

Abstract
Narcolepsy with cataplexy is a debilitating sleep disorder with an estimated prevalence of about 0.05%. Narcolepsy is caused by a selective loss of hypocretin (orexin) producing neurons in the perifornical hypothalamus. Based on the very strong association with the HLA subtype DQB1*0602, it is currently hypothesized narcolepsy is caused by an autoimmune mediated process directed at the hypocretin neurons. So far however, studies focussing on general markers of (auto)immune activation, as well as humoral immunity against the hypocretin system have not yielded consistent results supporting this hypothesis.
Keywords: narcolepsy, cataplexy, autoimmunity, antibody, antigen, hypocretin, orexin, HLA
INTRODUCTION
Over the last decade, the understanding of the pathophysiology of narcolepsy has increased greatly. A string of discoveries was initiated by the identification of the responsible gene in the dog model for narcolepsy; coding for the hypocretin (orexin) receptor 2 . It is now clear that human narcolepsy is caused by deficiencies in hypothalamic hypocretin neurotransmission, most likely through a selective loss of hypocretin producing neurons. Mainly based on the tight association of narcolepsy with a specific HLA subtype (DQB1*0602), many authors have postulated that the disorder may be autoimmune in nature. Obviously, the hypocretin system forms a very likely target for such a pathophysiological mechanism, and this revamped the research in this area. In this review we will describe the available knowledge on (possible) immune mechanisms in the pathophysiology of narcolepsy.
CLINICAL FEATURES OF NARCOLEPSY
Narcolepsy is a disabling sleep disorder that severely interferes with normal daily activities, interpersonal relations, education- and job opportunities.1–3 Narcolepsy with cataplexy is defined as a separate category in the recently updated International Classification of Sleep Disorder.4 The main reason for this refining is the intimate relation between the occurrence of cataplexy and hypocretin deficiency, a relation that is hardly present in narcolepsy without cataplexy, and absent in other causes of hypersomnia such as idiopathic hypersomnia.
Excessive daytime sleepiness (EDS) is the principal complaint. It will be present each and every day from disease onset on. Patients report an almost continuous feeling of sleepiness throughout the day, together with the occurrence of episodes of irresistible sleep. Automatic behavior may occur as another expression of EDS. EDS in narcolepsy is best described as a ‘continiously waxing and waining of attention’ and an ‘inability to stay awake for longer periods of time’ rather than a disorder with an increased need of sleep. The inability to stay awake during daytime, is mirrored during the night: nocturnal sleep in narcolepsy is characterized by sleep fragmentation and an inability to remain asleep. As a consequence, the total amount of sleep over 24 hours is virtually unaltered.5,6
Cataplexy is the only specific symptom of narcolepsy. It is defined as sudden and transient episodes of bilateral loss of muscle tone of brief duration (less than two minutes), triggered by emotions – most reliably laughing or joking- with preserved consiousness.1,7 In fact, narcolepsy patients may become literally ‘weak with laughter’. All striated muscles may be involved during cataplexy, except the extraocular and respiratory musculature. Although laughter is the strongest trigger, various emotions may induce a cataplectic attack such as unexpectedly meeting an acquaintance, scoring a goal in sports or merely thinking about the punch line of a joke. The last two features of the classical symptom ‘tetrad’ are sleep paralysis and hypnagogic hallucinations. Besides the aforementioned symptoms, narcoleptics are frequently obese.8
The sleep complaints of narcoleptics are reflected in the findings on polysomnography. Nocturnal sleep studies show a lowered sleep efficiency with frequent stage shifts and arousals. Multiple Sleep Latency Testing reveals a very short sleep latency during the day. Furthermore, narcoleptics typically have multiple sleep-onset REM periods (i.e. the occurrence of REM sleep within 15 minutes after sleep onset). These findings have also diagnostic value, although the specificity is not optimal. In the absence of cataplexy, sleep registrations are mandatory to make a diagnosis of narcolepsy.4 Hypocretin measurement is a newly developed diagnostic tool, as the majority of patient with narcolepsy-cataplexy are hypocretin deficient (see below). This has also important consequences for research; pathophysiological studies gain a lot of value when homogenous patient groups are included, and hypocretin measurements provide this opportunity.
EPIDEMIOLOGY
Narcolepsy has an estimated prevalence of 20–60 per 100,000 in Western countries.9–11 There have been reports of a much higher prevalence in Japan, and a much lower prevalence in Israel, although these discrepancies may be due to different diagnostic criteria and differences in study design. There seems to be a slight male predominance. Narcolepsy may start before puberty; in about 6% of cases it starts before the age of 10. In most cases, age at onset lies between 15 and 40 years. One study found a bimodal distribution in the age at onset, with the biggest peak around 15 years and a second peak around 36 years.12 Age at onset may be genetically determined, as the patients with a more early onset were more severely affected and more often had a positive family history. Symptoms usually develop gradually; it may take several years for each subsequent symptom to occur. However, in some patients symptoms may develop over a short period over several weeks. If cataplexy is the second symptom to occur, it appears on average six years after the onset of excessive daytime sleepiness.10 Once the symptoms are present there are usually only minor fluctuations in severity.
A number of studies examined the timing of birth of narcoleptic patients. If a seasonal birth pattern in narcolepsy is present, this may provide clues to specific environmental factors, such as infectious agents, nutrion effects or temperature. In two larger studies, an excess number of March births, and a decreased number of birth in September was found.13,14 In another study there was an increase in the first half of the year, and a decrease in the second half of the year.15 However, we performed a study in 283 Dutch patients with narcolepsy-cataplexy and compared the birth months to the expected proportion of births per month of all live-births in the Netherlands from 1924 to 1996, and did not find significant differences (Donjacour, Fronczek, le Cessie, Lammers and van Dijk; unpublished data).
Due to the considerable methodological difficulties, there have been only few studies specifically focusing on environmental factors. Using the ‘Schedule of Recent Experiences’ questionnaire, Orellana et al found that more than half of the patients report important life events in the time just before the first onset of symptoms. Often mentioned events include major psychological stress, major familial events, sleep deprivation or other changes in sleep/wake schedules, and head trauma.16 However, these types of studies are hampered by several methodological issues, such as recall-bias.
Studies on diseases associated with narcolepsy are also rare.17 There have been reports on an increased frequency of migraine. Furthermore, as HLA-DQB1*0602 is protective for type I diabetes and there is a high prevalence of obesity in narcoleptic patients, it is likely that there is an large discrepancy between the co-occurrence of type I versus type II diabetes with narcolepsy. However, this has not been studied.
There have been several case reports of secundary narcolepsy in patients with auto-immune neurological disorders such as multiple sclerosis and acute disseminated encephalomyelitis.18– 21 These disorders caused focal lesions in the hypothalamic area, showing that autoimmune mechanisms can damage the hypocretin system. Large epidemiological studies on co-occurrence with other proven autoimmune disorders, including non-neurological ones, are lacking however.
FAMILY AND TWIN STUDIES
Depending on the exact definition of narcolepsy, in about 1–4 % of cases, narcolepsy occurs in families. Although some early reports might have mistaken other sleep disorders for narcolepsy, numerous familial cases have been reported. Genuine multiplex (i.e. more affected generations) families are very rare. If narcolepsy runs in families, it typically shows an autosomal dominant mode of inheritance.22–24
The majority of patients suffer from non-familial (sporadic) narcolepsy, but genetic factors are still important in those cases. Prevalence studies have shown that the risk for a first-degree relative of a patient with narcolepsy is 1 to 2 percent.25 This risk, although small, is still 30 to 40 times higher than the estimated prevalence in the general population, suggesting the existence of genetic factors that predispose to the development of narcolepsy.
Although genetic factors play a role in sporadic narcolepsy, these are neither necessary, nor sufficient to cause narcolepsy. Twin studies showed that only 25 to 31% of monozygotic twins are concordant for the disease (for references see 26), suggesting a major contribution of environmental factors. In familial cases and concordant twin pairs, there is a much less strong association with either HLA-DQB1_*0602 or hypocretin deficiency, indicating the presence of additional disease influencing genes.
