

Abstract
The effects of estrogen on gene expression in mammary cells are mediated by interaction of the estrogen receptor (ER) with estrogen response elements in target DNA. Whereas the ER is the primary initiator of transcription, the recruitment of coregulatory proteins to the DNA-bound receptor influences estrogen responsiveness. To better understand how estrogen alters gene expression, we identified proteins associated with the DNA-bound ERα. Surprisingly, the antioxidant enzyme Cu/Zn superoxide dismutase (SOD1), which is known primarily as a scavenger of superoxide, was associated with the DNA-bound receptor. We have now demonstrated that SOD1 interacts with ERα from MCF-7 cell nuclear extracts and with purified ERα and that SOD1 enhances binding of ERα to estrogen response element-containing DNA. Although SOD1 decreases transcription of an estrogen-responsive reporter plasmid in transiently transfected U2 osteosarcoma cells, RNA interference assays demonstrate that SOD1 is required for effective estrogen responsiveness of the endogenous pS2, progesterone receptor, cyclin D1, and Cathepsin D genes in MCF-7 breast cancer cells. Furthermore, ERα and SOD1 are associated with regions of the pS2 and progesterone receptor genes involved in conferring estrogen-responsive gene expression. Interestingly, when MCF-7 cells are exposed to 17β-estradiol and superoxide generated by addition of potassium superoxide (KO2) to the cell medium, SOD1 levels are increased and tyrosine nitration, which is an indicator of oxidative stress-induced protein damage, is significantly diminished. Our studies have identified a new role for SOD1 in regulating estrogen-responsive gene expression and suggest that the 17β-estradiol- and KO2-induced increase in SOD1 may play a role in the survival of breast cancer cells and the progression of mammary tumors.
EUKARYOTIC CELLS PRODUCE reactive oxygen species (ROS) as by-products of normal cellular metabolism. The levels of ROS (superoxide, hydrogen peroxide, and hydroxyl radical) fluctuate in cells and can accumulate with increased metabolism or exposure to UV light, ionizing radiation, or chemotherapeutic agents (1,2). Accumulation of ROS can damage proteins, lipids, and DNA and has been linked to age-related degeneration and disease (1,3,4). It has been estimated that a single rat liver mitochondrion generates 3 × 107 superoxide anions/d (5). Thus, in the face of extensive daily superoxide production, the proteins involved in oxidative stress response must function effectively to sustain cell viability and avoid the cellular damage, disease, and death that can ensue if ROS are not appropriately dissipated.
The first line of defense against ROS-induced damage is the conversion of superoxide to hydrogen peroxide by the antioxidant enzyme superoxide dismutase (SOD). The two forms of cellular SOD contain no significant amino acid homology (6) and include Cu/Zn SOD (SOD1), which has largely been described as a cytoplasmic protein, and Mn SOD (SOD2), which is localized in the mitochondria (7). Regardless of their location, the function of these two proteins is to dismutate superoxide to generate hydrogen peroxide. If SOD is ineffective in decreasing superoxide levels, the accumulated superoxide can react with cellular nitric oxide and generate the more toxic peroxynitrite, which in turn initiates nitration of tyrosine residues in cellular proteins and alteration in protein function (8,9). If SOD effectively converts superoxide to hydrogen peroxide and the hydrogen peroxide comes into contact with ferrous iron, hydroxyl radical is formed and can cause extensive damage to cellular macromolecules. Just as SOD is needed to reduce superoxide levels in cells,catalase and glutathione and thioredoxin perioxidases are required to convert hydrogen peroxide to water and molecular oxygen. The collaborative actions of these antioxidant enzymes in regulating the various forms of ROS help to limit oxidative stress in cells.
Many transcription factors containing zinc fingers are susceptible to the deleterious effects of oxidative stress. Oxidation of the cysteine thiol group initiates the release of the coordinating zinc molecule, destroys secondary structure, and eliminates the ability of zinc finger proteins to bind to DNA (10). For example, nuclear receptors and Sp1, which possess zinc fingers that interact with DNA, are susceptible to oxidation and display significantly reduced DNA binding after exposure to oxidizing agents (11,12,13,14,15,16).
Estrogen receptor α (ERα) is a ligand-activated transcription factor responsible for mediating the effects of estrogen in target cells. ERα possesses two zinc fingers, the second of which is particularly vulnerable to oxidation (12,14,16). The biological effects of estrogen are initiated by binding of hormone to the receptor and interaction of the zinc finger region of the receptor with specific DNA sequences, estrogen response elements (EREs), residing in target genes. The interaction of ERα with DNA and the recruitment of numerous coregulatory proteins to the DNA-bound receptor lead to changes in gene expression.
To better understand how ERα regulates transcription of estrogen-responsive genes, we developed an agarose-based gel mobility shift assay to isolate proteins associated with the DNA-bound receptor, which has been described in previous publications (16,17,18,19,20). This method utilizes full-length ERα and endogenously expressed nuclear proteins and takes into consideration DNA- and ligand-induced changes in receptor conformation. An important aspect of this approach is that it segregates proteins on the basis of their abilities to associate with the ERE-bound receptor. Using this method, we isolated a number of proteins involved in DNA repair, oxidative stress response, protein degradation, transcription regulation, signal transduction, and translation initiation. Thus far we have shown that a number of these proteins interact with ERα and influence estrogen-responsive gene expression (16,17,18,19,20,21,22,23). Because we had identified a number of proteins involved in oxidative stress response and we had already determined that one of these proteins, protein disulfide isomerase, influenced estrogen responsiveness and functioned as a molecular chaperone for ERα, we were interested in determining whether another of these ERα-associated proteins involved in oxidative stress response, Cu/Zn superoxide dismutase (SOD1), might affect the activity of ERα. Herein we define the effect of SOD1 on estrogen-responsive gene expression and examine the role of 17β-estradiol (E2) and oxidative stress on SOD1 expression in breast cancer cells.
