

Abstract
Although studies have shown that 17β-estradiol (estradiol) normalized Kupffer cell function following trauma-hemorrhage, the mechanism by which E2 maintains immune function remains unclear. Activation of Toll-like receptor 4 (TLR4) initiates an inflammatory cascade, involving activation of p38 mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and nuclear factor-κB (NF-κB). This leads to the release of proinflammatory cytokines. Thus, we hypothesized that the salutary effects of estradiol on Kupffer cell function following trauma-hemorrhage are mediated via negative regulation of TLR4-dependent p38 MAPK and NF-κB. TLR4 mutant (C3H/HeJ) and wild type (C3H/HeOuJ) mice were subjected to trauma-hemorrhage (mean BP 35±5 mmHg ~90 min, then resuscitation) or sham operation. Administration of estradiol following trauma-hemorrhage in wild type mice decreased Kupffer cell TLR4 expression as well as prevented the phosphorylation of p38 MAPK and NF-κB. This was accompanied by normalization of Kupffer cell production capacities of IL-6, TNF-α, macrophage inflammatory protein (MIP)-1α, and MIP-2 and the decrease in plasma cytokine levels. In contrast, TLR4 mutant mice did not exhibit the increase in Kupffer cell p38 MAPK and NF-κB activation, cytokine production, or the increase in circulating cytokine levels following trauma-hemorrhage. No difference was observed in activation of PI3K among groups. These results suggest that the protective effect of estradiol on Kupffer cell function is mediated via downregulation of TLR4-dependent p38 MAPK and NF-κB signaling following trauma-hemorrhage, which prevents the systemic release of cytokines.
Keywords: Hemorrhagic shock, Estrogen, NF-κB, MIP-1α, MIP-2
INTRODUCTION
Despite numerous advances in intensive care medicine, ischemia-reperfusion, sepsis, and organ dysfunction leading to multiple organ failure remain the major causes of death in trauma patients, as well as in patients following major surgery (Angele et al., 2000; Ayala and Chaudry, 1996; Hsieh et al., 2004; Jarrar et al., 2000; Kher et al., 2005; Murphy et al., 2004; Nelson et al., 1999). A number of studies have shown that immune function is markedly depressed in males following trauma-hemorrhage and those changes persist for several days despite adequate fluid resuscitation (Xu et al., 1998; Zellweger et al., 1995). There is increasing evidence that hormonal mechanisms are involved in regulating the posttraumatic immune response. In this regard, studies have shown that depletion of 5α-dihrdrotestosterone by castration of male mice two weeks prior to trauma-hemorrhage prevented the depression of immune function (Wichmann et al., 1997). Moreover, females in the proestrus state have a maintained/enhanced immune function following trauma-hemorrhage (Chaudry et al., 2003). Additionally, administration of 17β-estradiol (estradiol) in males following trauma-hemorrhage normalized Kupffer cell, splenic, and peritoneal macrophage function compared to vehicle-treated males (Knoferl et al., 2000; Yokoyama et al., 2003). However, the mechanism by which estradiol mediates the salutary effects on immune function remains unknown.
Toll-like receptors (TLRs) are a group of pattern recognition receptors that recognize conserved molecular motifs found in a variety of organisms including bacteria, viruses and fungi (Bowie and Haga, 2005; Janeway and Medzhitov, 2002). TLR4 is the dominant mammalian receptor for the microbial product lipopolysaccharide (LPS) (Qureshi et al., 1999). TLR4-deficient mice were found to be less prone to hepatic ischemia/reperfusion injury (Tsung et al., 2005) and the expression of TLR4 mRNA and protein was increased in rat Kupffer cells following ischemia/reperfusion injury (Peng et al., 2004). Moreover, activation of TLR4 initiates an inflammatory cascade in macrophages, involving activation of p38 mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and activation of nuclear factor-κB (NF-κB) (Arndt et al., 2004; O'Neill, 2002; Olsson and Sundler, 2006). Such activation subsequently leads to the release of proinflammatory cytokines including TNF-α and IL-6 (Arndt et al., 2004; O'Neill, 2002). It has also been shown that Kupffer cells produce TNF-α and IL-6 under stress conditions which contribute to hepatocellular damage (Koo et al., 1999). Macrophage inflammatory protein (MIP)-1α and MIP-2 are members of a growing family of proinflammatory cytokines with chemotactic activity for inflammatory and immune effector cells. MIP-1α and MIP-2 were previously identified in the mouse and shown to stimulate inflammatory cell recruitment (Driscoll et al., 1993). It is well known that Kupffer cells, the largest pool of macrophages in the body, influence liver function following various adverse circulatory conditions (Carini and Albano, 2003). The aim of this study therefore was to determine the role of TLR4-dependent signaling in mediating the salutary effects of estradiol on Kupffer cell function and the systemic inflammatory responses following trauma-hemorrhage.
MATERIALS AND METHODS
Trauma-hemorrhagic shock model
Male TLR4 wild-type (C3H/HeOuJ) mice and TLR4 mutant (C3H/HeJ) mice (8-12 wk old) were purchased from the Jackson Laboratories (Bar Harbor, ME). Mice were fasted overnight but allowed water ad lib. They were anesthetized with isoflurane (Minrad, Bethlehem, PA) and restrained in a supine position. A midline laparotomy (2 cm) was performed, which was closed in two layers with sutures (Ethilon 6/0, Ethicon, Somerville, NJ). Both femoral arteries and the right femoral vein were cannulated with polyethylene tubing (Becton Dickinson, Sparks, MD). Blood pressure was measured via one of the arterial lines using a blood pressure analyzer (Micro-Med, Louisville, KY). Upon awakening, animals were bled rapidly through the other arterial catheter to a mean arterial blood pressure of 35±5 mmHg within 10 min, which was then maintained for 90 min. At the end of that interval, the animals were resuscitated via the venous line with four times the shed blood volume using Ringer's lactate. After ligating the vessels, catheters were removed; the incisions were flushed with lidocaine and closed with sutures. Sham-operated animals underwent the same surgical procedures, but were neither hemorrhaged nor resuscitated. The animals were sacrificed at 4 h after resuscitation or sham operation. In the treatment group, 17β-estradiol (50 μg/25 g) or vehicle (10% DMSO) (Sigma, St. Louis, MO) was administered subcutaneously at the onset of resuscitation. All the experiments were carried out in accordance to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Preparation of Kupffer Cells
Kupffer cells were isolated as previously described (Hildebrand et al., 2006; Rana et al., 1990). In brief, the portal vein was catheterized with a 27-gauge needle and the liver was perfused with 20 ml Hanks' balanced salt solution (HBSS, Gibco, Grand Island, NY) at 37° C, which was immediately followed by perfusion with 15 ml of 0.05% collagenase IV (Worthington, Lakewood, NJ) in HBSS with 0.05 mM CaCl2 (Sigma) at 37°C. The liver was then removed and transferred to a Petri dish containing the above mentioned collagenase IV solution. The liver was minced, incubated for 15 min at 37° C and passed through a sterile stainless steel mesh screen into a beaker containing 10 ml of cold HBSS with 10% FBS. The hepatocytes were removed by centrifugation at 50 × g for 3 min at 4° C. The residual cell suspension was washed twice by centrifugation at 800 × g for 10 min at 4° C in HBSS. The cells were resuspended in complete RPMI1640 medium containing 10% FBS and antibiotics (50 U/ml penicillin, 50 μg/ml streptomycin and 20 μg/ml gentamycin, all from Gibco) and layered over 16% Histodenz (Sigma) in RPMI1640 medium and centrifuged at 2300 × g for 45 min at 4° C. After removing the non-parenchymal cells from the interface, the cells were washed twice by centrifugation (800 × g, 10 min, 4° C) in complete RPMI1640 medium. The cells were then resuspended in complete RPMI1640 medium, and plated in a 6-well plate at a cell density of 1×106 cells/ml. After 2 h of incubation (37° C, 95% humidity and 5% CO2), non-adherent cells were removed by washing with RPMI1640 medium. We determined the number of adherent cells at the end of 2 h and found no significant difference in the number of adherent cells in sham and trauma-hemorrhage animals.