THE ROLE OF THE HYPOCRETIN SYSTEM
Animal models
A cloning effort was initiated in the early 1990s to identify the gene responsible for the autosomal recessive form of narcolepsy in the dog, designated canarc-1. Nine years later, a critical region was finally established containing 1 gene (Hcrtr2), coding for the hypocretin-receptor 2. Truncating mutations in the Hcrtr2 gene were found in both narcoleptic Labradors and Dobermans.27 Both mutations result in a receptor lacking the intracellular end, and therefore an absence of function.28 Interestingly, in dogs with a sporadic form of narcolepsy, low hypocretin levels were later reported.29 Shortly after canarc-1 was cloned, Yanagisawa’s group reported on the phenotype of preprohypocretin knockout mice. Using a combination of infrared video recordings during the active period and sleep recordings, they convincingly showed that these animals develop narcolepsy, including cataplexy-like behavior, sleep fragmentation and sleep-onset REM periods.30 More recent rodent models for narcolepsy include transgenic mice in which the hypocretin promotor drives the expression ataxin-3, a truncated Machado-Joseph disease gene, causing a post-natal degeneration of the hypocretin neurons and consequently symptoms of narcolepsy.31
Hypocretin defects in human narcolepsy
A large body of evidence shows that the hypocretin system is also critically involved in human narcolepsy. Shortly after the seminal papers on the pathophysiology of narcolepsy in dogs and mice, it was reported that the sporadic form of narcolepsy in humans is associated with absent levels of the hypocretin-1 peptide in the cerebrospinal fluid (CSF).32 Subsequent studies confirmed the association, and also showed that hypocretin defiency in the CSF is highly specific for HLA-DQB1*0602 positive narcolepsy with cataplexy.33,34 Besides case-reports,35,36 so far only a few disorders have been found in which undetectable CSF hypocretin levels can be present; two of them are autoimmune in nature: Guillain-Barré syndrome and anti-Ma2 associated limbic encephalitis.37 In healthy subjects CSF hypocretin-1 levels in the CSF are highly constant over a large age-range.38 In contrast, hypocretin levels are clearly decreased even shortly after disease onset in young narcoleptics.39
Post-mortem pathology studies in human narcolepsy
Using in-situ hybridization in frozen brain tissue, Peyron et al showed a complete disappearance of hypocretin mRNA in the perifornical hypothalamus of narcoleptic patients.40 In contrast, MCH producing neurons which are located in the same area were normally present. In this study, there were no signs of recent inflammation in two brains examined for TNF-alpha and HLA-DR2 expression. Furthermore, there were no overt signs of gliosis or global cell loss in the hypothalamus. Thannickal et al used to stain for hypocretin peptide in fixed human brains, after application of antigen retrieval techniques.41 In all narcoleptic brains there was a dramatically reduced number of hypocretin neurons, although a small percentage was preserved. Furthermore, using GFAP as a marker for reactive astrocytes, there were signs of gliosis in the hypothalamus but also in hypocretin projection areas.42 It is likely that the reduction in hypocretin expression in the hypothalamus is due to a selective cellular loss, as two co-localizing markers in hypocretin neurons, dynorphin and NARP, were also absent in the hypothalamic area of narcoleptic brains.43,44
In-vivo imaging studies
Althought the (small) post-mortem studies suggest a loss of hypocretin producing neurons, a ‘dying hypocretin cell’ has never been seen, so a neurodegenerative process has nog clearly been proven. Various investigators have approached this issue using in-vivo neuroimaging techniques. The most promising technique in this respect was voxel-based morphometry, an unbiased sensitive MRI method that compares grey and white matter density on a voxel-by-voxel basis between groups of subjects. Unfortunately, VBM studies did not yield consistent results, neither in the hypothalamus nor in hypocretin projection areas.45–48 Other, more indirect techniques such as MR spectroscopy may shed some insight,49 but the final answer still needs to come from additional post-mortem studies. Sometime in the future, the possibility may hopefully arise to study brain tissue obtained close to disease onset, and this will be a crucial study.
GENETICS: HLA
One of the most important predisposing genetic factors is a specific human leukocyte antigen (HLA) subtype. The HLA complex is formed by a collection of genes on chromosome 6, including the HLA DR and DQ loci encoding the alpha and beta chains of the corresponding HLA protein. Variants of these highly polymorphic genes can be determined by serological techniques, or in much more detail at the DNA level. In the early 1980s, it was reported that all narcolepsy patients in Japan carried the specific serologically determined subtype DR2.50 This association was quickly confirmed in Caucasians at an almost equally strong level. With the advent of HLA genotyping/sequencing it became clear that the DNA-defined DQ subtype DQB1*0602 is the most specific marker for narcolepsy across all ethnic groups.51 The complete DR2 haplotype associated with narcolepsy is DRB1*1501 – DQA1*0102 – DQB1*0602.52 Patients with narcolepsy-cataplexy carry this haplotype in 85–95% of cases, compared to about 30% of the normal population. People homozygous for HLA DQB1*0602 have an even higher relative risk to acquire narcolepsy than people heterozygous at the locus.53 Detailed studies have now shown that the HLA association in narcolepsy is complex, and involves multiple modulating loci. HLA DQB1*0301 is an additional susceptibility factor in DQB1*0602 heterozygotes. On the other hand DQB1*0501 and 0601 are found to be protective.54
There has been discussion whether or not HLA DQB1*0602 is a narcolepsy susceptibility gene itself, or that the actual gene is only located very close to the HLA DQ region. Systematic haplotype and sequence analysis of the HLA-DR and -DQ regions revealed a four marker haplotype surrounding the DQB1*0602 gene in narcoleptic patients. So far, 86 kb of contiguous genomic sequencing across the region has not revealed new genes.55 Furthermore, single nucleotide polymorphism analysis has not revealed variation among DQB1*0602 chromosomes specific to narcolepsy.
Siebold et al studied the crystal structure of DQB1*0602 molecule in reference to both diabetes, in which it is protective, and narcolepsy.56,57 DQB1*0602 was found to bind the N-terminal part of the preprohypocretin molecule with high affinity. By comparing to DQB1*0601, these researchers demonstrated that DQB1*0602 has a larger P4 pocket which would facilitate bing of larger hydrophobic residues (Figure 1). Apparently, the fact that DQB*0601 differs only in 9 amino acids from DQB1*0602 may have important consequences for peptide binding abilities, which may explain the opposite effects on narcolepsy susceptibility of the 2 alleles.
Figure 1.
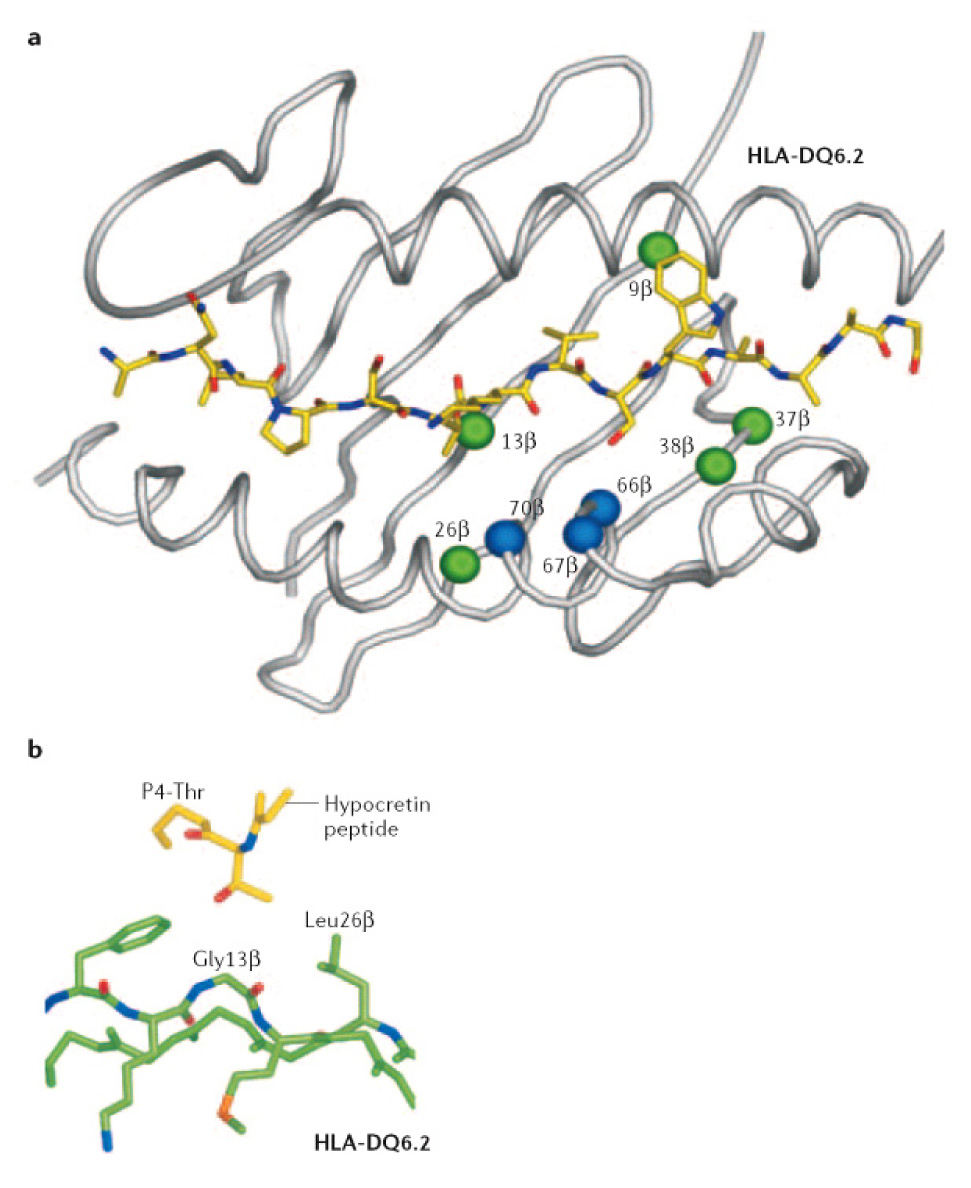
Structural representation of HLA-DQB1*0602 and 0601. A) Residues that are different between both subtypes are highlighted as spheres (green, residues contributing to peptide-binding pockets; blue, residues at the putative T-cell receptor (TCR)- recognition surface). B) Detail of the P4 binding pocket. Figure reprinted with permission from 57
In the majority of disorders with a proven autoimmune etiology, associations have been found with specific HLA subtypes, typically in a complex interaction between predisposing and protecting alleles. Examples of such disorders include type I diabetes, rheumatoid arthritis and systemic lupus erythematosus. The hypothesis that narcolepsy is autoimmune in nature is large based on the HLA association. It is important to note that in most autoimmune disorders the linkage with HLA is not as high as in narcolepsy.This should prompt us to keep an open mind towards other pathophysiological mechanisms. Co-segregating genes in the HLA region do not seem to be present.55 However, another mechanism could be a direct effect of HLA DQB1*0602 on sleep structure: even in the normal population, people positive for DQB1*0602 have a shorter REM sleep latency.58,59 Perhaps DQB1*0602 modulates the symptom expression of another pathophysiological process, in such a way that it is necessary in order to display a full narcolepsy phenotype.60
GENETICS: NON-HLA GENES
Although the vast majority of patients with the sporadic form of narcolepsy are HLA DQB1*0602 positive, there have been consistent reports on patients with unquestionable narcolepsy, being negative for HLA DQB1*0602.52,54 Furthermore, risk of family members to develop narcolepsy is not fully explained by the HLA association.61,62 Finally, more than a third of patients in true multiplex families are negative for HLA DQB1*0602.63 Taken together, these data point to a role for additional, non-HLA genes in the susceptibility to narcolepsy.