RESULTS
SOD1 Is Present in Nuclear Extracts of Cultured Cells
It is well established that SOD1 is highly expressed in the cytoplasm and, because of this, it has sometimes been referred to as cytoplasmic SOD (6). However, because we had identified SOD1 as a nuclear protein associated with the ERE-bound ERα, we first examined the expression of SOD1 in nuclear extracts prepared from a number of cultured human cell lines to determine whether SOD1 was present. As shown in Fig. 1, SOD1 was present in nuclear extracts from ERα-positive breast cancer cells (MCF-7) and ERα-negative cervical (HeLa), breast cancer (MDA-MB-231), and osteosarcoma (U2OS) cells. These findings are consistent with other studies that have identified SOD1 as a nuclear protein (24,25,26). The level of Sp1 in each nuclear extract was also examined to ensure that similar amounts of nuclear extracts were loaded.
Figure 1.
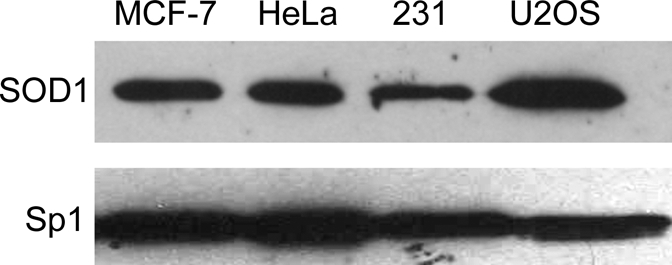
Expression of SOD1 in Cultured Cell Lines
The levels of SOD1 were examined in nuclear extracts (8 μg) from human ERα-positive breast (MCF-7) and ERα-negative cervical (HeLa), breast (MDA-MB-231) and osteosarcoma (U2OS) cells. Samples were fractionated on SDS-PAGE and subjected to Western analysis with an SOD1-specific antibody. Sp1 levels were also monitored to demonstrate that lanes had similar amounts of nuclear extract.
SOD1 Interacts with ERα
Because SOD1 was present in MCF-7 nuclear extracts, we determined whether it could interact with endogenously expressed ERα from these cells. MCF-7 nuclear extracts were incubated with immobilized glutathione-S-transferase (GST) or GST-SOD1. Endogenously expressed ERα from MCF-7 nuclear extracts bound to the immobilized GST-SOD1 in the absence and in the presence of E2 (Fig. 2A, lanes 4 and 6), but was unable to bind to the immobilized GST alone regardless of hormone exposure (lanes 3 and 5).
Figure 2.

Interaction of GST-SOD1 with ERα
GST or GST-SOD1 was immobilized on glutathione-sepharose beads and incubated with (A) nuclear extracts from MCF-7 cells that had been treated with ethanol (−) or E2 (+) or (B) purified ERα in the absence (−) or presence (+) of E2. Specifically bound proteins were eluted and subjected to Western analysis with an ERα-specific antibody. Ten percent of the MCF-7 nuclear extract or purified ERα included in each binding reaction is shown for comparison.
Although SOD1 bound to ERα from MCF-7 nuclear extracts, it was not clear whether this was a direct ERα-SOD1 interaction or whether other nuclear proteins were required. Thus, we assessed the ability of these two proteins to interact directly using purified SOD1 and ERα. Purified ERα bound directly to GST-SOD1 in the absence and in the presence of E2 (Fig. 2B, lanes 3 and 5), but failed to effectively interact with GST alone (lanes 2 and 4). These combined experiments demonstrate that SOD1 interacts with endogenously expressed ERα from MCF-7 cells and with purified ERα.
SOD1 Influences ERα-ERE Complex Formation
The interaction of SOD1 with ERα led us to investigate whether SOD1 might influence ERα-ERE complex formation. Gel mobility shift assays were carried out with 32P-labeled, ERE-containing oligos and purified ERα. Inclusion of increasing amounts of purified SOD1 elicited a dose-dependent increase in ERα-ERE complex formation (Fig. 3, lanes 3–8), but did not alter the migration of the protein-DNA complex. An ERα-specific antibody (lane 9) was able to supershift the receptor-DNA complex, indicating that ERα was present, but a SOD1-specific antibody (lane 10) failed to alter the migration of the receptor-DNA complex. The fact that neither purified SOD1 nor an SOD1 antibody altered migration of the ERα-ERE complex suggests that although SOD1 enhanced the receptor-DNA interaction, it was not present in this complex. The inability of SOD1 to form a trimeric complex with the DNA-bound receptor could indicate that other nuclear proteins are required to stabilize the complex formed with the purified ERα and SOD1 and/or that the trimeric complex is unable to withstand the extensive period of electrophoresis required for gel mobility shift assays. There are reports of other proteins that increase the interaction of a nuclear receptor with its cognate DNA-binding site, but are not present in the receptor-DNA complex (18,21,22,23,27,28,29). In contrast to these findings with GST-SOD1, GST alone had no effect on the ERα-ERE interaction (data not shown).
Figure 3.

Effect of SOD1 on the ERα-ERE Interaction
Radiolabled oligos containing the consensus ERE were incubated alone (lane 1) or with 50 fmol of baculovirus-expressed purified ERα (lanes 2–10). Purified SOD1 (lanes 3–10) and ERα (lane 9) or SOD1 (lane 10)-specific antibody (Ab) were added as indicated. Samples were fractionated on a nondenaturing polyacrylamide gel and subjected to autoradiography. Results are representative of three independent experiments.
SOD1 Influences Estrogen-Mediated Transcription
The fact that SOD1 interacted with ERα and enhanced the receptor-DNA interaction suggested that SOD1 might influence ERα-mediated gene expression. To determine whether this was the case, U2OS cells were transiently transfected with a luciferase reporter plasmid containing two EREs, a constant amount of ERα expression vector, and increasing amounts of an SOD1 expression vector. A Renilla luciferase expression vector was also used as an internal control. Compiled data from three independent experiments demonstrated that addition of 1 or 5 μg of SOD1 expression vector dramatically decreased transcription of the estrogen-responsive reporter plasmid (Fig. 4).
Figure 4.

Effect of Increasing SOD1 Expression on ERαMediated Transcription
Transient transfections were performed in U2OS cells with an ERα expression vector, a luciferase reporter plasmid containing two consensus EREs, and increasing amounts of an SOD1 expression vector. Cells were treated with ethanol vehicle (light gray bars) or 10 nm E2 (dark gray bars) as indicated. Luciferase activity, which was corrected for transfection efficiency, is shown as relative luciferase units (RLU). Data from three independent experiments, which had been performed in duplicate, were combined and analyzed by ANOVA. A significant decrease in transcription in response to the SOD1 expression vector is indicated by an asterisk (*, P ≤ 0.05).