Quantitative Real-Time PCR
Total RNA was prepared by TRIREAGENT (Life Technologies, Grand Island, NY) and 0.2 μg of total RNA was then reversed to cDNA by MMLV (Moloney murine leukemia virus) reverse transcriptase (Promega, Madison, WI). Real-time PCR was performed using TaqMan method on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA). The reaction mixture for TaqMan assay contained 5 μl of 2 × TaqMan Universal PCR Master Mix (Applied Biosystems), 0.2 μM primers and 0.25 μM probes, and 10 ng cDNA. All samples were tested in triplicate, and average values were used for quantification. 18S rRNA was used as an endogenous control. At each cycle, accumulation of PCR products was detected by monitoring the increase in fluorescence of the reporter dye from the TaqMan probe. Analysis was performed using SDS v2.2 software (Applied Biosystems) according to the manufacturer's instructions. The comparative cycle threshold (CT) method (ΔΔCT) was used for quantification of gene expression.
Cytokine Analysis
The adherent cells were cultured to assess their cytokine production capacity. After 24 h in culture, cell-free supernatants were harvested. The concentrations of IL-6 and TNF-α in these supernatants were measured by commercially available cytometric bead array (CBA) Mouse Inflammation Kits (BD Pharmingen, San Diego, CA), according to the manufacturer's instructions. Briefly, 50 μl of mixed capture beads were incubated with 50 μl of supernatant and 50 μl PE detection reagent for 2 h at room temperature. The immunocomplexes were then washed and analyzed using the LSRII flow cytometer (BD Biosciences, Mountain View, CA). Data analysis was carried out using the accompanying FACSDiva and FCAP Array software (BD Biosciences, Mountain View, CA). The concentrations of MIP-1α and MIP-2 in the cell supernatants were measured by ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
P38 MAPK and PI3K Total Protein and Phosphorylation
The relative amounts of phospho-p38 MAPK and PI3K, and total p38 MAPK and PI3K proteins in Kupffer cells were determined using Cellular Activation of Signaling ELISA Kit (SuperArray Inc, Frederick, MD) using a 96-well plate according to the manufacturer's instructions. The relative cell number of each well was then determined by the same kit with cell staining solution at 595 nm. The relative amount of phosphorylated and total protein was presented after normalizing with the corresponding relative cell number.
DNA Binding Activity of NF-κB
DNA binding activity was evaluated using the Trans-AM ELISA method (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. To prepare nuclear extract, 5×106 cells were washed with ice-cold phosphate-buffered saline (PBS) and incubated on ice under constant shaking with 400 μl of cytosolic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5% NP-40, 1 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride). After 15 min, nuclei were separated by centrifugation at 5000 rpm for 10 min. The supernatant which contained the cytosolic proteins was then removed. The pellet, containing nuclei, was resuspended in 25 μl of a second lysis buffer provided by the TransAM ELISA kit. After 30 min incubation, nuclei were clarified by high-speed centrifugation. Nuclear extract (2 μg) was assayed for the DNA binding activity of NF-κB according to the protocol of the TRANS AM Kit. Briefly the DNA binding motif of NF-κB (5′-GGGACTTTCC-3′) is coated to a 96-well plate. When nuclear extracts are added to the plate, transcriptionally active NF-κB binds to the DNA causing the exposure of an epitope which is recognized by a primary antibody directed against p65. A HRP-conjugated secondary antibody provides a sensitive colorimetric reaction which is quantified by spectrophotometry. Absorbances were read at 450 nm with a reference wavelength of 655 nM.
Statistical Analysis
Data are represented as mean ± standard error of the mean (SEM). Data analysis was carried out using the one-tailed Student t-test.
RESULTS
Kupffer Cell TLR4 mRNA Expression
Kupffer cells obtained from vehicle-treated trauma-hemorrhage mice exhibited a significant increase (p<0.05) in TLR4 mRNA expression regardless whether the cells were stimulated with or without LPS compared to sham-operated group (Fig. 1). Administration of estradiol following trauma-hemorrhage decreased TLR4 expression.
Fig. 1. Kupffer cell Toll-like receptor 4 (TLR4) expression following trauma-hemorrhage.
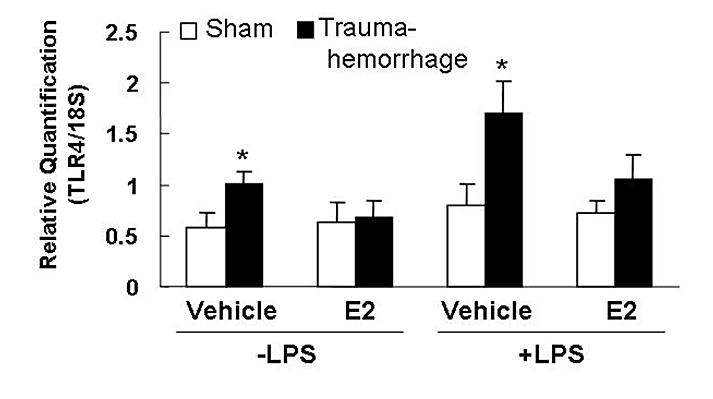
Kupffer cells were isolated 4 h following trauma-hemorrhage and cultured without or with LPS (1 μg/ml) for 24 h. Values are means ± SEM of 4 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. *p<0.05 vs. respective shams.
