

Abstract
Antiarrhythmic drug therapy remains the principal approach for suppression of atrial fibrillation (AF) and flutter (AFl) and prevention of their recurrence. Among the current strategies for suppression of AF/AFl is the development of antiarrhythmic agents that preferentially affect atrial, rather than ventricular electrical parameters. Inhibition of the ultrarapid delayed rectifier potassium current (IKur), present in the atria, but not in the ventricles, is an example of an atrial-selective approach. Our recent study examined the hypothesis that sodium channel characteristics differ between atrial and ventricular cells and that atrial-selective sodium channel block is another effective strategy for the management of AF. We have demonstrated very significant differences in the inactivation characteristics of atrial versus ventricular sodium channels and a striking atrial selectivity for the action of ranolazine, an inactivated-state sodium channel blocker, to produce use-dependent block of the sodium channels, leading to depression of excitability, development of post-repolarization refractoriness (PRR), and suppression of AF. Lidocaine and chronic amiodarone, both predominantly inactivated-state sodium channel blockers, also produced a preferential depression of sodium channel–dependent parameters (VMax conduction velocity, diastolic threshold of excitation, and PRR) in the atria. Propafenone, a predominantly open-state sodium channel blocker, produced similar changes of electrophysiological parameters, which were was not atrial-selective. The ability of ranolazine, chronic amiodarone, and propafenone to prolong the atrial action potential potentiated their ability to suppress AF in coronary-perfused canine atrial preparations. In conclusion: Our data demonstrate important differences in the inactivation characteristics of atrial versus ventricular sodium channels and a striking atrial selectivity for the action of agents like ranolazine to produce use-dependent block of sodium channels leading to suppression of AF. Our findings suggest that atrial-selective sodium channel block may be a valuable strategy to combat AF.
Keywords: electrophysiology, cardiac arrhythmias, lidocaine, amiodarone, ranolazine, propafenone
Introduction
Antiarrhythmic drug therapy remains the principal approach for suppression of atrial fibrillation (AF) and flutter (AFl) and their recurrences. Among the current strategies for suppression of AF/AFl is the development of antiarrhythmic agents that preferentially affect atrial, rather than ventricular electrical parameters. Inhibition of the ultrarapid delayed rectifier potassium current (IKur), present in atria but not in ventricles, is an example of an atrial-selective approach.1 We recently examined the hypothesis that sodium channel characteristics differ between atrial and ventricular cells and that atrial-selective sodium channel block is another effective strategy for the management of AF.2,3 Biophysical characteristics of sodium channels were measured in single myocytes isolated from canine atria and ventricles. Four agents capable of blocking cardiac sodium channels (ranolazine, lidocaine, propafenone, and chronically administrated amiodarone) were compared with regard to their ability to alter the electro-physiology of canine coronary artery–perfused atrial and ventricular preparations as well as their ability to suppress AF. This review contrasts the effects of these open- and closed-state channel blockers.
Methods and Results
Sodium Channel Inactivation Characteristics in Isolated Atrial versus Ventricular Myocytes
Whole-cell peak sodium currents were recorded at 37°C in low-sodium external solution from myocytes isolated from the right atrium and left ventricle (LV) of adult mongrel dogs. The half inactivation voltage (V0.5) in atrial myocytes was about 15 mV more negative than that recorded in ventricular myocytes, and the differences were increased after exposure to ranolazine (Fig. 1). These data indicate that a greater percentage of atrial versus ventricular sodium channels would be inactivated at a given resting or take-off potential and that inactivated-state sodium channel blockers may, therefore, be more effective in blocking sodium channels in atria than in ventricles. An intrinsically more positive resting membrane potential (RMP) in atria (−83 mV) versus ventricles (−87 mV) would further reduce the availability of sodium channels in atria and accentuate the atrial selectivity of sodium channel blockers.
FIGURE 1.
