

Abstract
The RP 10 form of autosomal dominant retinitis pigmentosa (adRP) is caused by mutations in the widely expressed protein inosine 5′-monophosphate dehydrogenase type 1 (IMPDH1). These mutations have no effect on the enzymatic activity of IMPDH1, but do perturb the association of IMPDH1 with nucleic acids. Two newly discovered retinal-specific isoforms, IMPDH1(546) and IMPDH1(595), may provide the key to the photoreceptor specificity of disease (S.J. Bowne, Q. Liu, L.S. Sullivan, J. Zhu, C.J. Spellicy, C.B. Rickman, E.A. Pierce, S.P. Daiger, Invest. Ophthalmol. Vis. Sci. 47 (2006) 3754–3765). Here we express and characterize the normal IMPDH1(546) and IMPDH1(595), together with their adRP-linked variants, D226N. The enzymatic activity of the purified IMPDH1(546) and IMPDH1(595) and the D226N variants is indistinguishable from the canonical form. The intracellular distribution of IMPDH1(546) and IMPDH1(595) is also similar to the canonical IMPDH1 and unaffected by the D226N mutation. However, unlike the canonical IMPDH1, the retinal specific isoforms do not bind significant fractions of a random pool of oligonucleotides. This observation indicates that the C-terminal extension unique to the retinal isoforms blocks the nucleic acid binding site of the IMPDH1, and thus uniquely regulates protein function within photoreceptors.
Keywords: retinitis pigmentosa, IMP dehydrogenase, retinal degeneration, nucleic acid binding protein
Introduction
Retinitis pigmentosa (RP)1 is an inherited retinal disease affecting approximately one in 3,500 individuals [1]. The progressive vision loss results from apoptosis of rod and cone photoreceptor cells [2, 3]. RP is often caused by mutations in photoreceptor-specific proteins, but can also result from mutations in widely expressed proteins such as IMPDH1 [4–6]. The most commonly occurring IMPDH1 mutation, D226N, accounts for 1–2.5% of all autosomal dominant RP cases [6–8]. Mutations in IMPDH1 also cause Leber congenital amaurosis (LCA), a more severe retinal degeneration [7]. The pathophysiological mechanism of IMPDH1-linked adRP and the tissue specificity of disease are not understood.
IMPDH catalyzes the controlling step in de novo synthesis of guanine nucleotide. Mammals express two isozymes, IMPDH1 and IMPDH2, which are 84% identical. To the first approximation, IMPDH1 is constitutively expressed and IMPDH2 is amplified in proliferating tissues [9, 10]. IMPDH1 predominates in the retina [11].
It is unlikely that adRP results from a loss of IMPDH1 function in the retina. First, the adRP-linked mutations of IMPDH1 have no effect on enzymatic activity [7, 12]. Moreover, most of the mutations are located in a subdomain that is not required for enzymatic function [13, 14]. Second, drugs targeting IMPDH are widely used as immunosuppressive, anticancer and antiviral therapy, yet no visual side effects have been reported. Lastly, IMPDH1 knocked-out mice display only mild retinopathy [15]. We have discovered that IMPDH binds nucleic acids, and that this interaction is mediated by the subdomain [16]. The adRP-linked mutations perturb this nucleic binding function, decreasing both affinity and specificity of this interaction [7, 12].
We have recently discovered that human retina contains two novel isoforms of IMPDH1, IMPDH1(546) (major) and IMPDH1(595) (minor) derived from alternative mRNA splicing [these proteins are also known as IMPDH1α/IMPDH1(13b) and IMPDHγ/IMPDH1(A+13b), respectively] [11, 17]. Both IMPDH1(546) and IMPDH1(595) contain a 32 residue C-terminal extension while IMPDH1(595) has an additional 49 residues on the N-terminus. Since poly-His and GFP tags do not perturb either the enzymatic or nucleic acid binding actitvities of IMPDH [12, 15], these N- and C-terminal extensions need not perturb function. The retinal-specific expression of IMPDH1 isoforms and the compensating presence of IMPDH2 in other tissues may account for the photoreceptor-specific effects of the adRP/LCA-causing mutations, but do not explain the pathological mechanism.