Candidate gene approaches
Obviously, the different hypocretin system genes are likely candidates to play a role in the pathophysiology of narcolepsy. Peyron and Faraco et al performed a direct sequencing study of the preprohypocretin, hypocreting receptor 1 and 2 genes, in a total of 74 patients, including patients with and without a family history of narcolepsy, and patients with and without the HLA DQB1*0602 antigen.40 Although 14 polymorphisms were detected in the three genes, none of these were associated with or linked to narcolepsy. These results were confirmed by other association studies.64,65 However, a mutation in the preprohypocretin gene was identified in a single, atypical patient (DQB1*0602 negative, very young age at onset, very severe clinical symptoms).40 This G-to-T transversion changes a leucine to arginine in the hypocretin signal peptide, impairing peptide trafficking and processing. In-vitro studies suggest that these results in cell death, and this patient indeed is CSF hypocretin deficient.
Other genetic predisposition factors are likely to play a role in narcolepsy, although their contribution is limited. Possible associations between narcolepsy and polymorphisms of genes involved in monoaminergic neurotransmission were reported.66,67 An interesting possibility in view of the autoimmune hypothesis is a possible association with tumor necrosis factor-alpha gene polymorphisms, which have been described in proven autoimmune disorders.68 Increased TNF-alpha levels were reported previously in narcolepsy. 69–71 Three population-based association studies analyzed a polymorphism in the TNFA gene promoter with conflicting results 72–74 but a recent study in 153 trio narcolepsy families, mostly from North European origins, showed a strong association with TNF-alpha 75 suggesting that TNFA may be another susceptibility gene, particularly in association with HLA DRB1*1501 and DQB1*0602.
Linkage and association studies
There are only two genome-wide linkage and one association studies available in narcolepsy. The low number of linkage studies is due to the rare familial forms with several subjects affected over several generations. One study included 8 small Japanese families with 21 HLA DR2-positive patients (14 narcoleptic cases with cataplexy and 7 with an incomplete form of narcolepsy) and found a suggestive linkage to 4p13-q21.76 In a second study, a large French family was included with 4 patients with clear-cut cataplexy and 10 others affected excessive daytime sleepiness without cataplexy. Genomewide linkage analysis identified a 5.15-Mb candidate region on chromosome 21q. 77 However, molecular analysis of several candidate genes in the region, revealed no pathogenic mutation. A recent genome-wide association study with a large number of microsatellite markers was performed in Japanese HLA DQB1*0602 positive patients and observed a strong association on chromosome 21q22.3.78 Further high-density mapping yielded a polymorphism in an unknown gene, which showed possible protective effects against narcolepsy.
THE AUTOIMMUNE HYPOTHESIS
As discussed in the section on HLA, the tight HLA association is the main fundament for the theory that narcolepsy is caused by an autoimmune process. This theory would also fit with the peripubertal age at onset, the complex susceptibility system with multiple genes interacting with environmental factors, and with the presumed selective destruction of hypocretin neurons. This model was further outlined by Longstreth et al, based on a comparable disease model as postulated in type I diabetes (Figure 2).10 In this model, the hypocretin cells can be targeted in individuals with a specific genetic susceptibility background, initiated by environmental factors. When the majority of hypocretin cells have disappeared, narcoleptic symptoms start to occur and the severity of symptoms will augment with an increasing number of degenerating hypocretin cells. The challenge now is to find direct evidence for this disease mechanism. Already before the involvement of the hypocretin system was discovered, various studies focussed on possible autoimmune mechanisms in narcolepsy. Over the last couple of years, researchers started to aim these efforts directly on the hypocretin system.
Figure 2.
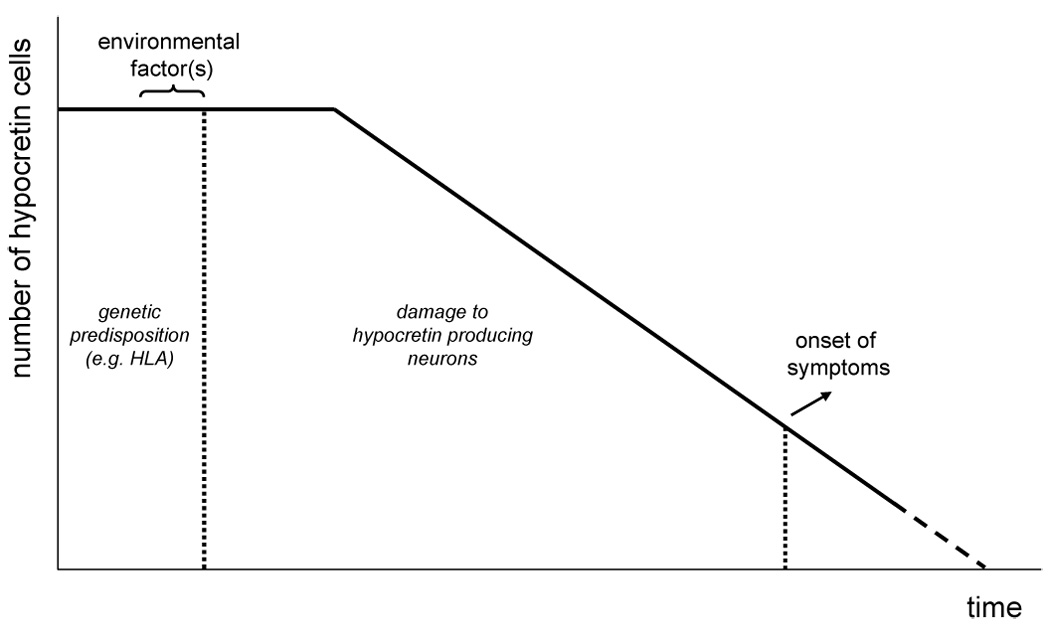
Theoretical model for the pathophysiology of narcolepsy, based on Longstreth et al.10 A similar model has been produced for autoimmune disorders such as type I diabetes. A specific genetic background is necessary to develop narcolepsy. Triggered by certain environmental factors, the disease process is initiated resulting in progressive damage to the hypocretin neurons. At some point, the number of remaining neurons has become so low that clinical symptoms become overt.