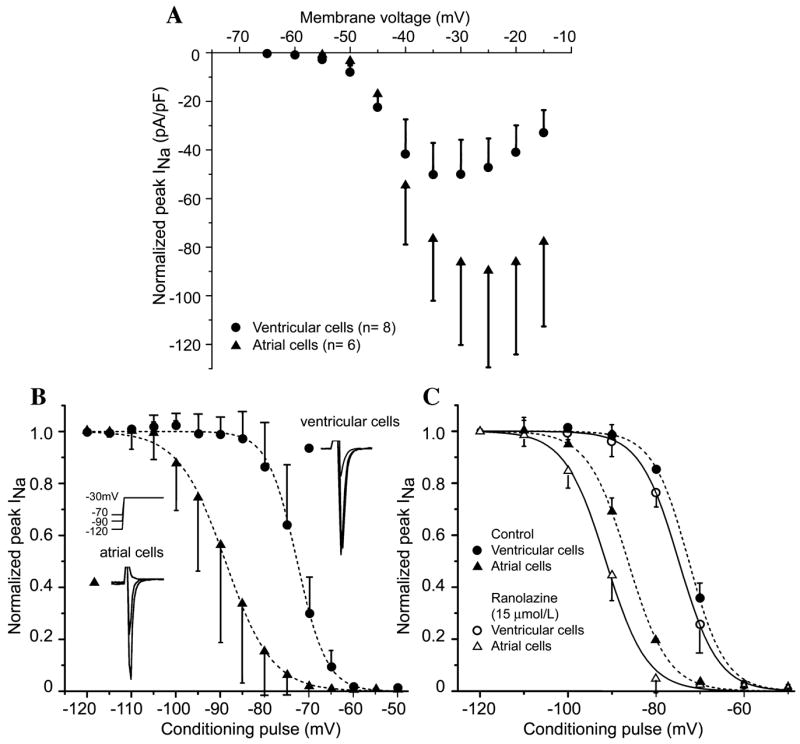
Activation and steady-state inactivation in canine atrial versus ventricular myocytes. (A) Current–voltage relation in ventricular and atrial myocytes. Voltage of peak INa is more positive and current density is larger in atrial versus ventricular myocytes. (B) Summarized steady-state inactivation curves. The half-inactivation voltage (V0.5) is −88.80 ± 0.19 mV in atrial cells (n = 9) and −72.64 ± 0.14 mV in ventricular cells (P < 0.001, n = 7). Insets show representative atrial and ventricular traces after 1-s conditioning pulses to the indicated potentials. (C) Steady-state inactivation curves before and after addition of 15 μM ranolazine. Ranolazine shifts V0.5 from −72.53 ± 0.16 mV to −74.81 ± 0.14 mV (P < 0.01) in ventricular myocytes (n = 4) and from −86.35 ± 0.19 to −91.38 ± 0.35 mV (P < 0.001) in atrial myocytes (n = 5). (From Burashnikov et al.2 Reproduced with permission.)
Because ranolazine has recently been identified as an inactivated sodium channel state blocker4 with little effect on peak INa or INa-mediated parameters in ventricular myocardium at therapeutic concentrations,5,6 we hypothesized that this agent may act as an atrial-selective sodium channel blocker. We contrasted the effects of ranolazine with those of other inactivated-state sodium channel blockers, such as lidocaine and amiodarone, as well as an open-state sodium channel blocker, propafenone, in atria and ventricles.
Sodium Channel–Dependent Parameters in Multicellular Atrial and Ventricular Preparations
Experiments were performed using isolated arterially perfused canine right atrial preparations and left ventricular arterially perfused wedge preparations.5,7,8 Therapeutic plasma concentrations of ranolazine (1–10 μM), lidocaine (2.1–21 μM), and propafenone (0.3–3.0 μM) were examined. Amiodarone was chronically administrated at a dose of 40 mg/kg/day for 6 weeks.
Sodium channel–mediated parameters, such as the maximum rate of rise of the AP upstroke (Vmax), conduction velocity (CV), diastolic threshold of excitation (DTE), and post-repolarization refractorines (PRR) were evaluated. PRR was defined as the difference between action potential duration (APD) and atrial effective refractory period (ERP). ERP normally coincides with APD70–90, but may extend well beyond APD70–90 or even APD100 (causing the appearance of PRR) under conditions associated with a reduction of excitability (ischemia, sodium channel block, etc.9).