Here we characterize the two retinal isoforms of IMPDH1 as well as their D226N variants. The enzymatic activity of these isoforms is indistinguishable from the canonical IMPDH1. In contrast, neither retinal isoform binds nucleic acids. This observation suggests that C-terminal extension blocks the nucleic acid binding site, perhaps by interacting with the subdomain. We propose that photoreceptors contain additional proteins that interact with the C-terminal extensions to regulate the nucleic acid binding properties of the retinal isoforms.
Materials and Methods
Materials
A pBluescript-based plasmid containing the promoter for human interphotoreceptor retinoid-binding protein (hIRBP) was the generous gift from Dr. Muna Naash (University of Oklahoma) [18]. HeLa cells were purchased from ATCC. HEK293 and COS cells were gifts from the laboraories of Drs. Daniel Oprian and Melissa Moore (Brandeis University).
Plasmids Construction and Mutagenesis
We expressed IMPDH1(546) and IMPDH1(595) with C-terminal GFP tags under the control of the hIRBP promoter. The 1.3 kb HindIII fragment containing the hIRBP promoter was cloned into the pCI mammalian expression vector (Promega) between the CMV promoter and the chimeric intron sequence. Kpn I/Not I fragments containing the cDNA for the retinal isoforms were inserted into this plasmid [11]. The TGA stop-codon for IMPDH1 was then changed to GGA encoding Gly using QuikChange® (Stratagene). A second round mutagenesis inserted an Xma I site at the end of the IMPDH1 gene, which was used to insert the GFP fragment. The D226N mutation was created with QuikChange® to obtain the RP-linked forms. The canonical IMPDH1-GFP was cloned into pCI using the Xho I/Not I fragment from pH1GFP [12] and was expressed under the control of the CMV promoter. The wild-type gene was also mutated to the D226N form in the cloned plasmid. The success of all cloning and mutagenesis procedures was confirmed by DNA sequencing (Genewiz).
Cell Culture and DNA Transfection
Cells were cultured at 37 °C in a humidified 5% CO2 incubator in RPMI 1640 medium (ATCC) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen). Cells were allowed to grow overnight to reach ~50% confluence prior to DNA transfection with SuperFect® reagent (Qiagen).
Fluorescence Microscopy
Cells were grown on 4-well chamber slides (Nagle Nunc) overnight, then fixed with 3.7% paraformaldehyde, washed twice with PBS and co-stained with DAPI reagent (Invitrogen). The slides were mounted using Prolong Gold antifade reagent (Invitrogen) and viewed with an Olympus IX70 inverted fluorescence microscopy (Melville, NY, USA).
Western Blot Analysis
HEK293 cells were transfected with the IMPDH1-GFP plasmids and allowed to grow for 48 h before being collected by trypsinization. Soluble cell extracts were prepared as described by the Clontech TransFactor Whole Cell Extraction Kit (PT3744-2). Proteins were resolved by SDS-PAGE and electro-blotted to PVDF membrane, which was then incubated with primary mouse monoclonal antibody AS37-P that recognizes both human IMPDH1 and IMPDH2 (Antibody Solutions). The membrane was subsequently incubated with goat anti-mouse IgG antibody conjugated with horseradish peroxidase (Upstate). The proteins (IMPDH1-GFP and native IMPDH) were detected by ECL Plus system (GE Healthcare) and visualized through exposure to Clear Blue X-ray film (Pierce).
Protein Purification
The pET-32Ek/LIC plasmids containing cDNA for retinal specific isoforms of IMPDH1 were used as templates to generate the corresponding D226N mutants using QuikChange [11]. These plasmids express the retinal isoforms as fusion proteins containing thioredoxin, His and RNase S peptide N-terminal tags. BL21 cells were grown in LB media in the presence of 50 μg/mL ampicillin at 37°C. Protein expression was induced by the addition of IPTG (0.6 mM). The cell pellets were collected by centrifugation and lysed through sonication. The cleared supernatants were applied to a Ni affinity column (HisTrap) and the fusion proteins were eluted with 300 mM imidazole followed by treatment with enterokinase (Roche) at 25°C for 12 h to remove the tags. The activity of the proteins did not change during the protease digestion, indicating that the tags do not affect protein structure. The protein preparation was applied to the HisTrap affinity column to remove the residual undigested His-tagged proteins and the tags. The purity of the final products was 95% as assessed by SDS-PAGE.