SOD1 Influences Transcription of Endogenous Estrogen-Responsive Genes
Because a supercoiled ERE-containing reporter plasmid might not necessarily reflect what occurs with an endogenous gene in its native chromatin environment, the expression of endogenous, estrogen-responsive genes was examined in MCF-7 cells using RNA interference experiments. MCF-7 cells were transfected with control small interfering RNA (siRNA), which targeted Renilla luciferase, or SOD1-specific siRNA duplex, which targeted the endogenously expressed SOD1 mRNA.
SOD1 siRNA, but not control siRNA, effectively reduced SOD1 protein and mRNA levels (Fig. 5, A and B). The increased pS2, progesterone receptor (PR), cyclin D1, and Cathepsin D mRNA levels in the presence of E2 and control siRNA were anticipated because the estrogen-responsiveness of these genes has been reported previously (30,31,32,33).
Figure 5.

Effect of Knocking Down Endogenous SOD1
MCF-7 cells were transfected with 50 pmol of control or SOD1-specific siRNA and treated with ethanol vehicle (light gray bars) or 10 nm E2 (dark gray bars) for 24 h and processed for protein or mRNA analysis. A, Protein samples were subjected to Western analysis with an SOD1-specific antibody. B, RNA was isolated and cDNA was synthesized for quantitative RT-PCR analysis with primers specific to SOD1, pS2, PR, cyclinD1, Cathepsin D and 36B4 (internal control) mRNA sequences. One of three independent experiments, each of which was performed in triplicate, is shown. ANOVA was used to detect significant differences in mRNA levels in response to E2 (*, P ≤ 0.05) or in response to SOD1-specific siRNA (#, P ≤ 0.05).
When SOD1 expression was knocked down with the SOD1-specific siRNA, the estrogen-induced increase in pS2, PR, cyclin D1, and Cathepsin D mRNA levels was significantly decreased. In contrast, 36B4 mRNA levels were unaffected by E2 or the SOD1 siRNA. These studies demonstrate that SOD1 is required for effective activation of estrogen-responsive genes.
SOD1 Is Associated with Endogenous Estrogen-Responsive Genes
Because SOD1 influenced the estrogen responsiveness of the pS2 and PR genes, we determined whether ERα and SOD1 associated with these two endogenous genes in MCF-7 cells. The association of ERα with the ERE-containing region of the pS2 gene has been reported by our laboratory and others (16,17,18,34,35). We have also shown that the +90 activator protein 1 (AP-1) and +571 ERE/Sp1 sites in the PR gene are associated with ERα using chromatin immunoprecipitation (ChIP) assays and are involved in conferring estrogen responsiveness in MCF-7 cells (36,37,38,39). Significantly more ERα (Fig. 6A) and SOD1 (Fig. 6B) were associated with these pS2 and PR gene regions in the presence than in the absence of E2. Thus, in addition to our siRNA experiments, which showed that SOD1 altered estrogen-responsive gene expression, these ChIP assays demonstrated that SOD1 associates with the regions of endogenous, estrogen-responsive genes involved in conferring hormone responsiveness. In contrast, there was no change in the association of ERα or SOD1 with the 36B4 gene or a region 3.5 kb upstream of the pS2 ERE, neither of which contains any apparent ERα-binding sites (data not shown).
Figure 6.

Association of SOD1 with Endogenous Estrogen-Responsive Genes
MCF-7 cells were treated with ethanol vehicle (light gray bars) or 10 nm E2 (dark gray bars) for 24 h and subjected to ChIP analysis with an (A) ERα- or (B) SOD1-specific antibody. Quantitative real-time PCR was used to examine the association of ERα and SOD1 with the ERE-containing region of the pS2 gene and the + 90 AP-1 and +571 ERE/Sp1 sites in the PR gene. Data are presented as the number of copies of each estrogen-responsive gene region pulled down relative to the number of copies of 36B4 gene region pulled down (occupancy units). A significant increase in the association of ERα and SOD1 with these genes in the presence of E2 is indicated (*, P ≤ 0.05).
Expression of SOD1 in MCF-7 Cells
We had noted what appeared to be an increase in SOD1 mRNA and protein levels when MCF-7 cells were treated with E2 (Fig. 5, A and B). To determine whether SOD1 expression was increased in MCF-7 cells that had been exposed to E2, immunocytochemistry was performed. As seen in Fig. 7A, treatment of MCF-7 cells with E2 increased SOD1 expression. An even greater increase in SOD1 expression was produced by exposure of MCF-7 cells to superoxide, which was generated by addition of KO2 to the medium (40,41). The greatest increase in SOD1 expression was observed when MCF-7 cells were exposed to both E2 and KO2. The differences in SOD1 expression were even more apparent in the pseudocolored images, in which the intensity of staining is reflected in the color of each pixel with red representing the most highly stained regions and black representing areas with little staining (Fig. 7B). Compiled data from three independent experiments demonstrated that E2 and superoxide increased SOD1 expression 24- and 109-fold, respectively, and that combined E2 and superoxide synergistically enhanced SOD1 expression 258-fold (Fig. 7C).
Figure 7.

Effect of E2 and KO2 on SOD1 Expression in MCF-7 Cells
A, MCF-7 cells were treated with ethanol vehicle or 10 nm E2 for 24 h. Potassium superoxide (KO2) was used to generate superoxide. Cells were exposed to KO2 for 1 h, media were removed, and samples were subjected to immunocytochemisty with an SOD1-specific antibody. B, The images shown in panel A were pseudocolored so that red corresponds to the highest intensity staining and black corresponds to the lowest intensity staining. C, The intensity of staining in 12 different fields (250–300 cells per field) from three independent experiments was analyzed by ANOVA. Each of the E2- and/or KO2-treated cells had significantly more SOD1 than cells that had been exposed to neither E2 nor KO2 (P ≤ 0.05).