Ranolazine and propafenone prolong APD90 selectively in atria (by 11% and 13%, respectively), with little change of APD90 in the ventricles (+ 2% and +3%, respectively; CL = 500 ms). Chronic amiodarone produced a greater prolongation of APD90 in atria than in ventricles (22 versus 12%, respectively; CL = 500 ms). In contrast, lidocaine abbreviates APD90 in both the atria and ventricles (6% and 9%, respectively; CL = 500 ms). Ranolazine, lidocaine, and chronic amiodarone lengthened ERP selectively (ranolazine) or predominantly (amiodarone and lidocaine) in atria in a rate-dependent manner, leading to the development of greater PRR in atria versus ventricles In contrast, propafenone induced prominent PRR in both the atria and ventricles (Table 1).
TABLE 1.
The effect of ranolazine, lidocaine, propafenone, and chronic amiodarone on sodium channel–dependent parameters in canine isolated coronary artery–perfused atrial and ventricular preparations
| Ranolazine (10 μM) 0.5/0.3 s | Lidocaine (21 μM) 0.5/03 s | Propafenone (1.5 μM) 0.5/0.3 s | Chronic Amiodarone 0.5/0.3 s | ||
|---|---|---|---|---|---|
| Vmax (%Δ) | Atria | −26/43 | −31/40 | −46/78 | −42/67 |
| Ventricles | −9/15 | −16/23 | −40/51 | −9/16 | |
| DTE (% Δ) | Atria | +18/139 | +30/105 | +112/172 | +109/148 |
| Ventricles | +3/8 | +8/40 | +84/125 | NA | |
| CV (% Δ) | Atria | −14/46 | −29/57 | −55/97 | +25/56 |
| Ventricles | −5/11 | −12/36 | −44/71 | +6/21 | |
| PRR (ms) | Atria | 51/79 | 71/84 | 68/94 | 48/107 |
| Ventricles | 3/7 | 47/69 | 52/83 | 31/36 |
Note: Data recorded at pacing cycle lengths of 0.5 and 0.3 s. Vmax = maximum rate of rise of the action potential upstroke; DTE = diastolic threshold of excitation; CV = conduction velocity; PRR = post-repolarization refractoriness. PRR was determined as the difference between ERP and APD75 in atria and APD90 in ventricles. (ERP is coincident with APD75 in atria and APD90 in ventricles.) CV was approximated from the duration of the P wave complex in atria and QRS complex in ventricles on the pseudo-ECG recordings. n = 3–18. (Modified from Burashnikov et al.2,3)
Ranolazine and chronic amiodarone caused a much greater rate-dependent reduction in Vmax, increase in DTE, and slowing of CV in atrial than ventricular preparations (Fig. 2; Table 1). Lidocaine also preferentially suppressed these parameters in atria, although to a lesser extent (Table 1). Propafenone depressed sodium channel–mediated parameters more potently than ranolazine, lidocaine, or chronic amiodarone, but without a sizable chamber selectivity at normal pacing rates (CL = 500 ms; Table 1). At a pacing CL of 300 ms, propafenone produced a potent depression of INa-mediated parameters in both atria and ventricles, but the effect in atria was more pronounced (Table 1). This atrial selectivity of propafenone at rapid activation rates was associated with atrial-selective prolongation of APD90, leading to elimination of diastolic intervals in atria but not in ventricles. Atrial selectivity of these agents to depress INa-dependent parameters derives in part from the agents’ ability to prolong atrial APD and thus leads to more positive take-off potential and elimination of the diastolic interval at rapid rates of activation (Fig. 3), potentiating the actions of the drug to depress INa.
FIGURE 2.