Enzyme Activity Assays
The steady-state kinetic parameters were determined at 25°C in 50 mM Tris-Cl buffered solution (pH 8.0) containing 100 mM KCl, 1 mM DTT and 3 mM EDTA. NADH formation was followed by the absorbance increase at 340 nm. The data were fitted into equation:
| (1) |
in assays with saturating concentration of NAD+ (500 μM) and varying concentrations of IMP. For reactions with saturating IMP (500 μM), the data were fitted to equation to account for substrate inhibition:
| (2) |
The initial reaction velocity and final enzyme concentration are designated as v and [E], kcat, Ka, Kb are the catalytic turnover number and Michaelis-Menten constants for IMP and NAD+, respectively, and Kii is the substrate inhibition constant for NAD+.
Filter Binding Assays
Binding of IMPDH1 to single-stranded DNA (ssDNA) was performed as previously described [12, 16]. The sequence of the DNA pool used was 5′-GGGAATGGATCCACATCTACGAATTC-N30-TTCACTGCAGACTTGACGAAGCTT-3′, where N30 denotes a 30 bases-long random sequence. Protein and the 5′-32P-labelled ssDNA (final concentration 0.15 nM) was mixed and incubated for 10 min at 25 °C in 10 mM Tris-Cl buffered solution (pH 8.0) containing 50 mM KCl and 1 mM DTT (binding buffer). The mixture was filtered through a nitrocellulose membrane (top layer) to trap protein and protein-DNA complexes followed by a Hybond™ (GE Healthcare) membrane (bottom layer) which binds free nucleic acid using a vacuum manifold. The membranes were washed twice with the binding buffer. The radioactivity on each membrane was quantified by PhosphorImager (Molecular Dynamics). The results were fitted into equation 3 using Kaleidagraph software:
| (3) |
where f is the fraction of protein-bound ssDNA, R is the maximum fraction of ssDNA bound to each protein and K is the binding constant.
Results and Discussion
Expression of GFP-tagged Retinal Isoforms of IMPDH1 in Mammalian Cells
The retinal specific isoforms of IMPDH1(546) and IMPDH1(595) were expressed into HEK293, HeLa and COS cells with similar results. The fusion protein was expressed at levels approximating that of the endogenous IMPDH (Figure 1). As was observed with the canonical IMPDH1 [12], the IMPDH1(546) and IMPDH1(595) are found predominately in the cytosol, with some protein also in the nucleus (Figure 2). The D226N mutation does not change the cellular distribution of either isoform. In all cases, the fluorescence is evenly distributed in the cytoplasm, no aggregation is observed and Western blots confirm that the proteins are soluble. These observations suggest that RP is unlikely to result from changes in the cellular localization of IMPDH1.
Figure 1.
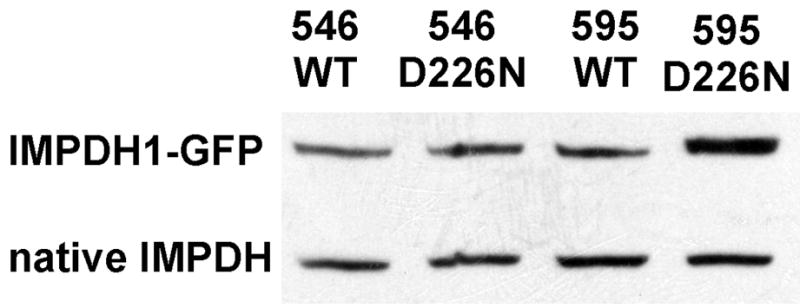
Expression of retinal IMPDH1 isoforms. Soluble extracts of HEK293 cells transiently transfected with plasmids encoding retinal IMPDH1-GFP were analyzed by Western blot. From left to right: IMPDH1(546), IMPDH1(546)-D226N, IMPDH1(595), IMPDH1(595)-D226N.