Kupffer Cell p38 and PI3K Phosphorylation
Phosphorylation of p38 was significantly increased in unstimulated or LPS-stimulated Kupffer cells obtained from vehicle-treated trauma-hemorrhage mice compared to sham animals (Fig. 2A; p<0.05). Administration of estradiol following trauma-hemorrhage decreased phospho-p38. In contrast, TLR4 mutant mice showed no increase in phospho-p38. Moreover, the amount of total p38 was not changed in any group (Fig. 2B). No difference was observed in phospho-PI3K between the groups (Fig. 3).
Fig. 2. Kupffer cell p38 phosphorylation (A) and total protein (B) following trauma-hemorrhage.
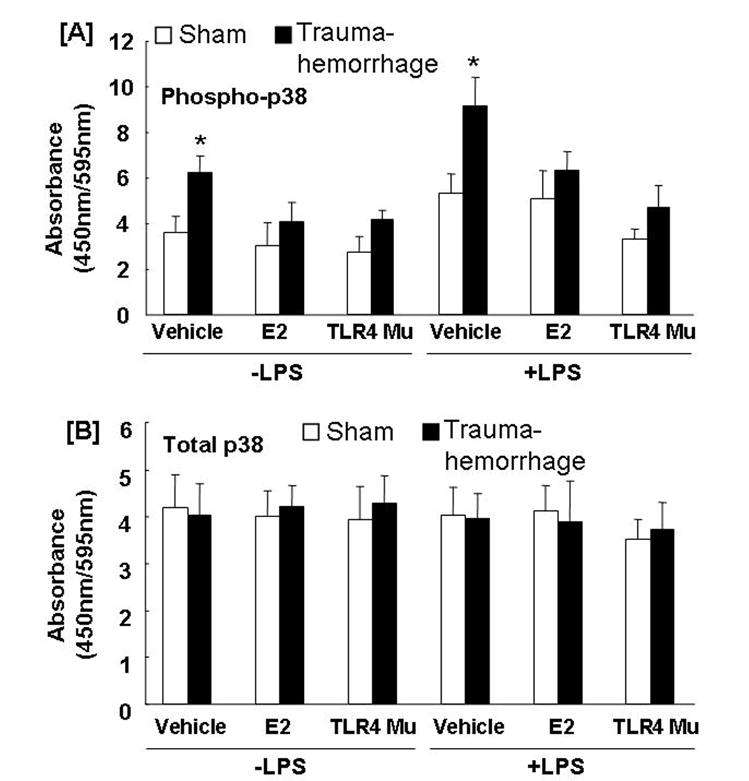
Kupffer cells were isolated 4 h after trauma-hemorrhage and stimulated without or with LPS (1 μg/ml) for 10 min. Values are means ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams.
Fig. 3. Kupffer cell phosphatidylinositol 3-kinase (PI3K) phosphorylation following trauma-hemorrhage.
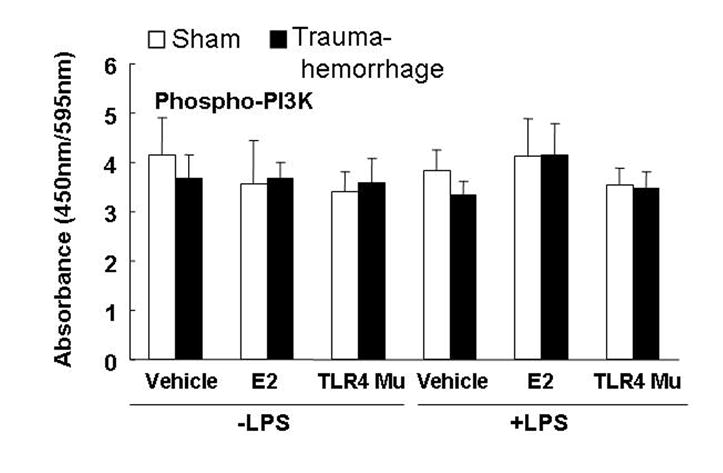
Kupffer cells were isolated 4 h following trauma-hemorrhage and stimulated without or with LPS (1 μg/ml) for 10 min. Values are means ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant.
Kupffer Cell NF-κB Activation
Trauma-hemorrhage resulted in a significant increase in DNA binding activity of NF-κB in unstimulated or LPS-stimulated Kupffer cells compared to Kupffer cells from sham animals (Fig. 4; p<0.05). Administration of estradiol following trauma-hemorrhage decreased the DNA binding activity of NF-κB in Kupffer cells and the values were not significantly different from the corresponding activity in Kupffer cells from sham animals. In TLR4 mutant mice, no difference was observed in NF-κB activity between sham-operated and trauma-hemorrhage groups.
Fig. 4. Kupffer cell nuclear factor-kB (NF-kB) binding activity following trauma-hemorrhage.
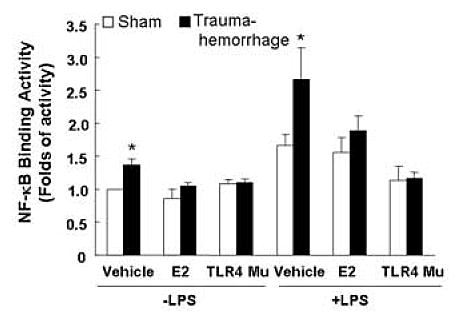
Kupffer cells were isolated 4 h after trauma-hemorrhage and stimulated without or with LPS (1 μg/ml) for 60 min. Values are means ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams.
Kupffer cell TNF-α and IL-6 production capacities
The production of TNF-α (Fig. 5A) by unstimulated Kupffer cells was increased (p<0.05) in the vehicle-treated trauma-hemorrhage mice compared to the sham-operated group, while IL-6 (Fig. 5B) levels were not affected. LPS-stimulated production of TNF-α and IL-6 by Kupffer cells was increased (p<0.05) following trauma-hemorrhage in the vehicle treatment group compared to sham-operated group. Administration of estradiol following trauma-hemorrhage decreased Kupffer cell IL-6 and TNF-α release and the levels were similar to those found in Kupffer cells from sham animals in the presence or absence of LPS (p<0.05). No significant difference was observed in Kupffer cell production of TNF-α and IL-6 in TLR4 mutant mice between sham-operated and trauma-hemorrhage groups.