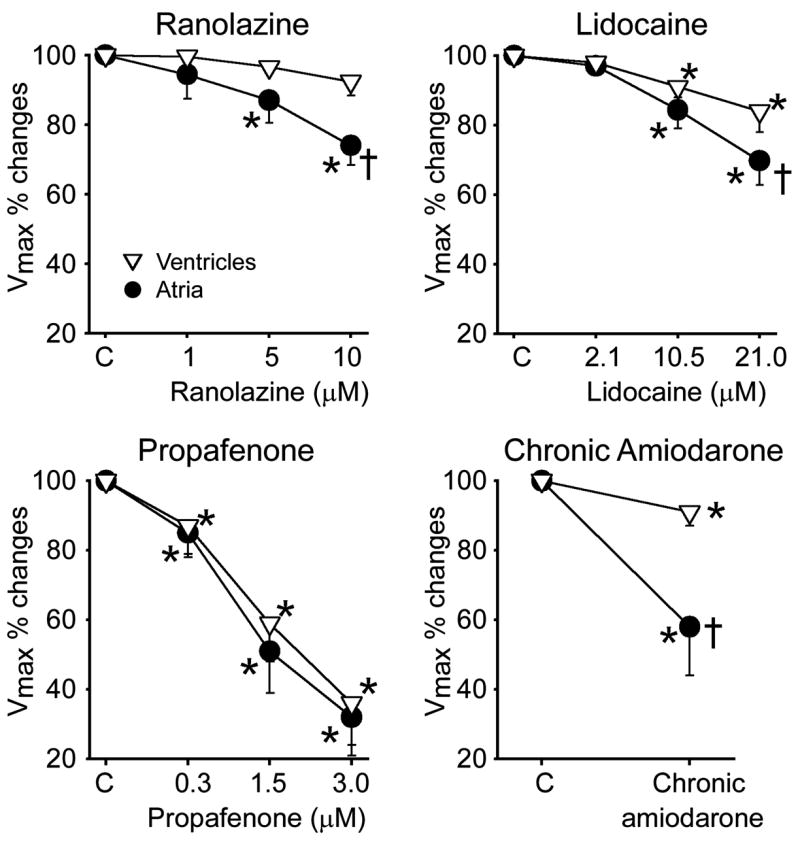
Atrial-selective suppression of Vmax by ranolazine, lidocaine, and chronic amiodarone, but not propafenone in canine coronary artery–perfused atrial and ventricular preparations. *P < 0.05 versus respective control (C); †P < 0.05 versus respective ventricular values. n = 8–15. CL = 500 ms.
FIGURE 3.
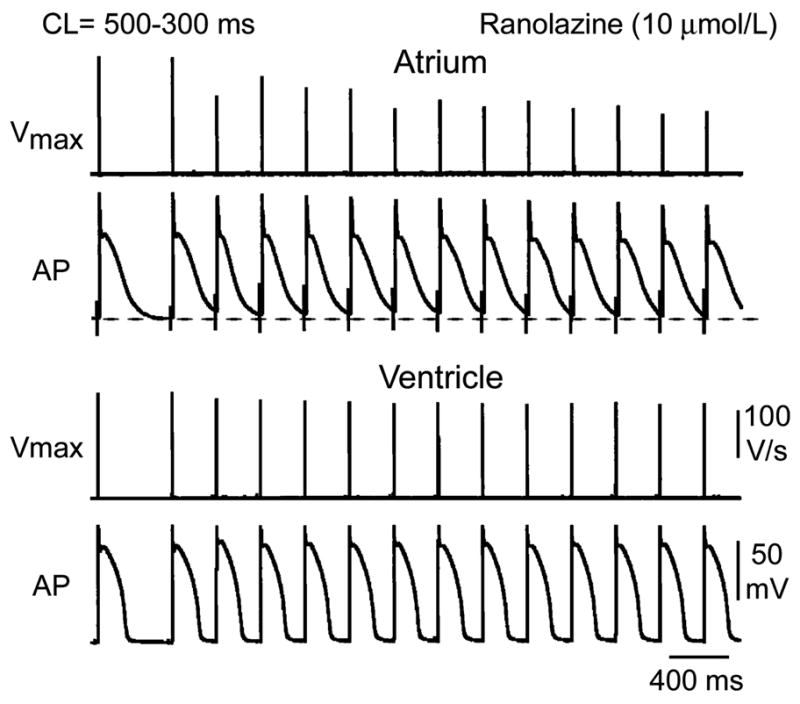
Mechanisms contributing to atrial selectivity of ranolazine in depressing Vmax at fast pacing rates: the role of atrial-selective APD prolongations. Shown are action potential tracings and corresponding Vmax values recorded during acceleration of pacing rate from a CL of 500 to 300 ms in atrial and ventricular preparations in the presence of ranolazine. Ranolazine prolongs late repolarization in atria, but not in ventricles. Acceleration of rate leads to elimination of the diastolic interval, resulting in a more positive take-off potential in atrium. The diastolic interval remains relatively long in ventricles. (Modified from Burashnikov et al.2)
Antiarrhythmic Effects of Ranolazine, Lidocaine, Propafenone, and Chronic Amiodarone in a Model of AF
Persistent AF is induced in 100% of canine coronary arterially perfused atrial preparations in the presence of acetylcholine (0.5 μM).8,10 Ranolazine was found to be more effective than lidocaine, but less effective than propafenone, in terminating acetylcholine-mediated persistent AF in coronary-perfused atria as well as in preventing the initiation of AF (Table 2). Persistent acetylcholine-mediated AF could be induced in only 1 of 6 atria isolated from dogs chronically treated with amiodarone (versus 10 of 10 untreated atria). Anti-AF actions of ranolazine, lidocaine, propafenone, and amiodarone were associated with the development of significant rate-dependent PRR.
TABLE 2.
Effectiveness of ranolazine, lidocaine, propafenone, and chronic amiodarone in terminating and preventing induction of acetylcholine-mediated AF in coronary artery–perfused right atrial preparations
| Ranolazine (10.0 μM) | Lidocaine (21.0 μM) | Propafenone (1.5–3.0 μM) | Chronic Amiodarone | |
|---|---|---|---|---|
| Termination of AF | 66% (4/6) | 33% (2/6) | 100% (7/7) | NA |
| Prevention of induction of AF | 80% (8/10) | 57% (4/7) | 100% (6/6) | 83% (5/6) |
Ranolazine (5–10 μM) also prevented the induction of AF in 4 of 5 atria in which self-terminating AF was induced by exposure to ischemia and β-adrenergic agonists.2,11 Ischemia/reperfusion coupled with iso-proterenol mimics the conditions that prevail during acute myocardial infarction or the substrate encountered postsurgically.
Discussion
Our recent studies demonstrate very significant differences in the inactivation characteristics of atrial versus ventricular sodium channels and a striking atrial selectivity for the action of ranolazine, an inactivated-state sodium channel blocker, to produce use-dependent block of the sodium channels, leading to depression of excitability, development of PRR, and suppression of AF.2,3 Lidocaine and amiodarone, two other predominantly inactivated-state sodium channel blockers,12 depressed sodium channel–dependent parameters (Vmax, CV, DTE, and PRR) predominantly in atria, providing support for the hypothesis that inactivated-state sodium channel blockers are