Figure 2.

Localization of the retinal IMPDH1 isoforms. HeLa cells transiently expressing IMPDH1-GFP were viewed under fluorescence microscopy. A) Distribution of various forms of IMPDH1 as revealed by the GFP fluorescence; B) Nucleic staining of the same cells by DAPI; C) Overlays of images in A and B.
Enzymatic Activity of the Retinal Isoforms of IMPDH1
IMPDH1(546) and IMPDH1(595) and their pathogenic D226N variants were expressed as C-terminal fusion proteins in E. coli and purified as described in Materials and Methods. The thioredoxin, His and RNAse S peptide tags were removed by enterokinase cleavage. Interestingly, the retinal isoforms display similar specific activities before and after enterokinase digestion, indicating that the N-terminal tags do not affect enzymatic activity. The enzymatic activities of these proteins are summarized in Table 1. The values of kcat, Km(IMP) and Km(NAD+) are indistinguishable from the canonical IMPDH1 in all the enzymes. Curiously, the retinal isoforms do not display inhibition at high NAD+ concentrations (Figure 3). However, since the intracellular concentrations of NAD+ are ~500 μM, this behavior is unlikely to be physiologically relevant. Therefore the N- and C-terminal segments do not influence the enzymatic activity of IMPDH1. As observed with the canonical IMPDH1 [12], the D226N mutation does not influence the enzymatic activity of the retinal isoforms.
Table 1.
Steady-state kinetic parameters for human IMPDH1 isoforms. Experiments on the retinal specific forms were conducted at 25 °C in 50 mM Tris-Cl buffered solution (pH 8.0) containing 100 mM KCl, 1 mM DTT and 3 mM EDTA. The conditions are described in Materials and Methods. N.A.: not applicable.
| Enzyme | kcat (s−1) | Km (IMP) (μM) | Km (NAD+) (μM) | Kii (NAD+) (mM) |
|---|---|---|---|---|
| IMPDH1(546) | 0.22 ± 0.03 | 15 ± 4 | 22 ± 3 | N.A. |
| IMPDH1(546)-D226N | 0.24 ± 0.03 | 11 ± 1 | 24 ± 3 | 6 ± 4 |
| IMPDH1(595) | 0.18 ± 0.03 | 11 ± 1 | 26 ± 4 | N.A. |
| IMPDH1(595)-D226N | 0.22 ± 0.03 | 10 ± 1 | 16 ± 2 | N.A. |
| IMPDH1(514) | 0.31 ± 0.03 | 4 ± 1 | 14 ± 2 | 0.51 ± 0.07 |
| IMPDH1(514)* | 1.2 ± 0.4 | 17 ± 2 | 70 ± 10 | 2.0 ± 0.6 |
| IMPDH1(514)-D226N* | 0.7 ± 0.2 | 12 ± 2 | 50 ± 10 | 4 ± 2 |
Data from Ref. [12], measured at 37°C.
Figure 3.