Fig. 5. Kupffer cell production capacities of tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 following trauma-hemorrhage.
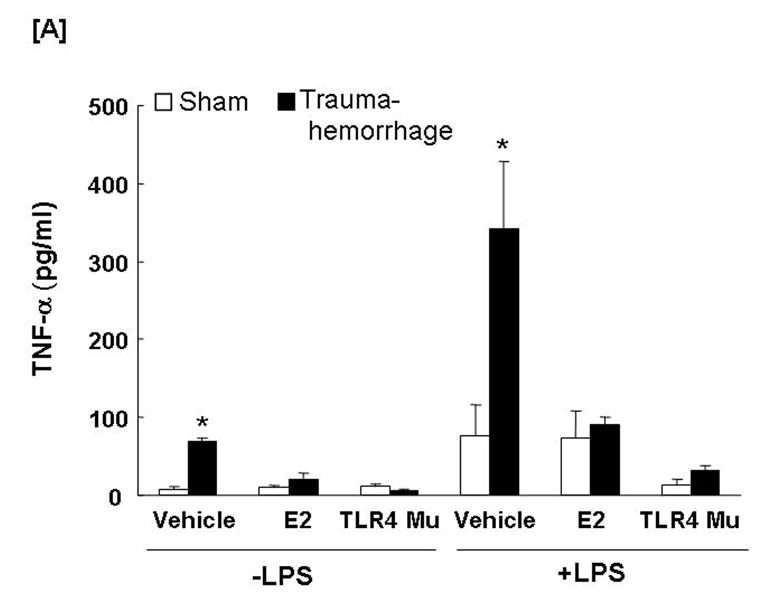
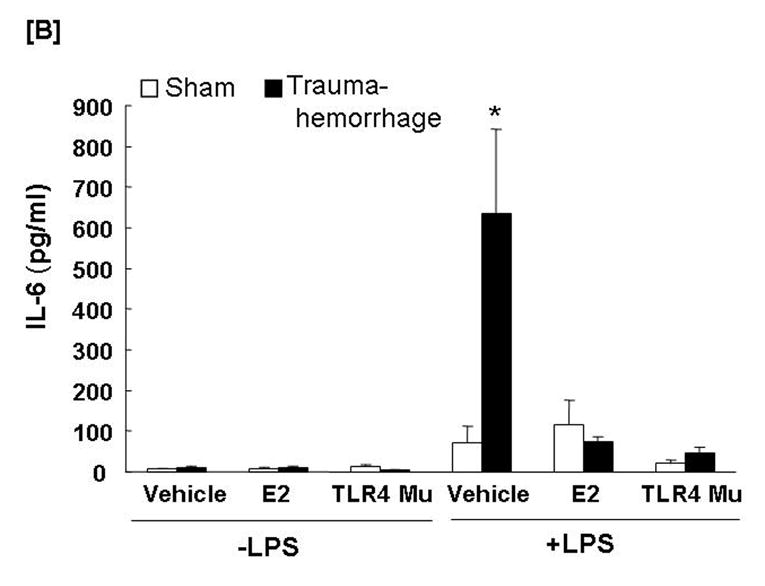
Kupffer cells were isolated 4 h following trauma-hemorrhage and cultured without or with LPS (1 μg/ml) for 24 h. Cytokine concentrations are shown as mean ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams.
Kupffer cell MIP-1α and MIP-2 production capacities
The production of MIP-1α (Fig. 6A) by unstimulated Kupffer cells following trauma-hemorrhage was not affected (Fig. 6A), however, the production of MIP-2 (Fig. 6B) by unstimulated Kupffer cells was increased (p<0.05) in the vehicle-treated trauma-hemorrhage mice compared to the sham-operated group. The LPS-stimulated production of MIP-1α and MIP-2 by Kupffer cells was increased (p<0.05) following trauma-hemorrhage in the vehicle treatment group compared to the sham-operated group. Administration of estradiol following trauma-hemorrhage normalized MIP-1α and MIP-2 release by Kupffer cells in the presence or absence of LPS (p<0.05). In TLR4 mutant mice, trauma-hemorrhage-induced production of MIP-1α and MIP-2 by Kupffer cells was not observed (Figs. 6A-6B).
Fig. 6. Kupffer cell production capacities of macrophage inflammatory protein (MIP)-1α and MIP-2 following trauma-hemorrhage.
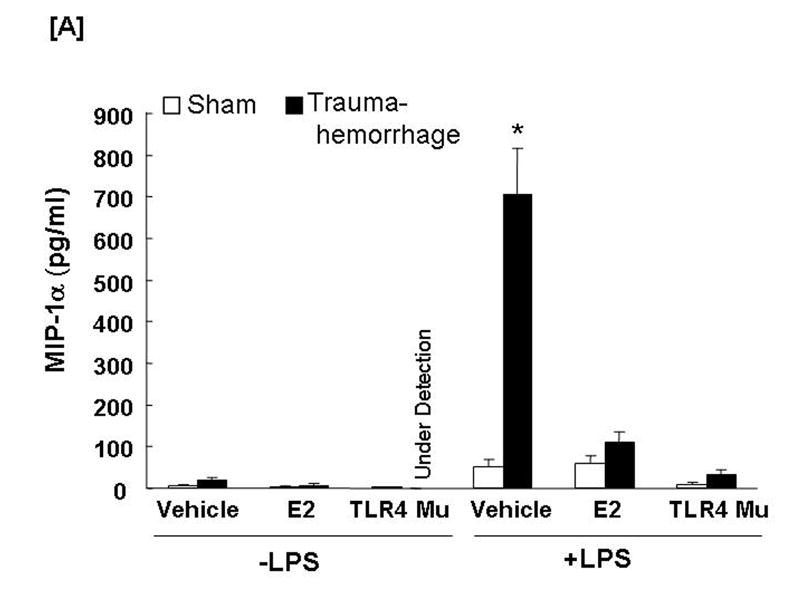
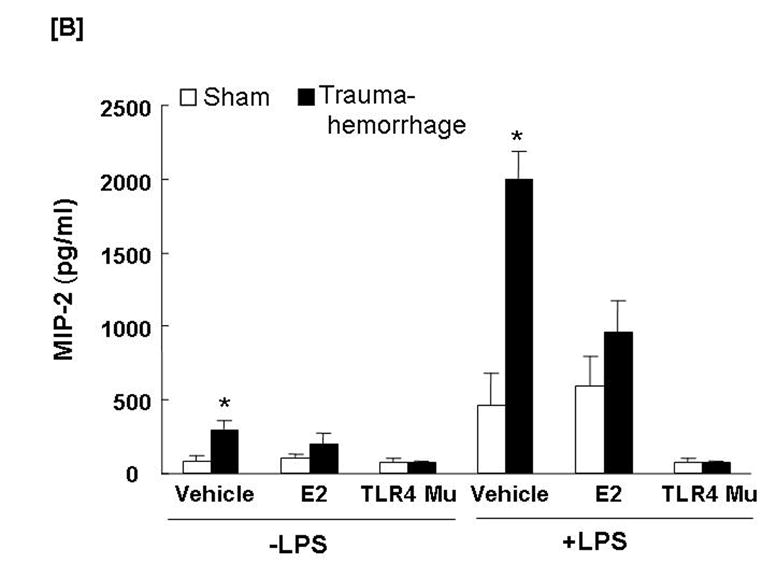
Kupffer cells were isolated 4 h after trauma-hemorrhage and cultured without or with LPS (1 μg/ml) for 24 h. Cytokine concentrations are shown as mean ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams.