likely to be atrial-selective. It is noteworthy that lidocaine was much less atrial-selective than ranolazine or chronic amiodarone (Table 1). Further evidence in support of the hypothesis derives from the demonstration that propafenone, a predominantly open-state sodium channel blocker,12 is not atrial-selective. Of interest, lidocaine causes tonic block in atrial, but not ventricular preparations, while quinidine and prajmaline (predominantly open-state sodium channel blockers) produce tonic block in both atrial and ventricular preparations.13 The atrioventricular differences in RMP are likely to contribute to the atrial selectivity of inactivated-state sodium channel blockers. When RMP is depolarized by high potassium in ventricular tissues (increasing the fraction of inactivated sodium channels), inactivated-state sodium channel blockers produce a greater reduction of Vmax than do open-state sodium channel blockers.14
In canine ventricular myocytes, ranolazine has been shown to inhibit late INa with an IC50 of 6 μM,5 but to inhibit peak INa with an IC50 of 294 μM.4 Consistent with the latter, ranolazine has been reported to suppresses Vmax with an IC50 of >100 μM in ventricular Purkinje fibers and M cell preparations paced at a CL of 500 ms.5,6 In sharp contrast, ranolazine causes a prominent use-dependent reduction of INa (estimated based on changes in Vmax) in atrial preparations at concentrations within the therapeutic range of ranolazine (2–10 μM).2
Sodium channel blockers generally bind more effectively to open and/or inactivated sodium channels (i.e., during the action potential) than to resting sodium channels (i.e., during the diastolic interval). Unblocking occurs largely during the resting state.12 Rapid activation rates contribute to the development of sodium channel block by increasing the proportion of time that the sodium channels are in the open/inactivated state and reducing the time that the channels are in the resting state. Agents that prolong APD selectively in atria but not ventricles are expected to display atrial-selective INa block, particularly at rapid activation rates on account of their ability to reduce or eliminate the diastolic interval and depolarize take-off potential in an atrial-selective manner (Fig. 3). The more depolarized RMP in atria potentiates the effects of INa blockers by increasing the fraction of channels in the inactivated state, which reduces the availability of sodium channels and prolongs the time needed for the sodium channels to recover from inactivation.
Ranolazine was more atrial-selective than was lidocaine and more effective than lidocaine in terminating and preventing recurrence of AF. This may be due to the fact that ranolazine prolongs only atrial APD because of its ability to also block the rapidly activating delayed rectifier potassium current (IKr,, IC50 = 12 μM),5 whereas lidocaine, a more selective INa blocker, abbreviates both atrial and ventricular APD. It is noteworthy that IKr blockers preferentially prolong atrial versus ventricular APD (see below). The selective prolongation of APD in atria by ranolazine leads to elimination of diastolic intervals and more depolarized take-off potentials at rapid rates in atria but not ventricles (Fig. 3). The more negative h-curve in atria