Non-specific immune markers
Matsuki et al studied 30 HLA-DR2 positive narcoleptic patients and compared them to 55 controls for differences in cellular subsets measured by the following markers: OKT3+ (mature T cell), OKT4+ (TH cells), OKT8+ (suppressor/cytotoxic T cells), Leu7 (large granular lymphocytes), OKB7 (mature B cells), OKIa1 (HLA-DR, DQ framework), and OKM1 (monocyte, C3b receptor).79 OKIa1 and OKM1 cells were increased significantly. Hinze-Selch et al found no difference in T-cell subsets with CD4 (TH marker), CD8 (TC marker), CD45 (T-, B-, TC, and NK cell marker), and CD29 (leukocyte marker) antigens and natural killer cell activity in 9 DR2 positive narcoleptics and HLA matched normal controls.71 In another study, non-specific CSF markers of immune mediated inflammation were examined in 15 narcoleptic patients.80 Two patients showed CSF oligoclonal bands, and one had an increased IgG index. These findings were within the range for other neurological, non-inflammatory diseases however.
Strohmaier et alattempted to detect disease-associated genetic variations or exogenously induced alterations in DR2 haplotypes of narcolepsy patients by studying primed lymphocytes generated in nine families against DR2 haplotypes of narcolepsy patients and healthy family members.81 24 primed lymphocyte reagents were obtained and restimulated by cells of unrelated patients with narcolepsy and DR2/Dw2-positive healthy individuals. The restimulation rates triggered by cells of patients or healthy controls never differed significantly. Therefore, the researchers did not observe cellular reaction patterns that would discriminate narcolepsy-associated haplotypes from those of healthy DR2/Dw2-positive controls.
Humoral immunity: non-hypocretin candidate antigens
Multiple autoimmune markers are sometimes detected in patients with autoimmune diseases and this subject has been reviewed in relation to narcolepsy by Black.82 Investigators have searched for evidence of serum autoantibodies to many antigens in patients with narcolepsy. Early work focused upon evidence of an infectious etiology for narcolepsy, presumably mediated through molecular mimicry in which bacterial or viral proteins lead to generation of autoantibodies cross reacting with an unspecified brain protein, causing narcolepsy. In one study elevated streptolysin titers were found in almost half of narcoleptic patients, and elevated DNase B titers in a quarter. 83 However, Monplaisir et al found titers higher than the normal population, but not increased compared to patients with EDS without cataplexy. 84 Rubin et al studied a large number of autoimmune markers, including rheumatoid factor, Sjogren's syndrome B antigen, Smith antigen, antinuclear antibodies, anti-DNA and anti-histone antibodies, but found no differences between patients and controls. 85 Furthermore, there was no reactivity against primate brainstem or rat whole brain.
Black et al analyzed serum of 43 patients with narcolepsy for neuron-specific and non-neuron-specific antibodies 86. The neural antigens tested were N-type and P/Q-type voltage-gated calcium channels, nicotinic acetylcholine receptors containing α3 subunit, Purkinje cell-type 1 (anti-Yo), anti-neuronal nuclear-type 1 (anti-Hu), anti-neuronal nuclear-type 2 (anti Ri), glutamic acid decarboxylase 65, and amphiphysin. Non-neuron-specific antigens tested were striated muscle, smooth muscle, thyroid microsomal (peroxidase) and thyroglobulin. Thirty percent of the patients had 1 or more antibodies present but no single antibody predominated. The frequency of positive studies was highter than would be predicted in performing 12 tests on each patient.
Guillain-Barré syndrome 87 and anti-Ma mediated limbic encephalitis are 2 known autoimmune disorders that may result in undetectable CSF hypocretin levels, and in the latter disorder even secondary narcolepsy with cataplexy.88 Overeem et al therefore screened a cohort of hypocretin deficient sporadic narcolepsy patients for antibodies known to be involved in these 2 disorders. Low titers of ganglioside-specific IgM antibodies were found in about 15% of patients, a similar percentage to that reported in healthy controls. 89 Anti-Ma antibodies were not detected. 37
Humoral immunity: the hypocretin system as a target
Recent studies directly focussed on (parts of) the hypocretin system as possible autoantibody targets. Taheri et al used 1D non-gradient gel western blots to screen serum immunoreactivity of 40 DQB1*0602 positive hypocretin-deficient patients and 120 healthy controls to proteins from the lateral hypothalamus of dogs, rats, and mice. 90 For 10 patients and 10 controls IgG was isolated by protein G method and tested on western blots. Intrathecal production of antibodies was tested using CSF from 20 HLA positive hypocretin-deficient patients and 10 controls. No specific or shared binding to any protein from the lateral hypothalamic blots of any animal species was noted with any of these tests.
Black et al also screened for circulating IgG antibodies to human preprohypocretin, hypocretin 1 and 2, and N-terminal leader and the C-terminal tail peptides of preprohypocretin using various techniques including ELISA and immunoprecipitation assays, and western blots.91,92 The results did not support the hypothesis that narcolepsy is due to autoantibodies directed against preprohypocretin or its peptide cleavage products. Comparable results were obtained by Tanaka et al who developed a sensitive radioligand assay to screen for antibodies against hypocretin-1 and -2, and the two known hypocretin receptors.93 However, in a screen using rat as well as baboon hypothalamic protein extract in an ELISA, significant differences were detected in CSF from narcolepsy versus control subjects.35,94 Overeem et al used yet another screening technique: immunohistochemistry with serum and CSF on human lateral hypothalamic slices.95 Autoantibodies against lateral hypothalamic neurons were found in 2 patients, but in 2 control subjects as well (Figure 4). In summary, several extensive studies have not provided evidence for a humoral immune response against the hypocretin system in narcolepsy, although reactivity to an unknown antigen in hypothalamic extracts may be present.
Figure 4.
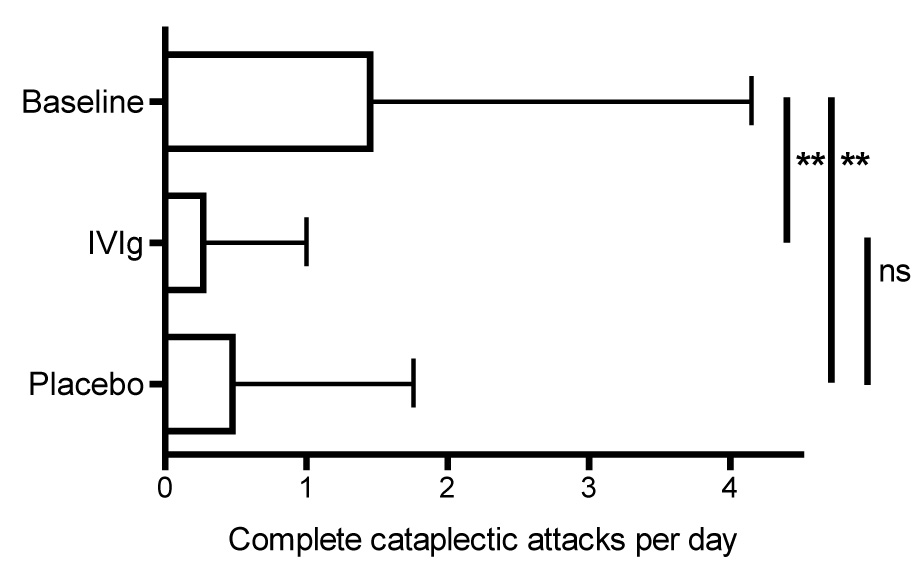
Results of a n=1 double-blind placebo-controlled study of IVIG in a patient with severe treatment refractory narcolepsy with cataplexy. Shown are the number of complete cataplexy attack per day during the baseline period, IVIG and placebo treatment. Error bars represent standard deviations. **: P < 0.001
Smith et al used a entirely different approach and passively transferred immunoglobulins from patients with narcolepsy with mice. 96 The animals were then sacrificed and their bladder tested for contractile responses to the muscarinic agonist carbachol. In the mice treated with patient IgG, this response was significantly increased. This effect was indirect, as direct application of purified IgG on the bladder preparation did not yield the same results. Furthermore, the authors reported behavioral changes in the mice treated with patient immunoglobulins. 96 The mechanism through which a putative antibody affecting periferal cholinergic transmission can lead to narcolepsy is unclear however. Moreover, extensive attempts to replicate the findings using different mice strains and longer-term administration are negative have been negative so far.97
IMMUNE-MODULATING THERAPY
The autoimmune hypothesis with a gradual destruction of hypocretin neurons (Figure 2) provide a rationale for the initiation of immune-modulating therapies near disease onset: preventing the progression of cell-loss may induce a long-lasting improvement. In 2003, prednisone was tried in Stanford in an eight year-old boy within two months after the onset of EDS. 98 He was HLA DQB1*0602 positive, had typical findings on the MSLT and an undetectable level of hypocretin in the CSF, but no cataplexy. He was treated with 1 mg/kg prednisone per day for 3 weeks. However, he did not improve and 1 year later cataplectic attacks started to occur. After this negative study, no other attempts to use corticosteroids have been published. There is one case report in which plasmapheresis was used, yielding only a very short lasting effect. 99 However, the patient had atypical narcolepsy, displaying an acute onset with very frequent attacks of muscle weakness after an upper respiratory infection.