The interaction of superoxide with cellular nitric oxide generates peroxynitrite, which in turn initiates nitration of tyrosine residues and alterations in protein structure and function (8,9,42). Thus, the accumulation of nitrotyrosines can be used as an indicator of superoxide-induced protein damage (43). To determine whether the E2- and/or KO2-mediated increases in SOD1 expression in MCF-7 cells altered tyrosine nitration, the level of nitrotyrosine was monitored in MCF-7 cells that had or had not been exposed to E2 and/or superoxide. The presence of nitrotyrosine in MCF-7 cells that had not been exposed to hormone or oxidative stress (Fig. 8A) likely reflects the effects of superoxide produced during basal cell metabolism. The decreased nitrotyrosine levels in MCF-7 cells that had been treated with E2 are consistent with the increased SOD1 expression observed in E2-treated cells. However, when MCF-7 cells were pretreated with E2 and exposed to KO2, which produced the greatest increase in SOD1 expression, the level of nitrotyrosine was dramatically reduced. The pseudocolored images highlight the combined effects of E2 and superoxide on tyrosine nitration (Fig. 8B). Data from four independent experiments demonstrated that concomitant exposure of MCF-7 cells to E2 and superoxide reduced nitrotyrosine levels 14-fold (Fig. 8C). Our studies suggest that the E2- and KO2-induced increases in SOD1 help to limit peroxinitrite-induced protein damage in MCF-7 breast cancer cells.
Figure 8.

Effect of E2 and KO2 on Nitrotyrosine Levels in MCF-7 Cells
A, MCF-7 cells were treated as described in Fig. 7 and subjected to immunocytochemistry with a nitrotyrosine-specific antibody. B, The images shown in panel A were pseudocolored so that red corresponds to the highest intensity staining and black corresponds to the lowest intensity staining. Images are representative of four independent experiments. C, The intensity of staining in 16 different fields (250–300 cells per field) from four independent experiments was analyzed by ANOVA. Cells that had been exposed to E2 and KO2 had significantly lower nitrotyrosine levels than cells that had been exposed to neither E2 nor KO2 (P ≤ 0.05).
DISCUSSION
SOD1 has been extensively characterized as a cytoplasmic protein responsible for the conversion of superoxide to hydrogen peroxide (6). We have now identified a new role for SOD1 in the nucleus in regulating gene expression. We show that SOD1 interacts with ERα, associates with ERα at endogenous estrogen-responsive genes, and is required for effective activation of estrogen-responsive genes.
Role of SOD1 in Regulating Gene Expression
The original intent of this study was to determine whether SOD1 influenced estrogen-responsive gene expression. Although transient transfection experiments demonstrated that increased expression of SOD1 decreased transcription of an ERE-containing reporter plasmid, RNA interference experiments established the role of SOD1 in enhancing activation of endogenous, estrogen-responsive genes. Thus, whereas transient transfection assays provided evidence that SOD1 influences ERα-mediated transcription, the supercoiled reporter plasmid with a simple promoter was unable to recapitulate the full spectrum of environmental cues required for regulating endogenous, estrogen-responsive genes.
We have considered potential mechanisms by which SOD1 might enhance ERα-mediated gene expression. First, SOD1 may influence transcription directly by interacting with ERα and stabilizing the receptor-DNA complex. Second, SOD1 could influence estrogen-responsive gene expression indirectly by helping to protect nuclear proteins from oxidative damage. A number of studies have shown that oxidation of ERα decreases its ability to bind to ERE-containing DNA and that reducing agents or proteins involved in oxidative stress response can partially or fully restore the receptor's functional activity (10,12,16). Like ERα, Sp1 is sensitive to oxidative stress, is unable to bind to DNA when oxidized, and is involved in regulating a number of estrogen-responsive genes (10,44,45). By limiting oxidative stress, SOD1 may help to maintain the structural integrity of ERα, Sp1, and other coregulatory proteins involved in modulating estrogen-responsive gene expression. Finally, in addition to its more immediate effects on protein structure, SOD1 may also have long-term effects on cell function by helping to limit DNA damage.
One serendipitous observation from our RNA interference experiments was that SOD1 mRNA and protein levels were increased when MCF-7 cells were treated with E2. The E2-mediated increase in SOD1 expression is most likely due to the interaction of the E2-occupied receptor with an ERα-binding site, which is far removed (∼135 kb) from the SOD1 transcription start site (46). The association of RNA polymerase II and the coregulatory protein amplified in breast cancer 1 with this regulatory region provides additional evidence that the site is functional.
Effect of Oxidative Stress Proteins on Estrogen Responsiveness
We recently characterized the effects of another protein involved in oxidative stress response, protein disulfide isomerase (PDI), which functions as a molecular chaperone for ERα and influences ERα-mediated transactivation (16). In addition to interacting with ERα, PDI interacts with SOD1 and helps to prevent SOD1 misfolding, which in turn limits protein aggregation involved in neurodegenerative disease and cell death (1,2,3,16,47). The ability of PDI to maintain the structural integrity of SOD1 may allow these two redox regulators to work in concert, presumably with other oxidative stress proteins, to maintain protein structure. The association of SOD1 and PDI with ERα and with each other suggests that these two oxidative stress proteins may cooperate to maintain ERα structure and function and ensure that estrogen-responsive genes are appropriately regulated.
Biological Effects of SOD1
As might be expected by its effects at the molecular and cellular levels, SOD1 plays a role in a number of physiological processes. Given its ability to augment estrogen-responsive gene expression, it is not surprising that the reproductive function of SOD1-null mice would be adversely affected. Female SOD1-null mice have implantation defects and decreased fertility (26,48). The increased susceptibility of motor neurons in SOD1-null mice to oxidative stress (49) and the reduced brain injury in transgenic mice overexpressing SOD1 after an ischemic event (50) demonstrate the critical role of SOD1 in neuronal function. Furthermore, mutations in SOD1 account for 10–20% of the cases of the familial form of amyotrophic lateral sclerosis or Lou Gehrig's disease (51,52). The increased vulnerability of the myocardium of SOD1-null mice to ischemia-reperfusion injury provides evidence that SOD1 also plays a role in cardiac function (53).