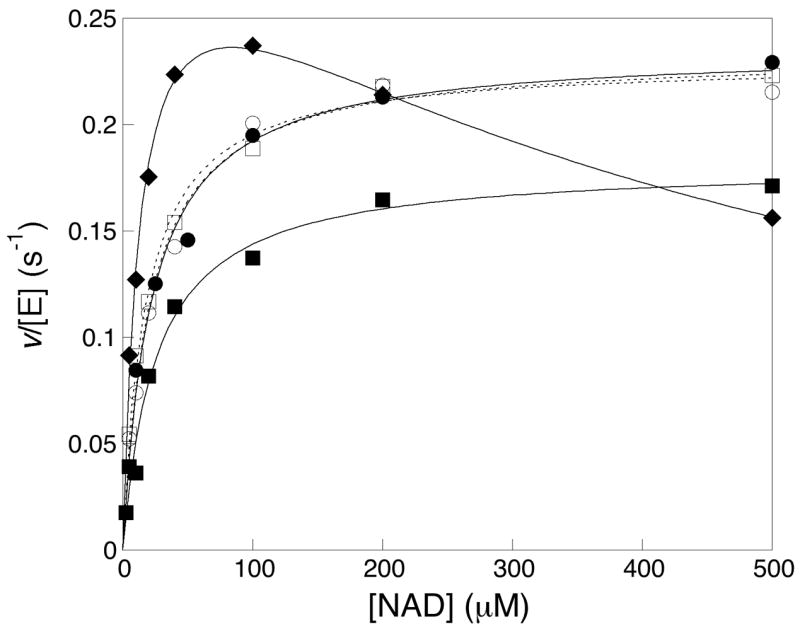
Steady state kinetics of retinal IMPDH1 isoforms. The rate of NADH formation in the presence of saturating IMP (final concentration 500 μM) was divided by the monomeric concentration of IMPDH1 to obtain v/[E]. The values were plotted against the concentration of NAD+. The results obtained on the canonical IMPDH(514) were designated with closed diamonds (◆); IMPDH1(546), closed circles (●); IMPDH1(546)-D226N, open circles (○); IMPDH1(595), closed squares (■); IMPDH1(595)-D226N, open squares (□)]. Each data point represents the average value from at least three independent experiments.
The Retinal Isoforms of IMPDH1 Are Poor Nucleic Acid Binding Proteins
The nucleic acid binding properties of the retinal isoforms are very different from the canonical IMPDH1. The standard assay measures the fraction of a random oligonucleotide pool associated with protein on a nitrocellulose filter. The canonical IMPDH1 binds ~7% of the pool with Kd = 6 nM [7, 12]. However, 50 nM IMPDH1(546) binds only ~0.3% of the pool (Figure 4). Since the assays contain 0.15 nM of oligonucleotide, only 0.5 pM of a contaminating protein is required to account for this observed binding, which would represent a contamination of ~0.001%. Therefore we cannot eliminate the possibility that the observed binding is due to association with a contaminating protein. Nevertheless, these results clearly indicate that the nucleic acid binding site of the canonical IMPDH1 is blocked by the C-terminal extension of IMPDH1(595). Since the presence of a C-terminal GFP does not block nucleic acid binding [12], a specific interaction likely exists between the C-terminal extension and the nucleic acid binding site. Perhaps the C-terminal extension binds to the subdomain.
Figure 4.
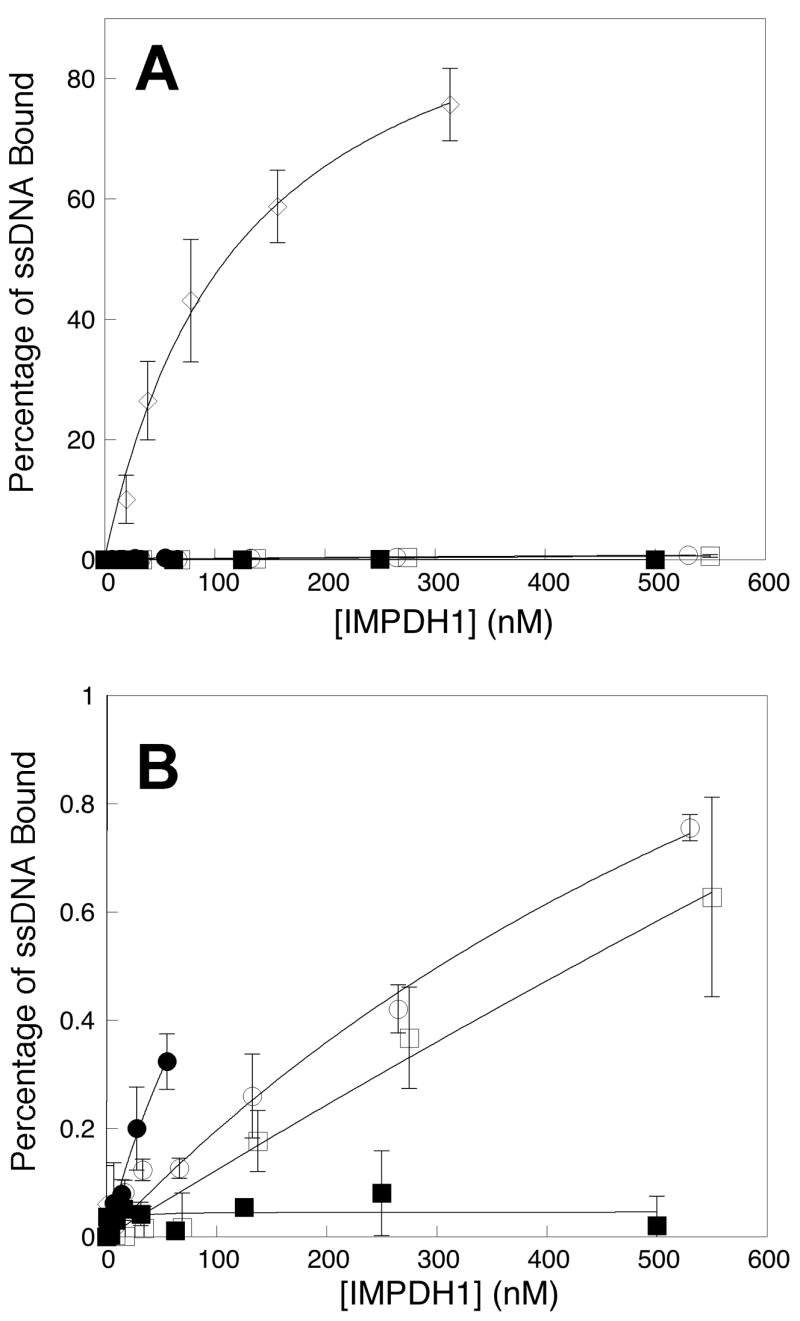
Binding of ssDNA to retinal IMPDH1 isoforms. A random pool of 32P-labeled oligonucleotides (0.15 nM) was incubated with varying concentrations of IMPDH1 (concentration is reported as tetramer). Protein-bound nucleic acid was measured in a filter-binding assay as described in details in Materials and Methods. A) The percentages of total ssDNA associated with IMPDH1 were plotted against