Circulating TNF-α and IL-6 Levels
Trauma-hemorrhage led to a significant increase in plasma TNF-α (Fig. 7A) and IL-6 (Fig. 7B) levels compared to shams (p<0.05). Administration of estradiol following trauma-hemorrhage resulted in a significant decrease in plasma TNF-α and IL-6 levels compared to trauma-hemorrhage mice treated with vehicle (p<0.05). However, the levels remained significantly higher than those observed in sham-operated animals. Similar to Kupffer cell cytokines production, the plasma levels of TNF-α and IL-6 were also not significantly different in TLR4 mutant mice following trauma-hemorrhage compared to shams.
Fig. 7. Plasma tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 concentrations following trauma-hemorrhage.
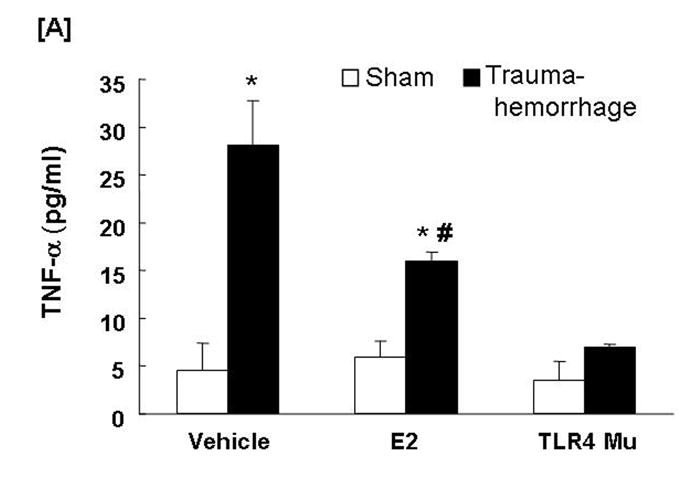
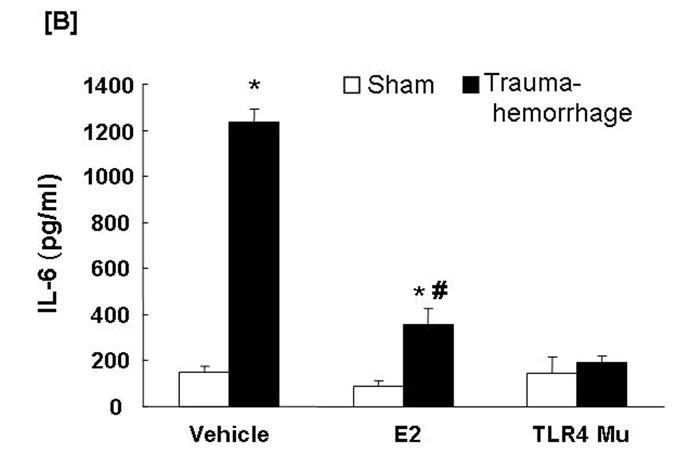
Animals were sacrificed 4 h following trauma-hemorrhage. Values are means ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams. #p<0.05 vs. trauma-hemorrhage+Vehicle.
Circulating MIP-1α and MIP-2 Levels
Trauma-hemorrhage led to a significant increase in plasma MIP-1α (Fig. 8A) and MIP-2 (Fig. 8B) levels compared to shams (p<0.05). Administration of estradiol following trauma-hemorrhage resulted in a significant decrease in plasma MIP-1α and MIP-2 levels compared to trauma-hemorrhage mice treated with vehicle (p<0.05). However, the MIP-1α levels remained significantly higher than those observed in sham-operated animals. In the TLR4 mutant mice, the plasma levels of MIP-1α and MIP-2 were not found to be significantly different following trauma-hemorrhage compared to shams.
Fig. 8. Plasma macrophage inflammatory protein (MIP)-1α and MIP-2 concentrations following trauma-hemorrhage.
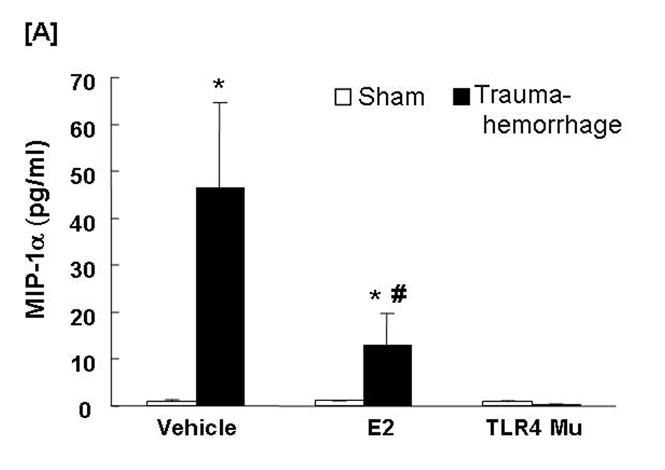
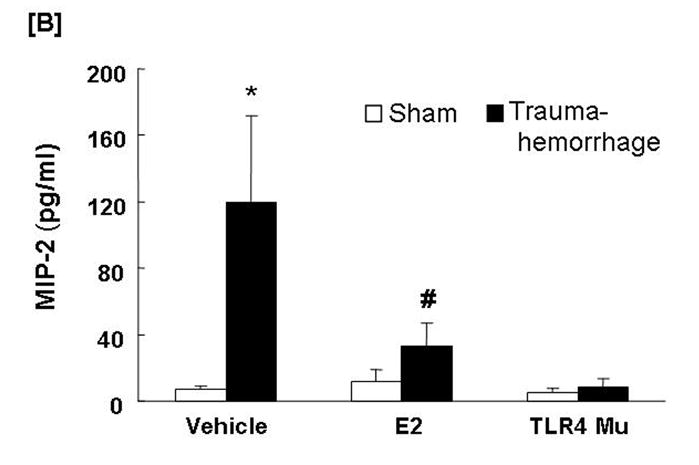
Animals were sacrificed 4 h after trauma-hemorrhage. Values are means ± SEM of 6 animals in each group. Data are compared by one-tailed Student t-test. E2: 17β-estradiol. Mu: TLR4 mutant. *p<0.05 vs. respective shams. #p<0.05 vs. trauma-hemorrhage+Vehicle.