The first case report on the use of intravenous immunoglobulings (IVIG) near disease onset was published shortly after the negative result with prednisone. 100 It concerned a 10 year-old French boy with an abrupt onset of EDS and the appearance of cataplexy two months later. He was DQB1*0602 positive and hypocretin-1 deficient. Treatment with IVIG was initiated (1 gram/kg per day for 2 days) within 5 months after disease onset. Subsequently, prednisolone was started for 3 weeks (1.3 mg/kg/day for 3 weeks). Clinical improvement started 3 days after therapy initiation of the therapy, and was dramatic for both EDS and cataplexy. At time of publication, the effects had lasted for more than 5 months. Repeated examination of the CSF, 3 weeks after the start of therapy, still showed an undetectable level of hypocretin. The only other published study, including a report on follow up 2 years later, is from Dauvilliers et al.101,102 They treated four patients, two of them within 4 months of disease onset, one after 8 months, and one after 9 years when a presumed auto-immune polyarthritis manifested. All patients suffered from EDS and cataplexy, were HLA DQ B1*0602 positive and had undetectable levels of hypocretin-1 in the CSF before and after treatment with IVIG (1 mg/kg per day for two days, repeated three times at 4-week intervals). Improvement of cataplexy was the most impressive result, although it did not completely disappear. EDS improved subjectively in two patients and objectively (MWT) in only the patient with the longest disease duration.
The question remains whether the available reports prove that IVIG indeed is an effective treatment or may at least prevent progression of the disease, in particular when applied shortly after disease onset. Unfortunately, there are several reasons for serious doubts. For one, the natural history of narcolepsy is not well known, and it is still very unusual to see and treat patients within months of symptom onset. It may be that cataplexy in particular is more severe around disease onset. More importantly, in none of the cases patients and physicians were blinded, and placebo was not used. We recently performed a n=1 double-blind placebo-controlled study 103 in a severely affected patient, 5 years after disease onset 104 She was almost unresponding on any accepted treatment for EDS as well as cataplexy. However, during the study, in which she received double-blind IVIG or placebo in a randomized order, she had a very dramatic response on intravenous placebo treatment in particular on cataplexy (Figure 4). This result shows the absolute need for a (multi-center) placebo-controlled study on the efficacy of IVIG before any conclusion can be drawn.
FUTURE PERSPECTIVES
Although no direct evidence has been provided after many years of research, the autoimmune theory remains a very attractive hypothesis for the pathophysiology of narcolepsy. The selective loss of hypocretin neurons, the complex genetic susceptibility with a strong HLA association, all are in favor for an autoimmune mechanism, but the search for proof so far has been ‘frustrating and mostly fruitless’.60 However -if present- it is most likely that the process targeting the hypocretin cells in the lateral hypothalamus is very focal and transient, making it very hard to detect. It will be of paramount importance to study patients just after disease onset, and applying the established antibody screening techniques on serum and CSF in these subjects. Up till now, no studies have looking at T-cell mediated responses direct towards the hypocetin system. Such studies are very much in need now. At the same time, extended studies focussing on association with non-HLA genes may provide further directions. Regardless of the ultimate cause, additional post-mortem studies will be crucial to definitively establish the presence of a neurodegenerative process affecting the hypocretin cells. There are many reasons to initate a world-wide network to collect and preserve brain material from deceased narcoleptics, again paying a lot of attention towards subjects with a short disease duration. At the same time, we must stay open towards other pathophysiological mechanisms that could underly the occurrence of narcolepsy, such as an infectious or toxic response to the hypocretin system. 60 Finally, whether or not it will provide pathophysiological clues, the clinical effect of immune-modulating therapy, at this stage IVIG in particular, should be determined in a properly designed controlled trial.
Figure 3.
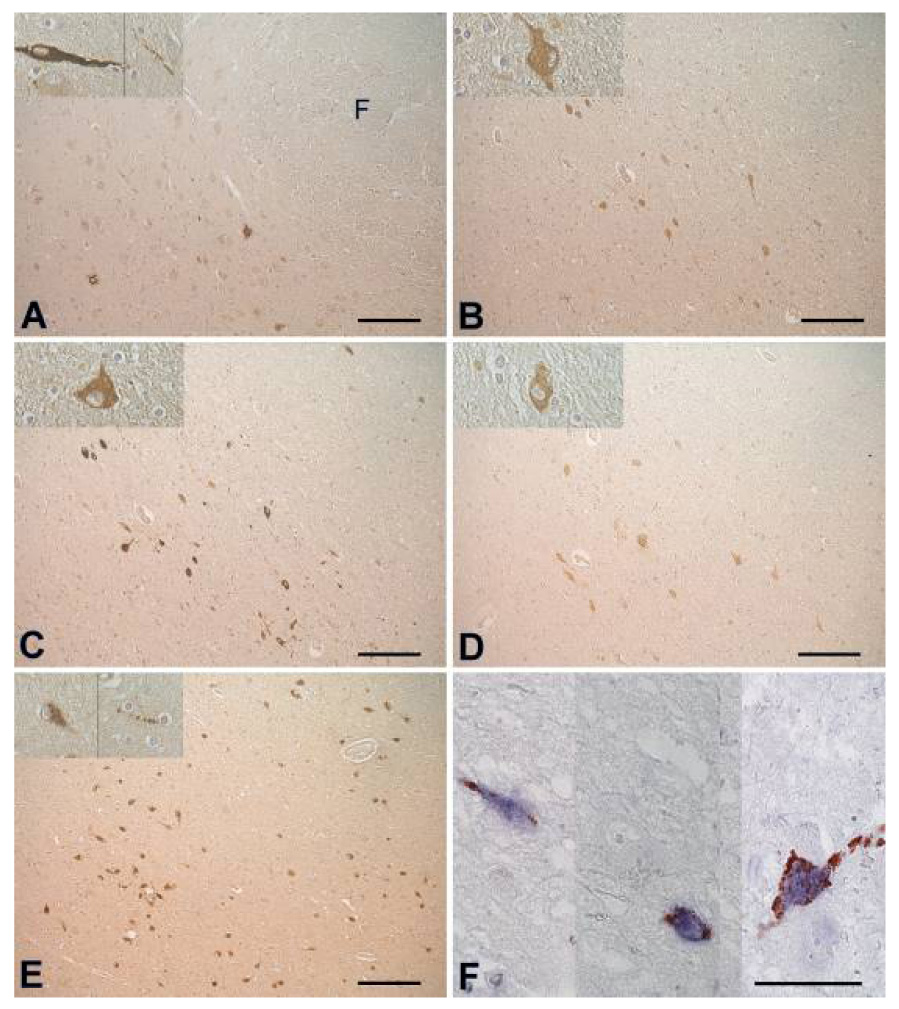
Adjacent sections from the lateral hypothalamus, stained with positive sera from 2 patients (A, B), 2 controls (C, D) and anti-hypocretin-1 (E). (F) 3 parts from sections double stained with anti-hypocretin-1 (blue) and serum of patient 1 (red). Note that no cell bodies are double stained. There are multiple bouton-like structures staining red, in close proximity of hypocretin cell bodies, suggesting nerve endings . Scale bars: (A–E): 200 µm, (F): 50 µm, (G): 1000 µm. Abbreviations: F = fornix, 3V = third ventricle. Figure reprinted with permission from 95
ACKNOWLEDGMENTS
S. Overeem is supported by a VENI grant from the Netherlands Organisation for Scientific Research (grant no. 916.56.103). The work by J. L. Black III was supported by NIMH R01 MH62599 "Autoimmune Diathesis in Human Narcolepsy" and the Piscopo Foundation.
ABBREVIATIONS
- CSF
cerebrospinal fluid
- EDS
excessive daytime sleepiness
- ELISA
enzyme linked immunosorbent assay
- GFAP
glial fibrillary acid protein
- HLA
human leucocyte antigen
- Hcrtr2
hypocretin receptor -2
- ICSD
international classification of sleep disorders
- IgG
immunoglobulin-G
- IVIG
intravenous immunoglobulins
- MSLT
multiple sleep latency test
- MWT
maintenance of wakefulness test
- NARP
neuronal activity-regulated pentraxin
- TNF
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
- In most cases, narcolepsy is a sporadic disorder, althoug first degree relatives have an increased risk.