It has been suggested that oxidative stress is involved in age-related degeneration and that ROS-mediated damage over time may be the cause, not a side effect, of aging (54,55). The decrease in SOD1 activity that occurs with aging may contribute to increased oxidative stress and incidence of disease (1,3,4).
SOD1 and Breast Cancer
One disease that significantly increases with age is breast cancer. Oxidative damage of DNA has been implicated in initiation of mammary tumors (56,57,58,59) and can alter the activity of proteins involved in regulating transcription. The ability of ERα to bind to DNA is compromised in nearly one third of all ERα-positive mammary tumors (13) and the ability of Sp1 to bind to DNA decreases with age (44). Thus, the oxidative stress that occurs with aging could lead to oxidation of ERα and Sp1, both of which are required for PR gene expression, and could result in the increased incidence of ERα-positive/PR-negative mammary tumors observed in women over 50 yr of age (44).
In addition to its potential effects on mammary tumorigenesis (60), SOD1 may also play a role in sustaining mammary tumors. The increased resistance of breast cancer cells to oxidative stress compared with normal mammary cells has been attributed, in part, to the increased activity of SOD1 in breast cancer cells (60,61). Our studies indicate that concomitant exposure of MCF-7 breast cancer cells to oxidative stress and E2 dramatically increases SOD1 expression and decreases tyrosine nitration. This increased expression of SOD1 in breast cancer cells induced by ROS combined with the exposure of these cells to circulating E2 may protect breast cancer cells from the detrimental effects of oxidative stress. Thus, E2 could enhance the survival of breast cancer cells that have left the primary tumor, but are not yet supported by an established blood supply, by enhancing SOD1 expression and allowing the colonizing cells to persist in spite of increased oxidative stress and limited vascular support.
MATERIALS AND METHODS
Isolation and Identification of SOD1
HeLa nuclear extracts were combined with 32P-labeled oligos containing a consensus ERE in the absence or presence of baculovirus-expressed, purified ERα. Agarose gel mobility shift assays were used to isolate proteins associated with the ERE-bound ERα, and these proteins were identified using mass spectrometry as previously described (16,17). Nine peptides with amino acid sequence identical to SOD1 were identified (AVCVLKGDGPVQGIINFEQK, DGVADVSIEDSVISLSGDHCIIGR, HVGDLGNVTADKNGVADV, GNGPVQGIINFEQKESNGPVKVWGSIK, TLVVHEKADDLGKGGNEESTK, AVCVLKGDGPVQGIINFEQKESNGPVKVWGSIK, GLTEGLHGFHVHEFGDNTAGCTSAGPHFNPL, HVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGR, LACGVIGIAQ). Together, these peptides account for 86% of the total SOD1 amino acid sequence.
Western Blot Analysis
Nuclear extracts from human osteosarcoma (U2OS), cervical (HeLa), and breast cancer (MCF-7 and MDA-MB-231) cells were prepared as described previously (62), and 8 μg of nuclear proteins were fractionated on a 15% sodium dodecyl sulfate polyacrylamide gel, transferred to a nitrocellulose membrane, and blotted with a SOD1-, ERα-, or Sp1-specific antibody (sc-17767, sc-8002, sc-59, respectively; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Blots were probed with a horseradish peroxidase-conjugated secondary antibody and detected by the SuperSignal West Femto Maximum Sensitivity Substrate chemiluminescent system (Pierce Chemical Co., Rockford, IL).
Transient Transfections
U2OS cells were maintained in phenol red-containing MEM supplemented with 10% fetal bovine serum. Cells were transferred to phenol red-free MEM supplemented with 5% charcoal dextran-treated calf serum 2 d before plating, seeded in 24-well plates, and transfected with 1 ng thymidine kinase-Renilla reporter plasmid, 5 ng cytomegalovirus-human ERα expression vector and 1 μg of a 2 ERE-thymidine kinase-luciferase reporter plasmid containing two copies of the consensus ERE (kindly provided by B. Katzenellenbogen, University of Illinois, Urbana, IL). Increasing amounts of the pcDNA3.1-SOD1 expression vector [kindly provided by M. Tortarolo, Istituto di Ricerche Farmacologiche, Milano, Italy (63)] were added as indicated. The parental expression vector pcDNA3.1 was included to maintain the total DNA concentration in each well. A constitutively active Renilla luciferase expression vector was also included to normalize for differences in transfection efficiency. Cells were treated with ethanol vehicle or 10 nm E2 for 24 h. Luciferase assays were performed using the Dual Luciferase Assay system (Promega Corp., Madison, WI).
Pull-Down Assays
GST-tagged SOD1 [a gift from C. Das, Brandeis University, Waltham, MA (64)] and pGEX-2T-GST (GE Healthcare, Piscataway, NJ) were expressed in BL21(DE3) pLysS cells (Invitrogen, Carlsbad, CA) and immobilized on glutathione-sepharose 4B beads that had been washed four times with TE (20 mm Tris, pH 7.4; 0.1 mm EDTA) containing 100 mm NaCl at 4 C. Samples were rotated in this same buffer for 1 h at 4 C. Nuclear extract (35 μg) or purified ERα (200 fmol) was added as indicated and rotated at 4 C for 1 h with ethanol vehicle or 10 nm E2. Samples were washed three times with TE containing 150 mm NaCl and 0.5% Nonidet P-40, and proteins were eluted with 3 mg glutathione. Samples were separated by SDS-PAGE and subjected to Western analysis.
Gel Mobility Shift Assays
Baculovirus-expressed purified ERα (50 fmol), which had been isolated as described (30), was incubated without or with increasing amounts of purified SOD1 in binding buffer (15 mm Tris, pH 7.9; 0.2 mm EDTA; 20 mm KCl; 4 mm dithiothreitol; 10% glycerol; and 50 ng polydeoxyinosinic deoxycytidylic acid) for 10 min at 4 C. 32P-labeled 50-bp oligos containing a consensus ERE (20,000 cpm) were added to all samples and incubated for 10 additional min at 25 C in a final volume of 20 μl. ERα and SOD1-specific antibodies were then added to the reactions as indicated and incubated for 10 min at 4 C. BSA was included to maintain constant protein concentrations. Samples were run on 6% nondenaturing polyacrylamide gels in low ionic strength buffer (65) and subjected to autoradiography.