DISCUSSION
This study was undertaken to determine the role of TLR4 in estradiol-mediated immunoprotective effects following trauma-hemorrhage. Our results showed that administration of estradiol following trauma-hemorrhage decreased Kupffer cell TLR4 and prevented the activation of p38 MAPK and NF-κB. This was accompanied by Kupffer cell cytokine production capacities which were similar to those observed in sham animals. Furthermore, the increased plasma levels of IL-6, TNF-α, MIP-1α, and MIP-2 following trauma-hemorrhage were decreased by estradiol treatment, however, the levels were still higher than shams. Moreover, TLR4 mutant mice did not exhibit the increases in Kupffer cell p38 MAPK and NF-κB activation, Kupffer cell cytokine production, as well as systemic release of cytokines following trauma-hemorrhage. No difference was observed in activation of PI3K among groups. These results indicate that the salutary effect of estradiol on immune function in Kupffer cells and systemic responses following trauma-hemorrhage is in part mediated via downregulation of TLR4 which is involved in the regulation of p38 MAPK and NF-κB signaling.
Kupffer cells are the largest population of macrophages in the body (Carini and Albano, 2003). Residing in the liver sinusoids, they are continuously exposed to possible blood-borne pathogens from the gastrointestinal tract through the portal vein or from the systemic circulation through the hepatic artery. Hence, the Kupffer cells represent an early line of defense against blood-borne infections. However, the Kupffer cells have also been suggested to be the cells that produce mediators which have deleterious inflammatory response under stress condition such as trauma-hemorrhage, ischemia reperfusion and sepsis (Ayala et al., 1997; Jaeschke et al., 1992; Koo et al., 1999; Yokoyama et al., 2004). In this regard, Kupffer cells represent the major source of inflammatory cytokine production and thus systemic release of proinflammatory mediators (Hildebrand et al., 2006; O'Neill et al., 1994). TLR4 has been implicated in such diverse processes as cytokine/chemokine production (Netea et al., 2002), phagocytic cell recruitment and function (Mattsby-Baltzer et al., 1996), and the generation of adaptive immunity (Medzhitov, 2001). Activation of TLR4 results in a stimulus-specific expression of genes required to control infection and injury, including the production of inflammatory cytokines and chemokines, complement products, and the recruitment of neutrophils to the site of injury (Mattsby-Baltzer et al., 1996; Medzhitov, 2001; Nelson et al., 1999). Although our recent studies indicate that administration of estradiol following trauma-hemorrhage downregulated TLR4 protein levels in Kupffer cells obtained from C3H/HeN mice (unpublished observation), the precise signaling pathway responsible for that effect remains unknown. The present study demonstrated that administration of estradiol following trauma-hemorrhage in mice with an intact TLR4 (C3H/HeJ) normalized Kupffer cell function and decreased systemic inflammatory responses, which was accompanied by downregulation of TLR4 expression in Kupffer cells. These findings therefore suggest that the salutary effects of estradiol on Kupffer cell function following trauma-hemorrhage are mediated via negative regulation of Kupffer cell TLR4 signaling.
The signaling transduction of TLR4 results in activation of p38 MAPK and NF-κB, which plays a significant role in the immune cell recruitment and function (Maung et al., 2005; Tsung et al., 2005; Yang et al., 2007). Both p38 MAPK and NF-κB are crucial to the signaling pathway that leads to the proinflammatory cytokine production (Baldwin, 1996; Raingeaud et al., 1995). P38 MAPK is a key mediator in organ dysfunction relating to inflammatory states, and acts as an important regulator of cell proliferation, differentiation, and production of proinflammatory cytokines (Carter et al., 1999; Thobe et al., 2006). NF-κB plays a key role in the transcriptional regulation of adhesion molecules, chemokines and cytokines involved in acute inflammatory responses (Baldwin, 1996). In this study, we found that treatment with estradiol following trauma-hemorrhage prevented the increase in Kupffer cell p38 and NF-κB activation, normalized Kupffer cell cytokine production, and decreased plasma cytokine levels. Moreover, trauma-hemorrhage activated p38 and NF-κB as well as cytokine production capacity of Kupffer cells and release in circulation was not observed in TLR4 mutant mice. These findings therefore suggest that the salutary effects of estradiol on immune function following trauma-hemorrhage are mediated via downregulation of TLR4 and that TLR4 is involved in the regulation of p38 MAPK and NF-κB signaling. Since treatment with estradiol following trauma-hemorrhage normalized Kupffer cell cytokine production capacity but systemic cytokine levels were not normalized, it suggests that cellular sources other than Kupffer cells may also contribute to circulating cytokine levels. It is also possible that enhanced cytokine production may occur during the trauma-hemorrhage procedure and circulating levels may still be high at 4 hr after resuscitation due to decreased clearance even though the Kupffer cell cytokine production is normalized by estradiol treatment after resuscitation.
p38 MAPK has been implicated in the activation of NF-κB (Carter et al., 1999). Other investigators have demonstrated that p38 MAPK-specific inhibitor SB203580 markedly attenuated NF-κB-dependent transcription (Conejo et al., 2001; Thirunavukkarasu et al., 2006). Thus, it can be postulated that TLR4-dependent activation of p38 MAPK regulates NF-κB signaling in Kupffer cells following trauma-hemorrhage. Alternatively, PI3K and the downstream serine/threonine kinase Akt also play important roles in host defense (Deane and Fruman, 2004). Although it has been suggested that the activation of PI3K by LPS is TLR4- and MyD88-dependent (Li et al., 2003; Ojaniemi et al., 2003), the signaling events between activation of TLR4 and PI3K have not been characterized. In our study, no change was observed in the phosphorylation of PI3K, suggesting that PI3K in Kupffer cells is not likely involved in TLR4-dependent pathway following trauma-hemorrhage.
