

Abstract
A series of carboranylporphyrins containing either amine or phosphonic acid functionalities and two to six closo-carborane clusters have been synthesized via a [2+2] condensation of a dimethylamino- or diethylphosphonate-substituted dipyrromethane with a dicarboranylmethyl-benzaldehyde. The X-ray structures of four key reaction intermediates (1, 2, 3 and 4a) and of two target porphyrins, the diphosphonate ester- and the diamino-tetracarboranylporphyrins 5b and 6a are presented and discussed. In vitro studies using human carcinoma HEp2 and human glioblastoma T98G cells show that these porphyrins are non-toxic in the dark up to 100 μM concentrations, and that a tetracarboranylporphyrin bearing two quaternary ammonium groups is the most efficiently taken up by cells at short times (up to 8 h), followed by a dicarboranylporphyrin bearing three phosphonic acid substituents. All carboranylporphyrins delivered therapeutic amounts of boron to T98G cells and localized mainly within the cell lysosomes.
Keywords: BNCT, carborane, porphyrin, cytotoxicity, cellular uptake
1. Introduction
Boron neutron capture therapy (BNCT) is a binary therapy that involves the activation of a tumor-localized 10B-containing sensitizer with low energy neutrons, resulting in the release of high linear energy transfer (high-LET) particles (4He2+ and 7Li3+) containing 2.4 MeV of kinetic energy.1–3 The short-range high-LET particles are highly cytotoxic and thus can selectively destroy 10B-containing malignant cells in the presence of boron-free normal tissues. BNCT is particularly attractive for the treatment of high-grade gliomas, malignant melanomas, meningiomas, head and neck tumors, and oral cancer.4 Two boron-containing compounds, mercapto-closo-dodecaborate (BSH) and 4-dihydroxyborylphenylalanine (BPA), are currently being used in BNCT clinical trials, in the USA, Europe and Japan.4–9 Since in the clinical studies performed to date there has been evidence of a therapeutic response to BNCT, the discovery of BNCT agents with higher biological efficacy than BSH and BPA has been an active area of research. Several boron-containing molecules have been synthesized and investigated as boron delivery agents for BNCT, including nucleosides, amino acids, phospholipids, monoclonal antibodies, dendrimers and porphyrins.10,11 Among these, porphyrins are very promising boron delivery vehicles because of their known tumor selectivity, low dark toxicity and long persistence within tumors.12,13 A promising BNCT delivery porphyrin agent should exhibit the following characteristics: (1) low dark toxicity, (2) high tumor cell selectivity, i.e. high tumor/normal tissue and tumor/blood boron concentration ratios, (3) deliver therapeutic concentrations of 10B to tumors, i.e. > 20 μg 10B/g tumor,4 (4) localize within tumor cells, preferentially within or near sensitive organelles such as nuclei,14,15 (5) have long retention times within tumors and rapid clearance from normal tissues and blood, and (6) be easily monitored by fluorescence techniques in order to facilitate radiation dosimetry. The major challenge in the development of BNCT delivery agents has been the selective delivery of therapeutic amounts of boron to targeted tumors with minimal normal tissue toxicity.4
Herein we report the syntheses of new amphiphilic closo-carboranylporphyrins (5d, 6c,d 7c,d and 8d) bearing either positively charged quaternary ammonium or negatively charged phosphonate water-solubilizing groups, and two to six closo-carborane cages.16 Only a few water-soluble closo-carboranylporphyrins have been previously reported in the literature, bearing carboxylates or a PEG as water-solubilizing groups.12 Our new closo-carboranylporphyrins contain either phosphonate groups that can form multiple hydrogen bonds with biological molecules, or quaternary ammonium salts that can potentially target highly vulnerable intracellular sites, such as the nuclei, and induce effective DNA and RNA damage.17–20 The X-ray molecular structures of four key intermediates (1, 2, 3 and 4a) and of two target porphyrins (5b and 6c) are discussed. We investigated and compared the in vitro toxicity, cellular uptake, and subcellular distribution properties of this series of compounds using both human carcinoma HEp2 and human glioblastoma T98G cells.