Small Interfering RNA (siRNA) Experiments
MCF-7 cells were maintained in phenol red-containing MEM with 5% calf serum and transferred to phenol red-free media 2 d before plating. Cells were seeded in 12-well plates 24 h before transfection with siLentFect (Bio-Rad Laboratories, Inc., Hercules, CA) and 50 pmol of SOD1-specific or control siRNA directed against Renilla luciferase (catalog nos. 16708 or 4630, respectively; Ambion, Inc., Austin, TX) in phenol red-free media in the absence of antibiotics for 24 h. Cells were then exposed to ethanol vehicle or 10 nm E2 for 24 h, harvested with 40 mm Tris (pH 7.5), 1.5 mm EDTA, and 140 mm NaCl, lysed in 20 mm Tris (pH 7.9), 1 mm EDTA, 200 mm NaCl, and 0.2% Nonidet P-40 and subjected to Western blot analysis. RNA was isolated using TRIzol (Invitrogen) and processed according to the manufacturer's instructions. cDNA was prepared using the Reverse Transcription System (Promega) and real-time PCR was performed using iQ SYBR Green Supermix and the iCycler PCR thermocycler (Bio-Rad). Standard curves were derived using cDNA equivalents of 0.02, 0.2, 2, and 20 ng of RNA and were run in duplicate for each primer set in each experiment.
ChIP Assays
MCF-7 cells were treated with ethanol vehicle or 10 nm E2 for 24 h and cross-linked with 1% formaldehyde. Cells were processed essentially as described (20). An ERα-(sc-8002)- or SOD1-(sc-8636)-specific antibody (Santa Cruz Biotechnology) was used to immunoprecipitate protein-DNA complexes. PCR primers specific to the pS2, +90 AP-1, and +571 ERE/Sp1 sites were used (16). Standard curves were generated using 1,000, 5,000, 10,000, 50,000, and 100,000 copies of each gene for each primer set in each experiment and subjected to real-time PCR using iQ SYBR Green Supermix and the iCycler PCR thermocycler.
Immunofluorescence
MCF-7 cells were plated on coverslips in six-well plates containing phenol red-free MEM with 5% charcoal-dextran-treated calf serum. Cells were treated with ethanol or 10 nm E2 for 24 h and then exposed to 0.5 μg/ml KO2, which generates 0.49 nmol of superoxide/ml for 1 h as previously described (41). The amount of KO2 used was carefully titrated so that cell death was not more than 10%. Samples were washed with PBS, fixed in PBS with 4% formaldehyde and 200 mm dextrose for 10 min, washed with PBS, permeabilized with PBS containing 0.2% Triton X-100 for 20 min, washed with PBS containing 0.1% Tween 20, and blocked with PBS containing 2% BSA and 2% fetal bovine serum, 0.1% Tween 20, and 0.02% NaN3 for 30 min. Cells were then incubated with SOD1 (sc11407, Santa Cruz Biotechnology)- or nitrotyrosine (06-284, Upstate Biotechnology, Lake Placid, NY)-specific antibodies for 1.5 h in a humidified chamber. Cells were washed with PBS containing 0.1% Tween 20 and incubated with donkey antirabbit fluorescein-conjugated antibody (711-096-152, Jackson ImmunoResearch, West Grove, PA) for 30 min, washed with PBS containing 0.1% Tween 20, mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA), and visualized on a Leica DM 2500 (Leica Microsystems, Inc., Bannockburn, IL) using a 20X objective. Images were obtained from QImaging Retiga 2000R (QImaging, Burnaby, British Columbia, Canada) using QCapture Pro software and black balanced using Adobe Photoshop CS2 software (Adobe Systems, Inc., San Jose, CA). For quantitation, original images were converted into 8-bit grayscale images using Adobe Photoshop CS2 software and a look up table developed by Frank F. Bartol [Auburn University, Auburn, AL (66)] was applied using ImageJ++ software (67) as described elsewhere (68). The look up table uses pixel data to assign each pixel to one of eight color categories (red, dark orange, light orange, yellow, green, light blue, blue, and black) based on the intensity of the staining. Red pixels indicate areas with a high intensity of immunostaining, and black pixels indicate areas of little or no immunostaining. The computer-generated color assignments provide uniform analysis across fields and eliminate user bias. Adobe Photoshop CS2 software was used to count the number of pixels of each color. Positive pixels were defined as pixels assigned to the top quartile of staining intensity. The number of cells counted in each of the 12 (SOD1) or 16 (nitrotyrosine) fields from three or four independent experiments ranged from 250–300 cells per field. Results are expressed as the number of positive pixels per cell.
Acknowledgments
We thank Lori Raetzman for the use of the imaging system and her expert advice; Kjirsten Walt and Rupesh Gupta for guidance in quantitating SOD1 and nitrotyrosine levels; and Carol Curtis for helpful discussions.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01 DK 53884 (to A.M.N.) and NIH Grant P41 RR11823-10 (to J.R.Y.).
Disclosure Summary: The authors have nothing to declare.
First Published Online February 7, 2008
Abbreviations: AP-1, Activator protein 1; ChIP, chromatin immunoprecipitation; E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen response element; GST, glutathione S-transferase; PR, progesterone receptor; RNAi, RNA interference; ROS, reactive oxygen species; siRNA, small interfering RNA; SOD, superoxide dismutase; SOD1, Cu/Zn SOD.