the protein concentration. The canonical IMPDH1(514)-D226N, open diamonds (◇); IMPDH1(546), closed circles (●); IMPDH1(546)-D226N, open circles (○); IMPDH1(595), closed squares (■); and IMPDH1(595)-D226N, open squares (□); B) Expanded view of the results on the retinal IMPDH1 isoforms with the same designations as in A. Each data point represents the average value from at least three independent experiments.
No oligonucleotide binding is observed to IMPDH1(595) (Figure 4). Although, given the caveats cited above, we cannot be confident that there is a significant difference between the nucleic acid binding properties of IMPDH1(546) and IMPDH1(595), it is possible that the N-terminal extension also blocks nucleic acid binding.
The D226N mutation clearly decreases the affinity and specificity of the canonical IMPDH1, so that the protein binds >70% of the pool with a Kd = 190 nM (Figure 4 and [12]). However, the D226N mutation has only subtle effects on the nucleic acid binding properties of the retinal isoforms (Figure 4; as noted above, these subtle effects could easily be due to the presence of a contaminating protein). Note that this result is not surprising if the C-terminal extension interacts with the nucleic acid binding site as proposed above: if the nucleic acid binding site is blocked, then the effects of the D226N mutation will be masked.
Summary
Here we report the characterization of novel isoforms of IMPDH1 that predominate in the retina. The enzymatic activity of these isoforms is indistinguishable from the canonical enzyme. The RP-linked mutation D226N does not affect enzymatic activity of the retinal isoforms. Likewise, these isoforms have similar cellular localization as the canonical form, and again the D226N mutation does not change the cellular distribution. Interestingly, unlike the canonical IMPDH1, the retinal isoforms do not bind a significant portion of a random pool of oligonucleotides. Therefore the C-terminal extensions unique to the retinal isoforms block the nucleic acid binding site, perhaps by interacting with the subdomain. We propose that photoreceptors contain additional factors that interact with the C-terminal extensions to regulate nucleic acid binding.