and acceleration-induced depolarization of take-off potential act in concert to increase the fraction of channels in the inactivated state, making sodium channels less available and more sensitive to block by ranolazine. The result is a greater atrial versus ventricular suppression of INa-dependent parameters such as Vmax, DTE, and CV, and the development of use-dependent PRR. The effect of ranolazine to prolong atrial repolarization potentiates but does not appear to be a determining factor in ranolazine’s atrial specificity and in antiarrhythmic efficacy. Propafenone (INa and IKr blocker), like ranolazine, selectively prolongs atrial APD90 but suppresses INa-dependent parameters in both the atrial and the ventricular preparations to a similar extent at a CL 500 ms,2 as does GE 68, a propafenone analogue.15 At faster pacing rates, propafenone more effectively depresses Vmax and CV in atria on account of atrial-selective APD90 prolongation (leading to elimination of diastolic interval in atria). Lidocaine abbreviates both atrial and ventricular APD90, but shows atrial specificity in depression of INa-dependent parameters. Chronic amiodarone produces depression of INa-dependent parameters pre-dominantly in atria via a similar mechanism, which includes preferential prolongation of atrial APD.
These results suggest that the IKr blocking effect of ranolazine, chronic amiodarone, and propafenone potentiates sodium channel inhibitory effect of these drugs in atria at fast pacing rates. Interestingly, IKr blockers generally produce a much greater APD prolongation in atria than in ventricles. Selective inhibition of IKr prolongs atrial ERP more than ventricular ERP at normal or moderately rapid activation rates,16,17 but not at slow rates. At relatively slow activation rates or following long pauses, IKr block preferentially prolongs ventricular versus atrial APD, leading to development of early afterdepolarization (EAD) and torsade de pointes arrhythmias in the ventricles, but not in atria.18–20
A number of antiarrhythmic agents have been shown to be effective in terminating and/or preventing clinical AF/AFl. Most of these agents have as a primary action the ability to reduce INa (e.g., propafenone or flecainide) and IKr (e.g., dofetilide) or to inhibit multiple ion channels, as in the case of amiodarone. An important limitation of these antiarrhythmic agents is their potential ventricular proarrhythmic actions and/or organ toxicity at therapeutically effective doses.6,18,21 This has prompted to the development of atrial-selective antiarrhythmic agents, such as those that block IKur.1,27,28 However, block of IKur alone may not be sufficient for the suppression of AF.22 In remodeled atria, IKur block selectively prolongs atrial APD90 (but only slightly) and, when combined with Ito (perhaps with IK–ACh) and/or INa inhibition, can suppress AF/AFl.22–24 In nonremodeled healthy atria, IKur inhibition abbreviates APD90, 7,22,25 and can promote AF.22 These data are consistent with the results of a recent study showing an association of loss-of-function mutations in KCNA5, which encodes the α-subunit of IKur channel, with familial AF.26 Our results suggest that atrial-selective sodium channel block may be another effective approach for the management of AF.