2. Results
2.1. Porphyrin syntheses
Porphyrins 5-8 containing two to six carborane cages (18–38% boron by weight) were synthesized via a [2+2] condensation of dicarboranyl-benzaldehyde 3 with an amino- or phosphonate-substituted dipyrromethane 4, as shown in Scheme 1. Due to acid-catalyzed scrambling in these reactions porphyrins 5-8 were obtained in a single-pot reaction and isolated in 2–8% yields, along with 2–9% of the corresponding octa-carboranylporphyrin (H2OCP)21, and <1% of other minor porphyrin products. The benzonitrile 1, prepared according to the procedure of Bodwell et al.,22 reacted with in-situ prepared 1-lithium-2-methyl-o-carborane producing the dicarboranyl-benzonitrile 2 in 51% yield. The cyano group of compound 2 was reduced to formyl using DIBAL-H, giving benzaldehyde 3 in 62% yield. We have previously reported a related synthesis of dicarboranyl-benzaldehyde 323 from 3,5-dimethylbromobenzene in 15% overall yield. Our new synthesis of 3 relies on the readily reduction of the cyano group of 2, as previously reported for other benzonitriles,24 rather than on a lithium/bromide exchange followed by DMF addition and acidic hydrolysis,23 thus involving less steps. Dipyrromethanes 4 were prepared from the reaction of the corresponding benzaldehyde with an excess of pyrrole using TFA as the catalyst, in 70–86% yields, as previously reported.25–27 The condensation of compounds 3 and 4a via a [2+2] methodology,27–29 using BF3•OEt2 for 1 h at room temperature, followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave porphyrins 6a, 7a and H2OCP in 2.1, 6.3 and 8.5% yields, respectively, as major products. On the other hand, the condensation of 3 and 4b under similar conditions but using BF3•OEt2 for only 4 min, afforded porphyrins 6b, 7b, 8b and H2OCP in 2.6, 8.0, 4.5 and 2.1% yields, respectively. Other porphyrins were also obtained as minor products (< 1% yield) as a result of the scrambling but were not isolated in sufficient amounts for characterization. In order to achieve water-solubility, the phosphonic esters were cleaved to the corresponding phosphonic acids using bromotrimethylsilane followed by hydrolysis30 in 78–99% yields, and the dimethylamino groups were quaternized with excess iodomethane in dichloromethane,19 in 83–92% yields. While the porphyrins containing either the diester phosphonate or dimethylamino functionalities are highly soluble in dichloromethane, the phosphonic acid-containing porphyrins are only soluble in DMSO and partially soluble in water, and the trimethylammonium-porphyrins are soluble in polar solvents (such as acetone and methanol) and also partially water-soluble. Porphyrin 5d exhibited particularly low solubility in all solvents, including DMSO, and therefore was not evaluated in in vitro studies.
Scheme 1.

(i) 1-Lithium-2-methyl-o-carborane, THF, LiI, rt, 20 h (51%); (ii) DIBAL, toluene followed by 10% H2SO4 (62%); (iii) pyrrole, TFA, CH2Cl2, rt (70–86%%); (iv) BF3•OEt2, CH2Cl2, rt, 4 min – 1 h; (v) DDQ, rt for 30 min (overall 17–22%).
2.2. Molecular structures
Crystals of intermediate dibromobenzonitrile 1 were grown from ethyl acetate/hexane, and those of carboranyl-benzonitrile 2, carboranyl-benzaldehyde 3, and dipyrromethane 4a were grown from CHCl3/hexane. Their molecular structures are shown in Figures 1, 2, 3 and 4, respectively, confirming their identities. Crystals of the toluene solvate of porphyrin 5b were grown from CHCl3/toluene, and those of the chloroform solvate of 6a were obtained by slow evaporation of a chloroform solution over a period of several days. The X-ray structures of porphyrins 5b and 6a are shown in Figures 5 and 6, respectively. In the crystal, porphyrin 5b lies on an inversion center, so the four N atoms are symmetry-required to be rigorously coplanar. The entire porphyrin ring deviates little from coplanarity, exhibiting a mean deviation of 0.029 Å for the 24-atom core and maximum deviation of 0.067(4) Å. The phenyl rings carrying the phosphonates form dihedral angles of 70.9(1)° with the porphyrin plane, and the phenyl rings carrying the carboranes form dihedral angles of 67.0(1)° with it. In porphyrin 6a, the four porphyrin N atoms in Figure 6 are coplanar to within 0.02(2) Å, but the 24-atom porphyrin core deviates somewhat from planarity, its atoms exhibiting a mean deviation of 0.12 Å from their best plane, with maximum deviation 0.26(3) Å. The distortion is mostly a twist about an axis bisecting the pyrrole between the two Me2NPh groups and the pyrrole between the two carborane-substituted phenyl groups. This twist places the two NMe2 nitrogen atoms 1.54(4) and −0.70(3) Å above and below the porphyrin best plane, as illustrated in Figure 6a, a view down the twist axis. The phenyl rings form dihedral angles which fall in the range 71.3(6) – 82.3(6)° with the porphyrin best plane.
Figure 1.