Acknowledgments
The authors wish to thank Dr. Melissa J. Moore for use of the confocal microscope.
Abbreviations used
- RP
retinitis pigmentosa
- adRP
autosomal dominant RP
- IMPDH1
inosine 5′-monophosphate dehydrogenase type 1
- IMPDH1(546)
retinal specific IMPDH1 with additional C-terminal residues
- IMPDH1(595)
retinal specific IMPDH1 with additional N- and C-terminal residues
- IMPDH1-514
canonical human IMPDH1
- D226N
the D226N mutant form
- GFP
green fluorescence protein
- ATCC
American Type Culture Collection
- PBS
phosphate-buffered saline
- RPMI
Roswell Park Memorial Institute
- CMV
cytomegalovirus
- hIRBP
human interphotoreceptor retinoid-binding protein
- ssDNA
single-stranded DNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heckenlively JR, Daiger SP. Principals and Practices of Medical Genetics. In: Rimoin DI, Connor JM, Pyeritz RE, editors. Churchill Livingston. New York: 2001; 2255. p. 2576. [Google Scholar]
- 2.Portera-Cailliau C, Sung CH, Nathans J, Adler R. Proc Natl Acad Sci USA. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang GQ, Hao Y, Wong F. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- 4.Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennan A, Aherne A, Palfi A, Humphries M, McKee A, Stitt A, Simpson DA, Demtroder K, Orntoft T, Ayuso C, Kenna PF, Farrar GJ, Humphries P. Hum Mol Genet. 2002;11:547–557. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- 6.Wada Y, Sandberg MA, McGee TL, Stillberger MA, Berson EL, Dryja TP. Invest Ophthalmol Vis Sci. 2005;46:1735–1741. doi: 10.1167/iovs.04-1197. [DOI] [PubMed] [Google Scholar]
- 7.Bowne SJ, Sullivan LS, Mortimer SE, Hedstrom L, Zhu J, Spellicy CJ, Gire AI, Hughbanks-Wheaton D, Birch DG, Lewis RA, Heckenlively JR, Daiger SP. Invest Ophthalmol Vis Sci. 2006;47:34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Invest Ophthalmol Vis Sci. 2006;47:3052–3064. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senda M, Natsumeda Y. Life Sci. 1994;54:1917–1926. doi: 10.1016/0024-3205(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 10.Nagai M, Natsumeda Y, Konno Y, Hoffman R, Irino S, Weber G. Cancer Res. 1991;51:3886–3890. [PubMed] [Google Scholar]
- 11.Bowne SJ, Liu Q, Sullivan LS, Zhu J, Spellicy CJ, Rickman CB, Pierce EA, Daiger SP. Invest Ophthalmol Vis Sci. 2006;47:3754–3765. doi: 10.1167/iovs.06-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mortimer SE, Hedstrom L. Biochem J. 2005;390:41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gan L, Petsko GA, Hedstrom L. Biochemistry. 2002;41:13309–13317. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- 14.Nimmesgern E, Black J, Futer O, Fulghum JR, Chambers SP, Brummel CL, Raybuck SA, Sintchak MD. Protein Expression and Purification. 1999;17:282–289. doi: 10.1006/prep.1999.1136. [DOI] [PubMed] [Google Scholar]
- 15.Aherne A, Kennan A, Kenna PF, McNally N, Lloyd DG, Alberts IL, Kiang AS, Humphries MM, Ayuso C, Engel PC, Gu JJ, Mitchell BS, Farrar GJ, Humphries P. Hum Mol Genet. 2004;13:641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- 16.McLean JE, Hamaguchi N, Belenky P, Mortimer SE, Stanton M, Hedstrom L. Biochem J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spellicy CJ, Daiger SP, Sullivan LS, Zhu J, Liu Q, Pierce EA, Bowne SJ. Mol Vis. 2007;13:1866–1872. [PubMed] [Google Scholar]
- 18.Liou GI, Geng L, al-Ubaidi MR, Matragoon S, Hanten G, Baehr W, Overbeek PA. J Biol Chem. 1990;265:8373–8376. [PubMed] [Google Scholar]