References
- Finkel T, Holbrook NJ 2000 Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247 [DOI] [PubMed] [Google Scholar]
- Schröder P, Krutmann J 2005 Environmental oxidative stress—environmental sources of ROS. In: The handbook of environmental chemistry. Berlin/Heidelberg: Springer; 19–31 [Google Scholar]
- Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M 2005 Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci USA 102:9754–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM 2002 Superoxide dismutase in aging and disease: an overview. Methods Enzymol 349:331–341 [DOI] [PubMed] [Google Scholar]
- Richter C 1995 Oxidative damage to mitochondrial DNA and its relationship to aging. In: Esser K, Martin G, eds. Molecular aspects of aging. Chichester, UK: John Wiley & Sons; 99–105 [Google Scholar]
- Zelko IN, Mariani TJ, Folz RJ 2002 Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33:337–349 [DOI] [PubMed] [Google Scholar]
- Fridovich I 1995 Superoxide radical and superoxide dismutases. Annu Rev Biochem 64:97–112 [DOI] [PubMed] [Google Scholar]
- Squadrito GL, Pryor WA 1998 Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med 25:392–403 [DOI] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH 1993 ALS, SOD and peroxynitrite. Nature 364:584 [DOI] [PubMed] [Google Scholar]
- Webster KA, Prentice H, Bishopric NH 2001 Oxidation of zinc finger transcription factors: physiological consequences. Antioxid Redox Signal 3:535–548 [DOI] [PubMed] [Google Scholar]
- Makino Y, Okamoto K, Yoshikawa N, Aoshima M, Hirota K, Yodoi J, Umesono K, Makino I, Tanaka H 1996 Thioredoxin: a redox-regulating cellular cofactor for glucocorticoid hormone action. Cross talk between endocrine control of stress response and cellular antioxidant defense system. J Clin Invest 98:2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Hajiro-Nakanishi K, Makino Y, Eguchi H, Yodoi J, Tanaka H 1997 Functional modulation of estrogen receptor by redox state with reference to thioredoxin as a mediator. Nucleic Acids Res 25:4035–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Lu B, Scott GK, Chang CH, Baldwin MA, Benz CC 1998 Oxidant stress impaired DNA-binding of estrogen receptor from human breast cancer. Mol Cell Endocrinol 146:151–161 [DOI] [PubMed] [Google Scholar]
- Whittal RM, Benz CC, Scott G, Semyonov J, Burlingame AL, Baldwin MA 2000 Preferential oxidation of zinc finger 2 in estrogen receptor DNA-binding domain prevents dimerization and, hence, DNA binding. Biochemistry 39:8406–8417 [DOI] [PubMed] [Google Scholar]
- Wu X, Bishopric NH, Discher DJ, Murphy BJ, Webster KA 1996 Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol Cell Biol 16:1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Norton JR, McDonald WH, Yates JR, Nardulli AM 2006 Protein disulfide isomerase serves as a molecular chaperone to maintain estrogen receptor α structure and function. Mol Endocrinol 20:1982–1995 [DOI] [PubMed] [Google Scholar]
- Schultz-Norton JR, Gabisi VA, Ziegler YS, McLeod IX, Yates JR, Nardulli AM 2007 Estrogen receptor α interaction with the DNA repair protein proliferating cell nuclear antigen (PCNA). Nucleic Acids Res 35:5028–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Norton JR, Walt KA, Ziegler YS, McLeod IX, Yates JR, Raetzman LT, Nardulli AM 2007 The DNA repair protein flap endonuclease-1 (FEN-1) modulates estrogen-responsive gene expression. Mol Endocrinol 21:1569–1580 [DOI] [PubMed] [Google Scholar]
- Marzouk S, Schultz-Norton J, McLeod I, Yates J, Nardulli A 2007 Rho GDP dissociation inhibitor α interacts with estrogen receptor α and influences estrogen responsiveness. J Mol Endocrinol 39:249–259 [DOI] [PubMed] [Google Scholar]
- Curtis CD, Likhite VS, McLeod IX, Yates JR, Nardulli AM 2007 Interaction of the tumor metastasis suppressor nonmetastatic protein 23 homologue H1 and estrogen receptor α alters estrogen-responsive gene expression. Cancer Res 67:10600–10607 [DOI] [PubMed] [Google Scholar]
- Loven MA, Muster N, Yates JR, Nardulli AM 2003 A novel estrogen receptor α associated protein, template activating factor I β, inhibits acetylation and transactivation. Mol Endocrinol 17:67–78 [DOI] [PubMed] [Google Scholar]
- Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM 2004 Interaction of estrogen receptor α with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J Biol Chem 279:16875–16882 [DOI] [PubMed] [Google Scholar]
- Loven MA, Davis RE, Curtis CD, Muster N, Yates JR, Nardulli AM 2004 A novel estrogen receptor α-associated protein alters receptor-deoxyribonucleic acid interactions and represses receptor-mediated transcription. Mol Endocrinol 18:2649–2659 [DOI] [PubMed] [Google Scholar]
- Casareno RL, Waggoner D, Gitlin JD 1998 The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem 273:23625–23628 [DOI] [PubMed] [Google Scholar]
- Chang LY, Kang BH, Slot JW, Vincent R, Crapo JD 1995 Immunocytochemical localization of the sites of superoxide dismutase induction by hyperoxia in rat lungs. Lab Invest 73:29–39 [PubMed] [Google Scholar]
- Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM 1998 Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology 139:4008–4011 [DOI] [PubMed] [Google Scholar]
- Landel CC, Kushner PJ, Greene GL 1994 The interaction of human estrogen receptor with DNA is modulated by receptor-associated proteins. Mol Endocrinol 8:1407–1419 [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, Melvin V, Prendergast P, Altmann M, Ronfani L, Bianchi ME, Taraseviciene L, Nordeen SK, Allegretto EA, Edwards DP 1998 High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol Cell Biol 18:4471–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine L, Wood J, Lamia L, Prendergast P, Edwards D, Nardulli A 1998 The high mobility group protein 1 enhances binding of the estrogen receptor DNA binding domain to the estrogen response element. Mol Endocrinol 12:664–674 [DOI] [PubMed] [Google Scholar]
- Kim J, Petz LN, Ziegler YS, Wood JR, Potthoff SJ, Nardulli AM 2000 Regulation of the estrogen-responsive pS2 gene in MCF-7 human breast cancer cells. J Steroid Biochem Mol Biol 74:157–168 [DOI] [PubMed] [Google Scholar]
- Nardulli AM, Greene GL, O'Malley BW, Katzenellenbogen BS 1988 Regulation of progesterone receptor messenger ribonucleic acid and protein levels in MCF-7 cells by estradiol: analysis of estrogen's effect on progesterone receptor synthesis and degradation. Endocrinology 122:935–944 [DOI] [PubMed] [Google Scholar]
- Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, Beato M, Sica V, Bresciani F, Weisz A 1996 17β-Estradiol induces cyclin D1 gene transcription, p36D1–p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene 12:2315–2324 [PubMed] [Google Scholar]
- Westley BR, May FE 1987 Oestrogen regulates cathepsin D mRNA levels in oestrogen responsive human breast cancer cells. Nucleic Acids Res 15:3773–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M 2000 Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843–852 [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F 2003 Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751–763 [DOI] [PubMed] [Google Scholar]
- Petz LN, Nardulli AM 2000 Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol Endocrinol 14:972–985 [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Loven MA, Nardulli AM 2002 Estrogen receptor α and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology 143:4583–4591 [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM 2004 Differential regulation of the human progesterone receptor gene by an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol 88:113–122 [DOI] [PubMed] [Google Scholar]
- Schultz JR, Petz LN, Nardulli AM 2005 Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors α and β. J Biol Chem 280:347–354 [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Lokesh BR 1983 Superoxide anion generated by potassium superoxide is cytotoxic and mutagenic to Chinese hamster ovary cells. Mutat Res 121:299–304 [DOI] [PubMed] [Google Scholar]
- Lokesh BR, Cunningham ML 1986 Further studies on the formation of oxygen radicals by potassium superoxide in aqueous medium for biochemical investigations. Toxicol Lett 34:75–84 [DOI] [PubMed] [Google Scholar]
- Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H 2001 Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci 62:212–220 [DOI] [PubMed] [Google Scholar]
- Gupta RK, Schuh RA, Fiskum G, Flaws JA 2006 Methoxychlor causes mitochondrial dysfunction and oxidative damage in the mouse ovary. Toxicol Appl Pharmacol 216:436–445 [DOI] [PubMed] [Google Scholar]
- Quong J, Eppenberger-Castori S, Moore III D, Scott GK, Birrer MJ, Kueng W, Eppenberger U, Benz CC 2002 Age-dependent changes in breast cancer hormone receptors and oxidant stress markers. Breast Cancer Res Treat 76:221–236 [DOI] [PubMed] [Google Scholar]
- Safe S 2001 Transcriptional activation of genes by 17β-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm 62:231–252 [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M 2005 Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122:33–43 [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK 2006 Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem 281:30152–30165 [DOI] [PubMed] [Google Scholar]
- Ho YS, Gargano M, Cao J, Bronson RT, Heimler I, Hutz RJ 1998 Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J Biol Chem 273:7765–7769 [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown Jr RH, Scott RW, Snider WD 1996 Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13:43–47 [DOI] [PubMed] [Google Scholar]
- Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH 1991 Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA 88:11158–11162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX 1993 Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62 [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I 2002 Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA 99:9010–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Maulik N, Engelman RM, Ho YS, Das DK 2000 Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ Res 86:264–269 [DOI] [PubMed] [Google Scholar]
- Harman D 1956 Aging: a theory based on free radical and radiation chemistry. J Gerontol 11:298–300 [DOI] [PubMed] [Google Scholar]
- Harman D 2001 Aging: overview. Ann NY Acad Sci 928:1–21 [DOI] [PubMed] [Google Scholar]
- Nyaga SG, Lohani A, Jaruga P, Trzeciak AR, Dizdaroglu M, Evans MK 2006 Reduced repair of 8-hydroxyguanine in the human breast cancer cell line, HCC1937. BMC Cancer 6:297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malins DC, Haimanot R 1991 Major alterations in the nucleotide structure of DNA in cancer of the female breast. Cancer Res 51:5430–5432 [PubMed] [Google Scholar]
- Malins DC 1993 Identification of hydroxyl radical-induced lesions in DNA base structure: biomarkers with a putative link to cancer development. J Toxicol Environ Health 40:247–261 [DOI] [PubMed] [Google Scholar]
- Hand RA, Craven RJ 2003 Hpr6.6 protein mediates cell death from oxidative damage in MCF-7 human breast cancer cells. J Cell Biochem 90:534–547 [DOI] [PubMed] [Google Scholar]
- Bianchi MS, Bianchi NO, Bolzan AD 1992 Superoxide dismutase activity and superoxide dismutase-1 gene methylation in normal and tumoral human breast tissues. Cancer Genet Cytogenet 59:26–29 [DOI] [PubMed] [Google Scholar]
- Starcevic SL, Diotte NM, Zukowski KL, Cameron MJ, Novak RF 2003 Oxidative DNA damage and repair in a cell lineage model of human proliferative breast disease (PBD). Toxicol Sci 75:74–81 [DOI] [PubMed] [Google Scholar]
- Wood JR, Likhite VS, Loven MA, Nardulli AM 2001 Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol Endocrinol 15:1114–1126 [DOI] [PubMed] [Google Scholar]
- Tortarolo M, Crossthwaite AJ, Conforti L, Spencer JP, Williams RJ, Bendotti C, Rattray M 2004 Expression of SOD1 G93A or wild-type SOD1 in primary cultures of astrocytes down-regulates the glutamate transporter GLT-1: lack of involvement of oxidative stress. J Neurochem 88:481–493 [DOI] [PubMed] [Google Scholar]
- Ray SS, Nowak RJ, Strokovich K, Brown Jr RH, Walz T, Lansbury Jr PT 2004 An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry 43:4899–4905 [DOI] [PubMed] [Google Scholar]
- Chodosh LA, Buratowski S 1989 Mobility shift DNA-binding assay using gel electrophoresis. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, eds. Current protocols in molecular biology. New York: Greene Publishing Associates and Wiley Interscience; 12.2.1–12.2.10 [Google Scholar]
- Masters RA, Crean BD, Yan W, Moss AG, Ryan PL, Wiley AA, Bagnell CA, Bartol FF 2006 Neonatal porcine endometrial development and epithelial proliferation affected by age and exposure to estrogen and relaxin. Domest Anim Endocrinol 33:335–346 [DOI] [PubMed] [Google Scholar]
- Abramoff MD, Magelhaes, PJ, Ram, SJ 2004 Image processing with Image J. Biophotonics Int 11:36–42 [Google Scholar]
- Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, Flaws JA 2007 The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol Reprod 76:1062–1070 [DOI] [PubMed] [Google Scholar]