–303. doi: 10.1097/00002371-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Schultze JL, Michalak S, Seamon MJ, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100:2757–65. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Bergwelt-Baildon MS, Vonderheide RH, Maecker B, et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical application. Blood. 2002;99:3319–25. doi: 10.1182/blood.v99.9.3319. [DOI] [PubMed] [Google Scholar]
- 21.Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63:2836–43. [PubMed] [Google Scholar]
- 22.von Bergwelt-Baildon MS, Schultze JL, Lapointe R, et al. Lapointe R, et al. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63:2836–43. Correspondence re. [PubMed] [Google Scholar]; Cancer Res. 2004;64:4055–7. [Google Scholar]
- 23.Kondo E, Topp MS, Kiem HP, et al. Efficient generation of antigen-specific cytotoxic T cells using retrovirally transduced CD40-activated B cells. J Immunol. 2002;169:2164–71. doi: 10.4049/jimmunol.169.4.2164. [DOI] [PubMed] [Google Scholar]
- 24.Coughlin CM, Vance BA, Grupp SA, Vonderheide RH. RNA-transfected CD40-activated B cells induce functional T-cell responses against viral and tumor antigen targets: implications for pediatric immuno-therapy. Blood. 2004;103:2046–54. doi: 10.1182/blood-2003-07-2379. [DOI] [PubMed] [Google Scholar]
- 25.Grube M, Rezvani K, Wiestner A, et al. Autoreactive, cytotoxic T lymphocytes specific for peptides derived from normal B-cell differentiation antigens in healthy individuals and patients with B-cell malignancies. Clin Cancer Res. 2004;10:1047–56. doi: 10.1158/1078-0432.ccr-03-0075. [DOI] [PubMed] [Google Scholar]
- 26.Waldrop SL, Pitcher CJ, Peterson DM, Maino VC, Picker LJ. Determination of antigen-specific memory/ effector CD4+ T cell frequencies by flow cytometry. J Clin Invest. 1997;99:1739–50. doi: 10.1172/JCI119338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hensel N, Melenhorst JJ, Bradstock K, et al. Flow cytometric quantitation and characterization of the T-lymphocyte memory response to CMV in healthy donors. Cytotherapy. 2002;4:29–40. doi: 10.1080/146532402317251509. [DOI] [PubMed] [Google Scholar]
- 28.Wang XM, Terasaki PI, Rankin GW, Jr, Chia D, Zhong HP, Hardy S. A new microcellular cytotoxicity test based on calcein AM release. Hum Immunol. 1993;37:264–70. doi: 10.1016/0198-8859(93)90510-8. [DOI] [PubMed] [Google Scholar]
- 29.Rezvani K, Grube M, Brenchley JM, et al. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood. 2003;102:2892–900. doi: 10.1182/blood-2003-01-0150. [DOI] [PubMed] [Google Scholar]
- 30.Heslop HE, Stevenson FK, Molldrem JJ. Immunotherapy of hematologic malignancy. Hematology (Am Soc Hematol Educ Program) 2003:331–49. doi: 10.1182/asheducation-2003.1.331. [DOI] [PubMed] [Google Scholar]
- 31.Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172:3422–7. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 32.Chen M, Shirai M, Liu Z, Arichi T, Takahashi H, Nishioka M. Efficient class II major histocompatibility complex presentation of endogenously synthesized Hepatitis C virus core protein by Epstein-Barr virus-transformed B lymphoblastoid cell lines to CD4+ Tcells. J Virol. 1998;72:8301–8. doi: 10.1128/jvi.72.10.8301-8308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonehill A, Heirman C, Tuyaerts S, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172:6649–57. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]