- The association with HLA-DQB1*0602 is the strongest of any disorder, but up to 35% of the normal population is positive for this allele.
- Narcolepsy is caused by a selective loss of hypocretin (orexin) producing neurons in the perifornical hypothalamus. This is reflected in undetectable hypocretin-1 levels in the CSF.
- An autoimmune mediated destruction of hypocretin neurons remains the most attractive mechanism, but no direct evidence for this theory is available as of yet.
- Autoantibody screenings in serum and CSF of narcoleptic patients, acquired around disease onset.
- Studies on T-cell mediated (auto)immunity in relation to the hypocretin system
- Extend the studies looking for non-HLA factors influencing susceptibility to narcolepsy
- Additional (post-mortem) histopathological studies in narcolepsy, again particularly around disease onset
- To search for alternative mechanisms that could result in a hypocretin deficiency.
REFERENCES
- 1.Overeem S, Mignot E, van Dijk JG, Lammers GJ. Narcolepsy: clinical features, new pathophysiologic insights, and future perspectives. J Clin Neurophysiol. 2001;18:78–105. doi: 10.1097/00004691-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Douglas NJ. The psychosocial aspects of narcolepsy. Neurology. 1998;50:S27–S30. doi: 10.1212/wnl.50.2_suppl_1.s27. [DOI] [PubMed] [Google Scholar]
- 3.Broughton WA, Broughton RJ. Psychosocial impact of narcolepsy. Sleep. 1994;17:S45–S49. doi: 10.1093/sleep/17.suppl_8.s45. [DOI] [PubMed] [Google Scholar]
- 4.ICSD. International Classification of Sleep Disorders. Second Edition. Westchester, Il: American Academy of Sleep Medicin; 2005. [Google Scholar]
- 5.Broughton R, Krupa S, Boucher B, et al. Impaired circadian waking arousal in narcolepsy-cataplexy. Sleep Res Online. 1998;1:159–165. [PubMed] [Google Scholar]
- 6.Broughton R, Dunham W, Newman J, et al. Ambulatory 24 hour sleep-wake monitoring in narcolepsy-cataplexy compared to matched controls. Electroencephalogr Clin Neurophysiol. 1988;70:473–481. doi: 10.1016/0013-4694(88)90145-9. [DOI] [PubMed] [Google Scholar]
- 7.Anic-Labat S, Guilleminault C, Kraemer HC, et al. Validation of a cataplexy questionnaire in 983 sleep-disorders patients. Sleep. 1999;22:77–87. [PubMed] [Google Scholar]
- 8.Kok SW, Overeem S, Visscher TL, et al. Hypocretin deficiency in narcoleptic humans is associated with abdominal obesity. Obes Res. 2003;11:1147–1154. doi: 10.1038/oby.2003.156. [DOI] [PubMed] [Google Scholar]
- 9.Hublin C, Partinen M, Kaprio J, et al. Epidemiology of narcolepsy. Sleep. 1994;17:S7–S12. doi: 10.1093/sleep/17.suppl_8.s7. [DOI] [PubMed] [Google Scholar]
- 10.Longstreth WT, Jr, Koepsell TD, Ton TG, et al. The epidemiology of narcolepsy. Sleep. 2007;30:13–26. doi: 10.1093/sleep/30.1.13. [DOI] [PubMed] [Google Scholar]
- 11.Ohayon MM, Priest RG, Zulley J, et al. Prevalence of narcolepsy symptomatology and diagnosis in the European general population. Neurology. 2002;58:1826–1833. doi: 10.1212/wnl.58.12.1826. [DOI] [PubMed] [Google Scholar]
- 12.Dauvilliers Y, Montplaisir J, Molinari N, et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57:2029–2033. doi: 10.1212/wnl.57.11.2029. [DOI] [PubMed] [Google Scholar]
- 13.Okun ML, Lin L, Pelin Z, et al. Clinical aspects of narcolepsy-cataplexy across ethnic groups. Sleep. 2002;25:27–35. doi: 10.1093/sleep/25.1.27. [DOI] [PubMed] [Google Scholar]
- 14.Dauvilliers Y, Carlander B, Molinari N, et al. Month of birth as a risk factor for narcolepsy. Sleep. 2003;26:663–665. doi: 10.1093/sleep/26.6.663. [DOI] [PubMed] [Google Scholar]
- 15.Dahmen N, Tonn P. Season of birth effect in narcolepsy. Neurology. 2003;61:1016–1017. doi: 10.1212/01.wnl.0000080371.15688.30. [DOI] [PubMed] [Google Scholar]
- 16.Orellana C, Villemin E, Tafti M, et al. Life events in the year preceding the onset of narcolepsy. Sleep. 1994;17:S50–S53. doi: 10.1093/sleep/17.suppl_8.s50. [DOI] [PubMed] [Google Scholar]
- 17.Mayer G, Kesper K, Peter H, et al. Comorbidity in narcoleptic patients. Dtsch Med Wochenschr. 2002;127:1942–1946. doi: 10.1055/s-2002-34207. [DOI] [PubMed] [Google Scholar]
- 18.Kanbayashi T, Goto A, Hishikawa Y, et al. Hypersomnia due to acute disseminated encephalomyelitis in a 5-year-old girl. Sleep Med. 2001;2:347–350. doi: 10.1016/s1389-9457(00)00082-4. [DOI] [PubMed] [Google Scholar]
- 19.Aldrich MS, Naylor MW. Narcolepsy associated with lesions of the diencephalon. Neurology. 1989;39:1505–1508. doi: 10.1212/wnl.39.11.1505. [DOI] [PubMed] [Google Scholar]
- 20.Gledhill RF, Bartel PR, Yoshida Y, et al. Narcolepsy caused by acute disseminated encephalomyelitis. Arch Neurol. 2004;61:758–760. doi: 10.1001/archneur.61.5.758. [DOI] [PubMed] [Google Scholar]
- 21.Schrader H, Gotlibsen OB, Skomedal GN. Multiple sclerosis and narcolepsy/cataplexy in a monozygotic twin. Neurology. 1980;30:105–108. doi: 10.1212/wnl.30.1.105. [DOI] [PubMed] [Google Scholar]
- 22.Guilleminault C, Mignot E, Grumet FC. Familial patterns of narcolepsy. Lancet. 1989;2:1376–1379. doi: 10.1016/s0140-6736(89)91977-6. [DOI] [PubMed] [Google Scholar]
- 23.Billiard M, Pasquie-Magnetto V, Heckman M, et al. Family studies in narcolepsy. Sleep. 1994;17:S54–S59. doi: 10.1093/sleep/17.suppl_8.s54. [DOI] [PubMed] [Google Scholar]
- 24.Nevsimalova S, Mignot E, Sonka K, Arrigoni JL. Familial aspects of narcolepsy-cataplexy in the Czech Republic. Sleep. 1997;20:1021–1026. doi: 10.1093/sleep/20.11.1021. [DOI] [PubMed] [Google Scholar]
- 25.Kadotani H, Faraco J, Mignot E. Genetic studies in the sleep disorder narcolepsy. Genome Res. 1998;8:427–434. doi: 10.1101/gr.8.5.427. [DOI] [PubMed] [Google Scholar]
- 26.Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50:S16–S22. doi: 10.1212/wnl.50.2_suppl_1.s16. [DOI] [PubMed] [Google Scholar]
- 27.Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 28.Hungs M, Fan J, Lin L, et al. Identification and functional analysis of mutations in the hypocretin (orexin) genes of narcoleptic canines. Genome Res. 2001;11:531–539. doi: 10.1101/gr.gr-1610r. [DOI] [PubMed] [Google Scholar]
- 29.Ripley B, Fujiki N, Okura M, et al. Hypocretin levels in sporadic and familial cases of canine narcolepsy. Neurobiol Dis. 2001;8:525–534. doi: 10.1006/nbdi.2001.0389. [DOI] [PubMed] [Google Scholar]
- 30.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 31.Hara J, Beuckmann CT, Nambu T, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 32.Nishino S, Ripley B, Overeem S, et al. Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381–388. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- *33.Mignot E, Lammers GJ, Ripley B, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–1562. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- 34.Dauvilliers Y, Baumann CR, Carlander B, et al. CSF hypocretin-1 levels in narcolepsy, Kleine-Levin syndrome, and other hypersomnias and neurological conditions. J Neurol Neurosurg Psychiatry. 2003;74:1667–1673. doi: 10.1136/jnnp.74.12.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silber M, Black JL, Krahn LE, Fredrickson PA. Narcolepsy and Hypersomnia, Anonymouspp. Informa Healthcare; 2006. Autoimmune studies in narcolepsy; pp. 451–457. [Google Scholar]