The activation of NF-κB-dependent iNOS (inducible nitric oxide synthase) expression has been shown to be mediated by TLR4 signaling (Akira and Takeda, 2004). Moreover, TLR4 mediates increased expression of iNOS which is associated with mitochondrial DNA damage and the decreased ATP production in the liver (Suliman et al., 2005). We have previously found that increased ATP levels following trauma-hemorrhage normalized the depressed splenocyte and peritoneal macrophages functions and also the circulating cytokines under those conditions (Meldrum et al., 1992; Meldrum et al., 1991; Wang et al., 1992). Our recent studies suggest that the restoration of Kupffer cell immune function following trauma-hemorrhage by estradiol may be mediated via downregulation of TLR4 and iNOS through increased ATP production and mitochondrial DNA synthesis (unpublished observation). Thus, it is likely that estradiol-mediated restoration of Kupffer cell function is via downregulation of TLR4, which in turn negatively regulates NF-κB-dependent iNOS and thus increases cellular ATP levels following trauma-hemorrhage.
In summary, our study demonstrated that administration of estradiol following trauma-hemorrhage downregulated Kupffer TLR4 as well as prevented p38 and NF-κB activation, which was accompanied by normalized Kupffer cell function and decreased systemic inflammatory responses. Moreover, trauma-hemorrhage-induced Kupffer cell activation of p38 and NF-κB, immune dysfunction, and the increase in circulating inflammatory cytokines were not observed in TLR4 mutant mice. These results indicate that the downregulation of TLR4-dependent signaling is critical to estradiol-mediated restoration of Kupffer cell function following trauma-hemorrhage.
Acknowledgments
The authors thank Bobbi Smith for her valuable help in editing the manuscript. This work was supported by NIH Grant R01 GM 37127. Dr. Schwacha is in part supported by NIH grant KO2 AI049960.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Angele MK, Schwacha MG, Ayala A, Chaudry IH. Effect of gender and sex hormones on immune responses following shock. Shock. 2000;14:81–90. doi: 10.1097/00024382-200014020-00001. [DOI] [PubMed] [Google Scholar]
- Arndt PG, Suzuki N, Avdi NJ, Malcolm KC, Worthen GS. Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-Kinase and Syk-mediated pathways. J Biol Chem. 2004;279:10883–10891. doi: 10.1074/jbc.M309901200. [DOI] [PubMed] [Google Scholar]
- Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6 Suppl 1:S27–38. [PubMed] [Google Scholar]
- Ayala A, O'Neill PJ, Uebele SA, Herdon CD, Chaudry IH. Mechanism of splenic immunosuppression during sepsis: key role of Kupffer cell mediators. J Trauma. 1997;42:882–888. doi: 10.1097/00005373-199705000-00019. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bowie AG, Haga IR. The role of Toll-like receptors in the host response to viruses. Mol Immunol. 2005;42:859–867. doi: 10.1016/j.molimm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Carini R, Albano E. Recent insights on the mechanisms of liver preconditioning. Gastroenterology. 2003;125:1480–1491. doi: 10.1016/j.gastro.2003.05.005. [DOI] [PubMed] [Google Scholar]
- Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-kappaB-dependent gene expression. The role of TATA-binding protein (TBP) J Biol Chem. 1999;274:30858–30863. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- Chaudry IH, Samy TS, Schwacha MG, Wang P, Rue LW, 3rd, Bland KI. Endocrine targets in experimental shock. J Trauma. 2003;54:S118–125. doi: 10.1097/01.TA.0000064511.14322.F1. [DOI] [PubMed] [Google Scholar]
- Conejo R, Valverde AM, Benito M, Lorenzo M. Insulin produces myogenesis in C2C12 myoblasts by induction of NF-kappaB and downregulation of AP-1 activities. J Cell Physiol. 2001;186:82–94. doi: 10.1002/1097-4652(200101)186:1<82::AID-JCP1001>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- Driscoll KE, Hassenbein DG, Carter J, Poynter J, Asquith TN, Grant RA, Whitten J, Purdon MP, Takigiku R. Macrophage inflammatory proteins 1 and 2: expression by rat alveolar macrophages, fibroblasts, and epithelial cells and in rat lung after mineral dust exposure. Am J Respir Cell Mol Biol. 1993;8:311–318. doi: 10.1165/ajrcmb/8.3.311. [DOI] [PubMed] [Google Scholar]
- Hildebrand F, Hubbard WJ, Choudhry MA, Frink M, Pape HC, Kunkel SL, Chaudry IH. Kupffer cells and their mediators: the culprits in producing distant organ damage after trauma-hemorrhage. Am J Pathol. 2006;169:784–794. doi: 10.2353/ajpath.2006.060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YC, Hsu C, Yang RC, Lee PY, Hsu HK, Sun YM. Isolation of bona fide differentially expressed genes in the 18-hour sepsis liver by suppression subtractive hybridization. Shock. 2004;21:549–555. doi: 10.1097/01.shk.0000126148.83935.6a. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J Leukoc Biol. 1992;52:377–382. doi: 10.1002/jlb.52.4.377. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Jarrar D, Wang P, Song GY, Knoferl MW, Cioffi WG, Bland KI, Chaudry IH. Metoclopramide: a novel adjunct for improving cardiac and hepatocellular functions after trauma-hemorrhage. Am J Physiol Endocrinol Metab. 2000;278:E90–95. doi: 10.1152/ajpendo.2000.278.1.E90. [DOI] [PubMed] [Google Scholar]
- Kher A, Wang M, Tsai BM, Pitcher JM, Greenbaum ES, Nagy RD, Patel KM, Wairiuko GM, Markel TA, Meldrum DR. Sex differences in the myocardial inflammatory response to acute injury. Shock. 2005;23:1–10. doi: 10.1097/01.shk.0000148055.12387.15. [DOI] [PubMed] [Google Scholar]
- Knoferl MW, Diodato MD, Angele MK, Ayala A, Cioffi WG, Bland KI, Chaudry IH. Do female sex steroids adversely or beneficially affect the depressed immune responses in males after trauma-hemorrhage? Arch Surg. 2000;135:425–433. doi: 10.1001/archsurg.135.4.425. [DOI] [PubMed] [Google Scholar]