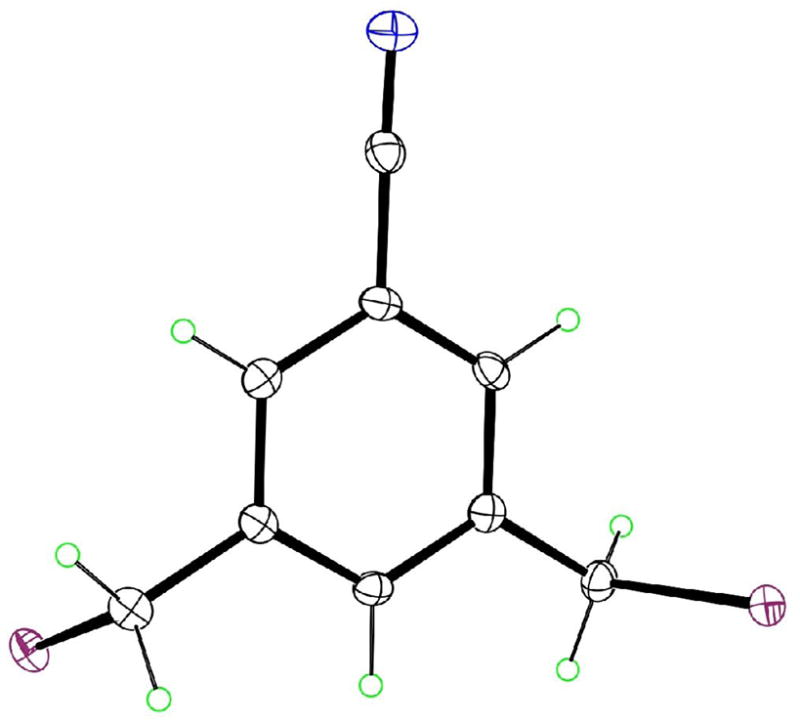
The molecular structure of 1 as determined by X-ray crystallography, with ellipsoids at the 50% level.
Figure 2.
The molecular structure of 2 with 50% ellipsoids. H atoms are not shown.
Figure 3.
The molecular structure of 3 with 50% ellipsoids. H atoms are not shown.
Figure 4.
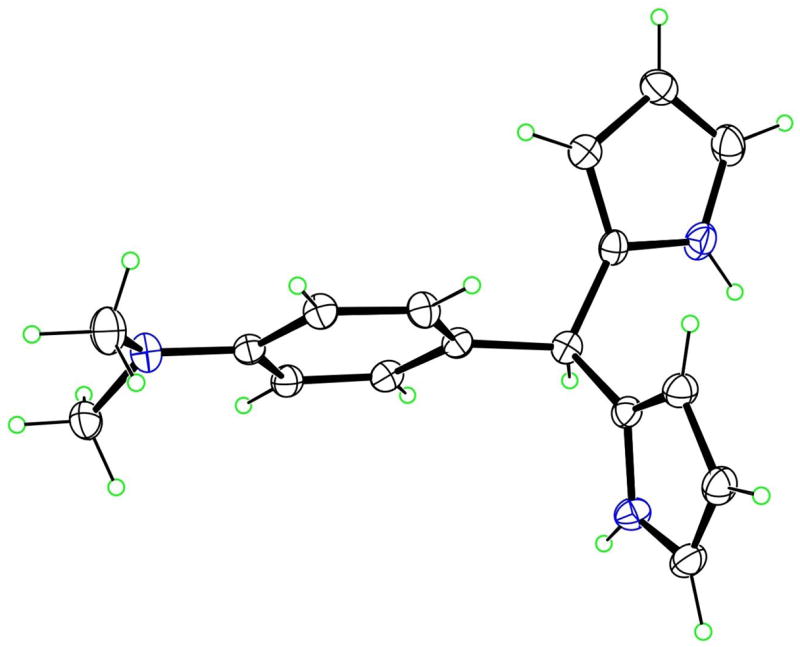
The molecular structure of 4a with 50% ellipsoids.
Figure 5.