- 36.Nishino S, Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9:269–310. doi: 10.1016/j.smrv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Overeem S, Dalmau J, Bataller L, et al. Hypocretin-1 CSF levels in anti-Ma2 associated encephalitis. Neurology. 2004;62:138–140. doi: 10.1212/01.wnl.0000101718.92619.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanbayashi T, Yano T, Ishiguro H, et al. Hypocretin-1 (orexin-A) levels in human lumbar CSF in different age groups: infants to elderly persons. Sleep. 2002;25:337–339. doi: 10.1093/sleep/25.3.337. [DOI] [PubMed] [Google Scholar]
- 39.Kubota H, Kanbayashi T, Tanabe Y, et al. Decreased cerebrospinal fluid hypocretin-1 levels near the onset of narcolepsy in 2 prepubertal children. Sleep. 2003;26:555–557. doi: 10.1093/sleep/26.5.555. [DOI] [PubMed] [Google Scholar]
- *40.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- *41.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thannickal TC, Siegel JM, Nienhuis R, Moore RY. Pattern of hypocretin (orexin) soma and axon loss, and gliosis, in human narcolepsy. Brain Pathol. 2003;13:340–351. doi: 10.1111/j.1750-3639.2003.tb00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *43.Blouin AM, Thannickal TC, Worley PF, et al. Narp immunostaining of human hypocretin (orexin) neurons: loss in narcolepsy. Neurology. 2005;65:1189–1192. doi: 10.1212/01.wnl.0000175219.01544.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *44.Crocker A, Espana RA, Papadopoulou M, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–1188. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenneis C, Brandauer E, Frauscher B, et al. Voxel-based morphometry in narcolepsy. Sleep Med. 2005;6:531–536. doi: 10.1016/j.sleep.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Kaufmann C, Schuld A, Pollmacher T, Auer DP. Reduced cortical gray matter in narcolepsy: preliminary findings with voxel-based morphometry. Neurology. 2002;58:1852–1855. doi: 10.1212/wnl.58.12.1852. [DOI] [PubMed] [Google Scholar]
- 47.Overeem S, Steens SC, Good CD, et al. Voxel-based morphometry in hypocretin-deficient narcolepsy. Sleep. 2003;26:44–46. [PubMed] [Google Scholar]
- 48.Draganski B, Geisler P, Hajak G, et al. Hypothalamic gray matter changes in narcoleptic patients. Nat Med. 2002;8:1186–1188. doi: 10.1038/nm1102-1186. [DOI] [PubMed] [Google Scholar]
- 49.Lodi R, Tonon C, Vignatelli L, et al. In vivo evidence of neuronal loss in the hypothalamus of narcoleptic patients. Neurology. 2004;63:1513–1515. doi: 10.1212/01.wnl.0000142259.94107.4c. [DOI] [PubMed] [Google Scholar]
- 50.Juji T, Satake M, Honda Y, Doi Y. HLA Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens. 1984;24:316–319. doi: 10.1111/j.1399-0039.1984.tb02144.x. [DOI] [PubMed] [Google Scholar]
- 51.Mignot E, Lin X, Arrigoni J, et al. DQB1*0602 and DQA1*0102 (DQ1) are better markers than DR2 for narcolepsy in Caucasian and black Americans. Sleep. 1994;17:S60–S67. doi: 10.1093/sleep/17.suppl_8.s60. [DOI] [PubMed] [Google Scholar]
- 52.Lin L, Hungs M, Mignot E. Narcolepsy and the HLA region. J Neuroimmunol. 2001;117:9–20. doi: 10.1016/s0165-5728(01)00333-2. [DOI] [PubMed] [Google Scholar]
- *53.Pelin Z, Guilleminault C, Risch N, et al. HLA-DQB1*0602 homozygosity increases relative risk for narcolepsy but not disease severity in two ethnic groups. US Modafinil in Narcolepsy Multicenter Study Group. Tissue Antigens. 1998;51:96–100. doi: 10.1111/j.1399-0039.1998.tb02952.x. [DOI] [PubMed] [Google Scholar]
- *54.Mignot E, Lin L, Rogers W, et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68:686–699. doi: 10.1086/318799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *55.Ellis MC, Hetisimer AH, Ruddy DA, et al. HLA class II haplotype and sequence analysis support a role for DQ in narcolepsy. Immunogenetics. 1997;46:410–417. doi: 10.1007/s002510050295. [DOI] [PubMed] [Google Scholar]
- 56.Siebold C, Hansen BE, Wyer JR, et al. Crystal structure of HLA-DQ0602 that protects against type 1 diabetes and confers strong susceptibility to narcolepsy. Proc Natl Acad Sci U S A. 2004;101:1999–2004. doi: 10.1073/pnas.0308458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6:271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 58.Mignot E, Young T, Lin L, Finn L. Nocturnal sleep and daytime sleepiness in normal subjects with HLA-DQB1*0602. Sleep. 1999;22:347–352. [PubMed] [Google Scholar]
- 59.Mignot E, Young T, Lin L, et al. Reduction of REM sleep latency associated with HLA-DQB1* 0602 in normal adults. Lancet. 1998;351:727. doi: 10.1016/S0140-6736(05)78496-8. [DOI] [PubMed] [Google Scholar]
- 60.Scammell TE. The frustrating and mostly fruitless search for an autoimmune cause of narcolepsy. Sleep. 2006;29:601–602. doi: 10.1093/sleep/29.5.601. [DOI] [PubMed] [Google Scholar]
- 61.Tafti M, Maret S, Dauvilliers Y. Genes for normal sleep and sleep disorders. Ann Med. 2005;37:580–589. doi: 10.1080/07853890500372047. [DOI] [PubMed] [Google Scholar]
- 62.Dauvilliers Y, Maret S, Tafti M. Genetics of normal and pathological sleep in humans. Sleep Med Rev. 2005;9:91–100. doi: 10.1016/j.smrv.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 63.Singh SM, George CF, Kryger MH, Jung JH. Genetic heterogeneity in narcolepsy. Lancet. 1990;335:726–727. doi: 10.1016/0140-6736(90)90842-s. [DOI] [PubMed] [Google Scholar]
- 64.Hungs M, Lin L, Okun M, Mignot E. Polymorphisms in the vicinity of the hypocretin/orexin are not associated with human narcolepsy. Neurology. 2001;57:1893–1895. doi: 10.1212/wnl.57.10.1893. [DOI] [PubMed] [Google Scholar]
- 65.Olafsdottir BR, Rye DB, Scammell TE, et al. Polymorphisms in hypocretin/orexin pathway genes and narcolepsy. Neurology. 2001;57:1896–1899. doi: 10.1212/wnl.57.10.1896. [DOI] [PubMed] [Google Scholar]
- 66.Wieczorek S, Jagiello P, Arning L, et al. Screening for candidate gene regions in narcolepsy using a microsatellite based approach and pooled DNA. J Mol Med. 2004;82:696–705. doi: 10.1007/s00109-004-0569-5. [DOI] [PubMed] [Google Scholar]
- 67.Dauvilliers Y, Neidhart E, Lecendreux M, et al. MAO-A and COMT polymorphisms and gene effects in narcolepsy. Mol Psychiatry. 2001;6:367–372. doi: 10.1038/sj.mp.4000911. [DOI] [PubMed] [Google Scholar]
- 68.Serrano NC, Millan P, Paez MC. Non-HLA associations with autoimmune diseases. Autoimmun Rev. 2006;5:209–214. doi: 10.1016/j.autrev.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 69.Okun ML, Giese S, Lin L, et al. Exploring the cytokine and endocrine involvement in narcolepsy. Brain Behav Immun. 2004;18:326–332. doi: 10.1016/j.bbi.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Vgontzas AN, Papanicolaou DA, Bixler EO, et al. Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J Clin Endocrinol Metab. 1997;82:1313–1316. doi: 10.1210/jcem.82.5.3950. [DOI] [PubMed] [Google Scholar]
- 71.Hinze-Selch D, Wetter TC, Zhang Y, et al. In vivo and in vitro immune variables in patients with narcolepsy and HLA-DR2 matched controls. Neurology. 1998;50:1149–1152. doi: 10.1212/wnl.50.4.1149. [DOI] [PubMed] [Google Scholar]
- 72.Hohjoh H, Nakayama T, Ohashi J, et al. Significant association of a single nucleotide polymorphism in the tumor necrosis factor-alpha (TNF-alpha) gene promoter with human narcolepsy. Tissue Antigens. 1999;54:138–145. doi: 10.1034/j.1399-0039.1999.540204.x. [DOI] [PubMed] [Google Scholar]