Summary
Recent studies point to major distinctions in the inactivation characteristics of atrial and ventricular cardiac sodium channels. Sodium channel blockers such as ranolazine and chronic amiodarone take advantage of these distinctions and are capable of inhibiting sodium channels more effectively in the atria of the heart. Our findings suggest that inactivated-state sodium channel blockers have a proclivity towards producing atrial-selective sodium channel inhibition. The results point to atrial-selective sodium channel block as a novel strategy for the management of AF and suggest that the additional presence of IKr block and APD prolongation can potentiate the atrial selectivity of INa blockers and thus enhance their effectiveness in suppressing and preventing the development of AF.
Acknowledgments
This study was supported by grants from CV Therapeutics (to C.A.), the NIH (HL-47687; to C.A.), and the New York State and Florida Grand Lodges of the Free and Accepted Masons.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Nattels S, Matthews C, de Blasio E, et al. Dose-dependence of 4-aminopyridine plasma concentrations and electrophysiological effects in dogs: potential relevance to ionic mechanisms in vivo. Circulation. 2000;101:1179–1184. doi: 10.1161/01.cir.101.10.1179. [DOI] [PubMed] [Google Scholar]
- 2.Burashnikov A, di Diego JM, Zygmunt AC, et al. Atrial-selective sodium channel block as a strategy for suppression of atrial fibrillation. Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burashnikov A, Belardinelli L, Antzelevitch C. Ranolazine and propafenone both suppress atrial fibrillation but ranolazine unlike propafenone does it without prominent effects on ventricular myocardium [abstract] Heart Rhythm. 2007;4:S163. [Google Scholar]
- 4.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S161–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antzelevitch C, Belardinelli L, et al. Electrophysiologic effects of ranolazine: a novel anti-anginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antzelevitch C, Belardinelli L, Wu L, et al. Electrophysiologic properties and antiarrhythmic actions of a novel anti-anginal agent. J Cardiovasc Pharmacol Therapeut. 2004;9(Suppl 1):S65–S83. doi: 10.1177/107424840400900106. [DOI] [PubMed] [Google Scholar]
- 7.Burashnikov A, Mannava S, Antzelevitch C. Transmembrane action potential heterogeneity in the canine isolated arterially-perfused atrium: effect of IKr and Ito/IKur block. Am J Physiol. 2004;286:H2393–H2400. doi: 10.1152/ajpheart.01242.2003. [DOI] [PubMed] [Google Scholar]
- 8.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 9.Davidenko JM, Antzelevitch C. Electrophysiological mechanisms underlying rate-dependent changes of refractoriness in normal and segmentally depressed canine Purkinje fibers: the characteristics of post-repolarization refractoriness. Circ Res. 1986;58:257–268. doi: 10.1161/01.res.58.2.257. [DOI] [PubMed] [Google Scholar]
- 10.Burashnikov A, Antzelevitch C. Role of repolarization restitution in the development of coarse and fine atrial fibrillation in the isolated canine right atria. J Cardiovasc Electrophysiol. 2005;16:639–645. doi: 10.1046/j.1540-8167.2005.40689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burashnikov A, Antzelevitch C. Beta-adrenergic stimulation is highly arrhythmogenic following ischemia/reperfusion injury in the isolated canine right atrium [abstract] Heart Rhythm. 2005;2:S179. [Google Scholar]