- Koo DJ, Chaudry IH, Wang P. Kupffer cells are responsible for producing inflammatory cytokines and hepatocellular dysfunction during early sepsis. J Surg Res. 1999;83:151–157. doi: 10.1006/jsre.1999.5584. [DOI] [PubMed] [Google Scholar]
- Li X, Tupper JC, Bannerman DD, Winn RK, Rhodes CJ, Harlan JM. Phosphoinositide 3 kinase mediates Toll-like receptor 4-induced activation of NF-kappa B in endothelial cells. Infect Immun. 2003;71:4414–4420. doi: 10.1128/IAI.71.8.4414-4420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsby-Baltzer I, Ahlstrom B, Edebo L, de Man P. Susceptibility of lipopolysaccharide-responsive and -hyporesponsive ItyS Mice to infection with rough mutants of Salmonella typhimurium. Infect Immun. 1996;64:1321–1327. doi: 10.1128/iai.64.4.1321-1327.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maung AA, Fujimi S, Miller ML, MacConmara MP, Mannick JA, Lederer JA. Enhanced TLR4 reactivity following injury is mediated by increased p38 activation. J Leukoc Biol. 2005;78:565–573. doi: 10.1189/jlb.1204698. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Meldrum DR, Ayala A, Chaudry IH. Energetics of defective macrophage antigen presentation after hemorrhage as determined by ultraresolution 31P nuclear magnetic resonance spectrometry: restoration with adenosine triphosphate-MgCl2. Surgery. 1992;112:150–156. discussion 156–158. [PubMed] [Google Scholar]
- Meldrum DR, Ayala A, Wang P, Ertel W, Chaudry IH. Association between decreased splenic ATP levels and immunodepression: amelioration with ATP-MgCl2. Am J Physiol. 1991;261:R351–357. doi: 10.1152/ajpregu.1991.261.2.R351. [DOI] [PubMed] [Google Scholar]
- Murphy TJ, Paterson HM, Mannick JA, Lederer JA. Injury, sepsis, and the regulation of Toll-like receptor responses. J Leukoc Biol. 2004;75:400–407. doi: 10.1189/jlb.0503233. [DOI] [PubMed] [Google Scholar]
- Nelson JL, Alexander JW, Mao JX, Vohs T, Ogle CK. Effect of pentoxifylline on survival and intestinal cytokine messenger RNA transcription in a rat model of ongoing peritoneal sepsis. Crit Care Med. 1999;27:113–119. doi: 10.1097/00003246-199901000-00038. [DOI] [PubMed] [Google Scholar]
- Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- O'Neill LA. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Curr Top Microbiol Immunol. 2002;270:47–61. [PubMed] [Google Scholar]
- O'Neill PJ, Ayala A, Wang P, Ba ZF, Morrison MH, Schultze AE, Reich SS, Chaudry IH. Role of Kupffer cells in interleukin-6 release following trauma-hemorrhage and resuscitation. Shock. 1994;1:43–47. doi: 10.1097/00024382-199401000-00008. [DOI] [PubMed] [Google Scholar]
- Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- Olsson S, Sundler R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol Immunol. 2006;43:607–612. doi: 10.1016/j.molimm.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Peng Y, Gong JP, Liu CA, Li XH, Gan L, Li SB. Expression of toll-like receptor 4 and MD-2 gene and protein in Kupffer cells after ischemia-reperfusion in rat liver graft. World J Gastroenterol. 2004;10:2890–2893. doi: 10.3748/wjg.v10.i19.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rana MW, Ayala A, Dean RE, Chaudry IH. Decreased Fc receptor expression on macrophages following simple hemorrhage as observed by scanning immunoelectron microscopy. J Leukoc Biol. 1990;48:512–518. doi: 10.1002/jlb.48.6.512. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. Faseb J. 2005;19:1531–1533. doi: 10.1096/fj.04-3500fje. [DOI] [PubMed] [Google Scholar]
- Thirunavukkarasu C, Watkins SC, Gandhi CR. Mechanisms of endotoxin-induced NO, IL-6, and TNF-alpha production in activated rat hepatic stellate cells: role of p38 MAPK. Hepatology. 2006;44:389–398. doi: 10.1002/hep.21254. [DOI] [PubMed] [Google Scholar]
- Thobe BM, Frink M, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Src Family Kinases Regulate p38 MAPK-Mediated IL-6 Production in Kupffer Cells Following Hypoxia. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00076.2006. [DOI] [PubMed] [Google Scholar]
- Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- Wang P, Ba ZF, Morrison MH, Ayala A, Dean RE, Chaudry IH. Mechanism of the beneficial effects of ATP-MgCl2 following trauma-hemorrhage and resuscitation: downregulation of inflammatory cytokine (TNF, IL-6) release. J Surg Res. 1992;52:364–371. doi: 10.1016/0022-4804(92)90117-i. [DOI] [PubMed] [Google Scholar]
- Wichmann MW, Ayala A, Chaudry IH. Male sex steroids are responsible for depressing macrophage immune function after trauma-hemorrhage. Am J Physiol. 1997;273:C1335–1340. doi: 10.1152/ajpcell.1997.273.4.C1335. [DOI] [PubMed] [Google Scholar]
- Xu YX, Ayala A, Chaudry IH. Prolonged immunodepression after trauma and hemorrhagic shock. J Trauma. 1998;44:335–341. doi: 10.1097/00005373-199802000-00018. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhou H, Yang Y, Li W, Zhou M, Zeng Z, Xiong W, Wu M, Huang H, Zhou Y, Peng C, Huang C, Li X, Li G. Lipopolysaccharide (LPS) regulates TLR4 signal transduction in nasopharynx epithelial cell line 5-8F via NFkappaB and MAPKs signaling pathways. Mol Immunol. 2007;44:984–992. doi: 10.1016/j.molimm.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Kitchens WC, Toth B, Schwacha MG, Rue LW, 3rd, Bland KI, Chaudry IH. Role of IL-10 in regulating proinflammatory cytokine release by Kupffer cells following trauma-hemorrhage. Am J Physiol Gastrointest Liver Physiol. 2004;286:G942–946. doi: 10.1152/ajpgi.00502.2003. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Kuebler JF, Matsutani T, Schwacha MG, Bland KI, Chaudry IH. Mechanism of the salutary effects of 17beta-estradiol following trauma-hemorrhage: direct downregulation of Kupffer cell proinflammatory cytokine production. Cytokine. 2003;21:91–97. doi: 10.1016/s1043-4666(03)00014-0. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Ayala A, DeMaso CM, Chaudry IH. Trauma-hemorrhage causes prolonged depression in cellular immunity. Shock. 1995;4:149–153. doi: 10.1097/00024382-199508000-00012. [DOI] [PubMed] [Google Scholar]