- 73.Kato T, Honda M, Kuwata S, et al. Novel polymorphism in the promoter region of the tumor necrosis factor alpha gene: No association with narcolepsy. Am J Med Genet. 1999;88:301–304. doi: 10.1002/(sici)1096-8628(19990820)88:4<301::aid-ajmg4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 74.Wieczorek S, Gencik M, Rujescu D, et al. TNFA promoter polymorphisms and narcolepsy. Tissue Antigens. 2003;61:437–442. doi: 10.1034/j.1399-0039.2003.00068.x. [DOI] [PubMed] [Google Scholar]
- 75.Rubio JP, Bahlo M, Stankovich J, et al. Analysis of extended HLA haplotypes in multiple sclerosis and narcolepsy families confirms a predisposing effect for the class I region in Tasmanian MS patients. Immunogenetics. 2007;59:177–186. doi: 10.1007/s00251-006-0183-5. [DOI] [PubMed] [Google Scholar]
- 76.Nakayama J, Miura M, Honda M, et al. Linkage of human narcolepsy with HLA association to chromosome 4p13-q21. Genomics. 2000;65:84–86. doi: 10.1006/geno.2000.6143. [DOI] [PubMed] [Google Scholar]
- 77.Dauvilliers Y, Blouin JL, Neidhart E, et al. A narcolepsy susceptibility locus maps to a 5 Mb region of chromosome 21q. Ann Neurol. 2004;56:382–388. doi: 10.1002/ana.20208. [DOI] [PubMed] [Google Scholar]
- 78.Kawashima M, Tamiya G, Oka A, et al. Genomewide association analysis of human narcolepsy and a new resistance gene. Am J Hum Genet. 2006;79:252–263. doi: 10.1086/505539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsuki K, Honda Y, Naohara T, et al. Lymphocyte subsets in HLA-DR2-positive narcoleptic patients. Folia Psychiatr Neurol Jpn. 1985;39:499–505. doi: 10.1111/j.1440-1819.1985.tb00803.x. [DOI] [PubMed] [Google Scholar]
- 80.Fredrikson S, Carlander B, Billiard M, Link H. CSF immune variables in patients with narcolepsy. Acta Neurol Scand. 1990;81:253–254. doi: 10.1111/j.1600-0404.1990.tb00978.x. [DOI] [PubMed] [Google Scholar]
- 81.Strohmaier P, Mueller-Eckhardt G, Meier-Ewert K. Cellular approach for detecting narcolepsy-specific alterations in DR2 haplotypes. Hum Immunol. 1988;22:221–225. doi: 10.1016/0198-8859(88)90001-8. [DOI] [PubMed] [Google Scholar]
- 82.Black JL., III Narcolepsy: a review of evidence for autoimmune diathesis. Int Rev Psychiatry. 2005;17:461–469. doi: 10.1080/02646830500381492. [DOI] [PubMed] [Google Scholar]
- 83.Billiard M, Laaberki MF, Reygrobellet C, et al. Elevated antibodies to streptococcal antigens in narcoleptic subjects. Sleep Res. 1989;18:201. [Google Scholar]
- 84.Montplaisir J, Poirier G, Lapierre O, Montplaisir S. Streptococcal antibodies in narcolepsy and idiopathic hypersomnia. Sleep Res. 1989;18:271. [Google Scholar]
- 85.Rubin RL, Hajdukovich RM, Mitler MM. HLA-DR2 association with excessive somnolence in narcolepsy does not generalize to sleep apnea and is not accompanied by systemic autoimmune abnormalities. Clin Immunol Immunopathol. 1988;49:149–158. doi: 10.1016/0090-1229(88)90104-3. [DOI] [PubMed] [Google Scholar]
- 86.Black JL, III, Krahn LE, Pankratz VS, Silber M. Search for neuron-specific and nonneuron-specific antibodies in narcoleptic patients with and without HLA DQB1*0602. Sleep. 2002;25:719–723. doi: 10.1093/sleep/25.7.719. [DOI] [PubMed] [Google Scholar]
- 87.Nishino S, Kanbayashi T, Fujiki N, et al. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology. 2003;61:823–825. doi: 10.1212/01.wnl.0000081049.14098.50. [DOI] [PubMed] [Google Scholar]
- 88.Rosenfeld MR, Eichen JG, Wade DF, et al. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001;50:339–348. [PubMed] [Google Scholar]
- 89.Overeem S, Geleijns K, Garssen MP, et al. Screening for anti-ganglioside antibodies in hypocretin-deficient human narcolepsy. Neurosci Lett. 2003;341:13–16. doi: 10.1016/s0304-3940(03)00085-5. [DOI] [PubMed] [Google Scholar]
- 90.Taheri S, Krempetz M, Jackson M, et al. Investigation of the autoimmune basis of narcolepsy using western blot analysis of lateral hypothalamus protein extract with serum and cerebrospinal fluid (abstract) Sleep. 2003;26:A285. [Google Scholar]
- 91.Black JL, III, Silber MH, Krahn LE, et al. Studies of humoral immunity to preprohypocretin in human leukocyte antigen DQB1*0602-positive narcoleptic subjects with cataplexy. Biol Psychiatry. 2005;58:504–509. doi: 10.1016/j.biopsych.2005.04.026. [DOI] [PubMed] [Google Scholar]
- *92.Black JL, III, Silber MH, Krahn LE, et al. Analysis of hypocretin (orexin) antibodies in patients with narcolepsy. Sleep. 2005;28:427–431. doi: 10.1093/sleep/28.4.427. [DOI] [PubMed] [Google Scholar]
- 93.Tanaka S, Honda Y, Inoue Y, Honda M. Detection of autoantibodies against hypocretin, hcrtrl, and hcrtr2 in narcolepsy: anti-Hcrt system antibody in narcolepsy. Sleep. 2006;29:633–638. doi: 10.1093/sleep/29.5.633. [DOI] [PubMed] [Google Scholar]
- 94.Black JL, III, Avula RK, Walker DL, et al. HLA DQB1*0602 positive narcoleptic subjects with cataplexy have CSF lgG reactive to rat hypothalamic protein extract. Sleep. 2005;28:1191–1192. [PubMed] [Google Scholar]
- 95.Overeem S, Verschuuren JJ, Fronczek R, et al. Immunohistochemical screening for autoantibodies against lateral hypothalamic neurons in human narcolepsy. J Neuroimmunol. 2006;174:187–191. doi: 10.1016/j.jneuroim.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 96.Smith AJ, Jackson MW, Neufing P, et al. A functional autoantibody in narcolepsy. Lancet. 2004;364:2122–2124. doi: 10.1016/S0140-6736(04)17553-3. [DOI] [PubMed] [Google Scholar]
- 97.Lin L, Mignot E. Narcolepsy and Hypersomnia. Informa Healthcare; 2006. Human Leukocyte Antigen and narcolepsy: present status and relationship with familial history and hypocretin deficiency. [Google Scholar]
- 98.Hecht M, Lin L, Kushida CA, et al. Report of a case of immunosuppression with prednisone in an 8-year-old boy with an acute onset of hypocretin-deficiency narcolepsy. Sleep. 2003;26:809–810. doi: 10.1093/sleep/26.7.809. [DOI] [PubMed] [Google Scholar]
- 99.Chen W, Black J, Call P, Mignot E. Late-onset narcolepsy presenting as rapidly progressing muscle weakness: response to plasmapheresis. Ann Neurol. 2005;58:489–490. doi: 10.1002/ana.20603. [DOI] [PubMed] [Google Scholar]
- 100.Lecendreux M, Maret S, Bassetti C, et al. Clinical efficacy of high-dose intravenous immunoglobulins near the onset of narcolepsy in a 10-year-old boy. J Sleep Res. 2003;12:347–348. doi: 10.1046/j.1365-2869.2003.00380.x. [DOI] [PubMed] [Google Scholar]
- 101.Dauvilliers Y, Carlander B, Rivier F, et al. Successful management of cataplexy with intravenous immunoglobulins at narcolepsy onset. Ann Neurol. 2004;56:905–908. doi: 10.1002/ana.20339. [DOI] [PubMed] [Google Scholar]
- *102.Dauvilliers Y. Follow-up of four narcolepsy patients treated with intravenous immunoglobulins. Ann Neurol. 2006;60:153. doi: 10.1002/ana.20892. [DOI] [PubMed] [Google Scholar]
- 103.Guyatt G, Sackett D, Taylor DW, et al. Determining optimal therapy--randomized trials in individual patients. N Engl J Med. 1986;314:889–892. doi: 10.1056/NEJM198604033141406. [DOI] [PubMed] [Google Scholar]
- 104.Fronczek R, Verschuuren J, Lammers GJ. Response to intravenous immunoglobulins and placebo in a patient with narcolepsy with cataplexy. J Neurol. 2007 doi: 10.1007/s00415-007-0594-8. in press. [DOI] [PubMed] [Google Scholar]