- 12.Whalley DW, Wendt DJ, Grant AO. Basic concepts in cellular cardiac electrophysiology. Part II: Block of ion channels by antiarrhythmic drugs. Pacing Clin Elecrophysiol. 1995;18:1686–1704. doi: 10.1111/j.1540-8159.1995.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 13.Langenfeld H, Weirich J, Kohler C, Kochsiek K. Comparative analysis of the action of class I antiar-rhythmic drugs (lidocaine, quinidine, and prajmaline) in rabbit atrial and ventricular myocardium. J Cardiovasc Pharmacol. 1990;15:338–345. doi: 10.1097/00005344-199002000-00023. [DOI] [PubMed] [Google Scholar]
- 14.Campbell TJ, Wyse KR, Hemsworth PD. Effects of hyperkalemia, acidosis, and hypoxia on the depression of maximum rate of depolarization by class-I antiarrhythmic drugs in guinea pig myocardium: differential actions of class-Ib and class-Ic agents. J Cardiovasc Pharmacol. 1991;18:51–59. doi: 10.1097/00005344-199107000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Lemmens-Gruber R, Marei H, Heistracher P. Electrophysiological properties of the propafenone-analogue GE 68 (1-[3-(phenylethyl)-2-benzofuryl]-2-(propylamino)-ethanol) in isolated preparations and ventricular myocytes of guinea-pig hearts. Naunyn Schmiedebergs. Arch Pharmacol. 1997;355:230–238. doi: 10.1007/pl00004937. [DOI] [PubMed] [Google Scholar]
- 16.Spinelli W, Parsons RW, Colatsky TJ. Effects of WAY-123,398, a new Class-III antiarrhythmic agent, on cardiac refractoriness and ventricular fibrillation threshold in anesthetized dogs: a comparison with UK-68798, e-4031, and DL- sotalol. J Cardiovasc Pharmacol. 1992;20:913–922. doi: 10.1097/00005344-199212000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Wiesfeld AC, de Langen CD, et al. Rate-dependent effects of the class III antiarrhythmic drug almokalant on refractoriness in the pig. J Cardiovasc Pharmacol. 1996;27:594–600. doi: 10.1097/00005344-199604000-00021. [DOI] [PubMed] [Google Scholar]
- 18.Antzelevitch C, Shimizu W, Yan GX, et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 19.Burashnikov A, Antzelevitch C. Late-phase 3 EAD: a unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol. 2006;29:290–295. doi: 10.1111/j.1540-8159.2006.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vincent GM. Atrial arrhythmias in the inherited lLong QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1034–1035. doi: 10.1046/j.1540-8167.2003.03365.x. [DOI] [PubMed] [Google Scholar]
- 21.Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 22.Burashnikov A, Antzelevitch C. IKUR block promotes atrial fibrillation in healthy canine atria [abstract] Heart Rhythm. 2007;4:S112. [Google Scholar]
- 23.Blaauw Y, Gogelein H, Tieleman RG, et al. Early” class III drugs for the treatment of atrial fibrillation: efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation. 2004;110:1717–1724. doi: 10.1161/01.CIR.0000143050.22291.2E. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein RN, Khrestian C, Carlsson L, Waldo AL. Azd7009: a new antiarrhythmic drug with predominant effects on the atria effectively terminates and prevents reinduction of atrial fibrillation and flutter in the sterile pericarditis model. J Cardiovasc Electrophysiol. 2004;15:1444–1450. doi: 10.1046/j.1540-8167.2004.04354.x. [DOI] [PubMed] [Google Scholar]
- 25.Wettwer E, Hala O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- 26.Olson TM, Alekseev AE, Liu XK, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 27.Wang ZG, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes: evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- 28.Amos GJ, Wettwer E, Metzger F, et al. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996;491(Pt 1):31–50. doi: 10.1113/jphysiol.1996.sp021194. [DOI] [PMC free article] [PubMed] [Google Scholar]