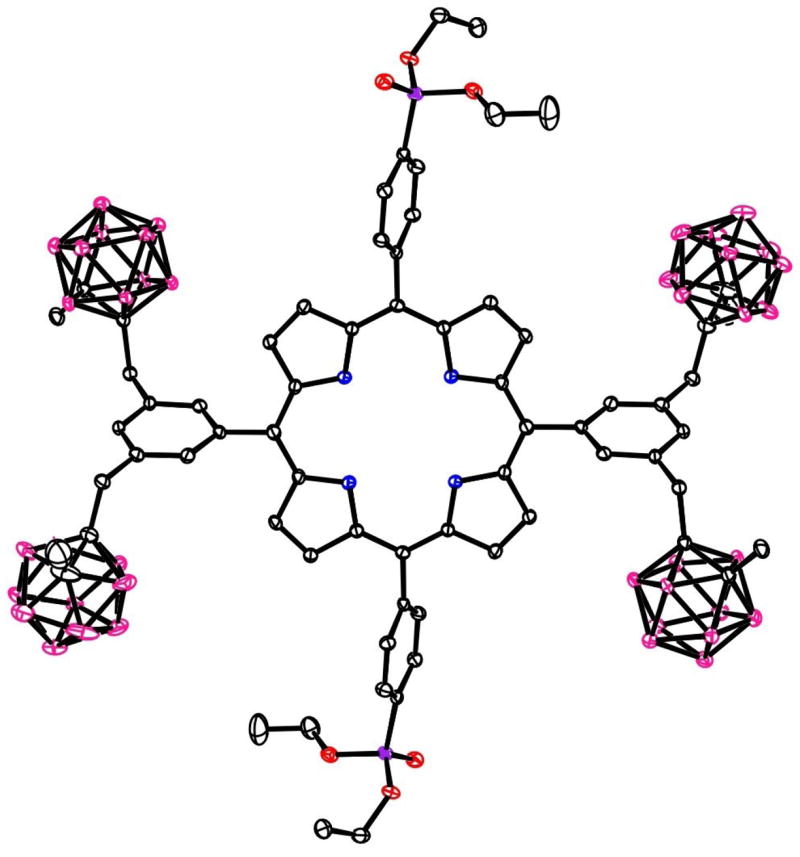
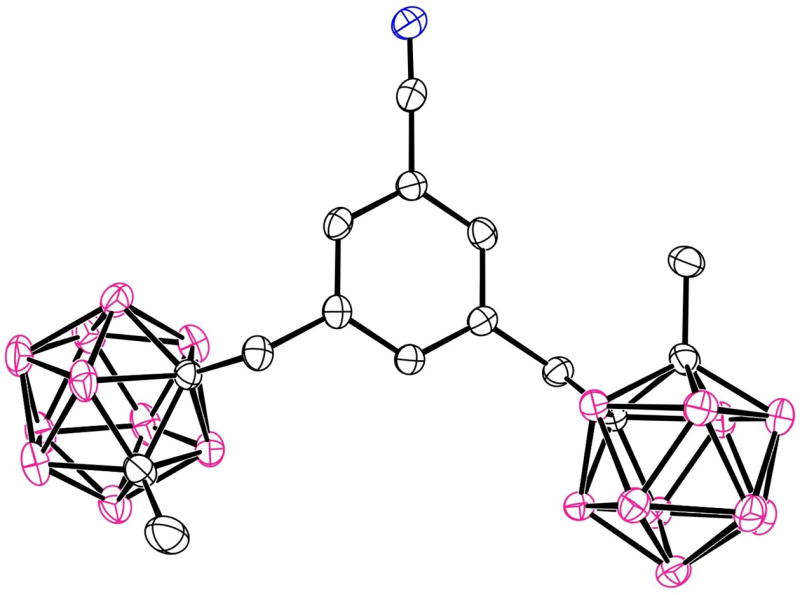
The molecular structure of porphyrin 5b with 20% ellipsoids. Solvent molecules and H atoms are not shown.
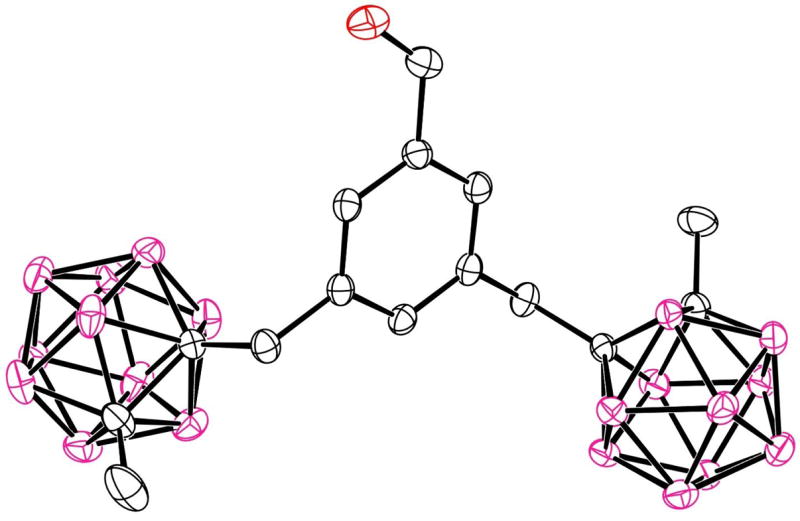
Figure 6.
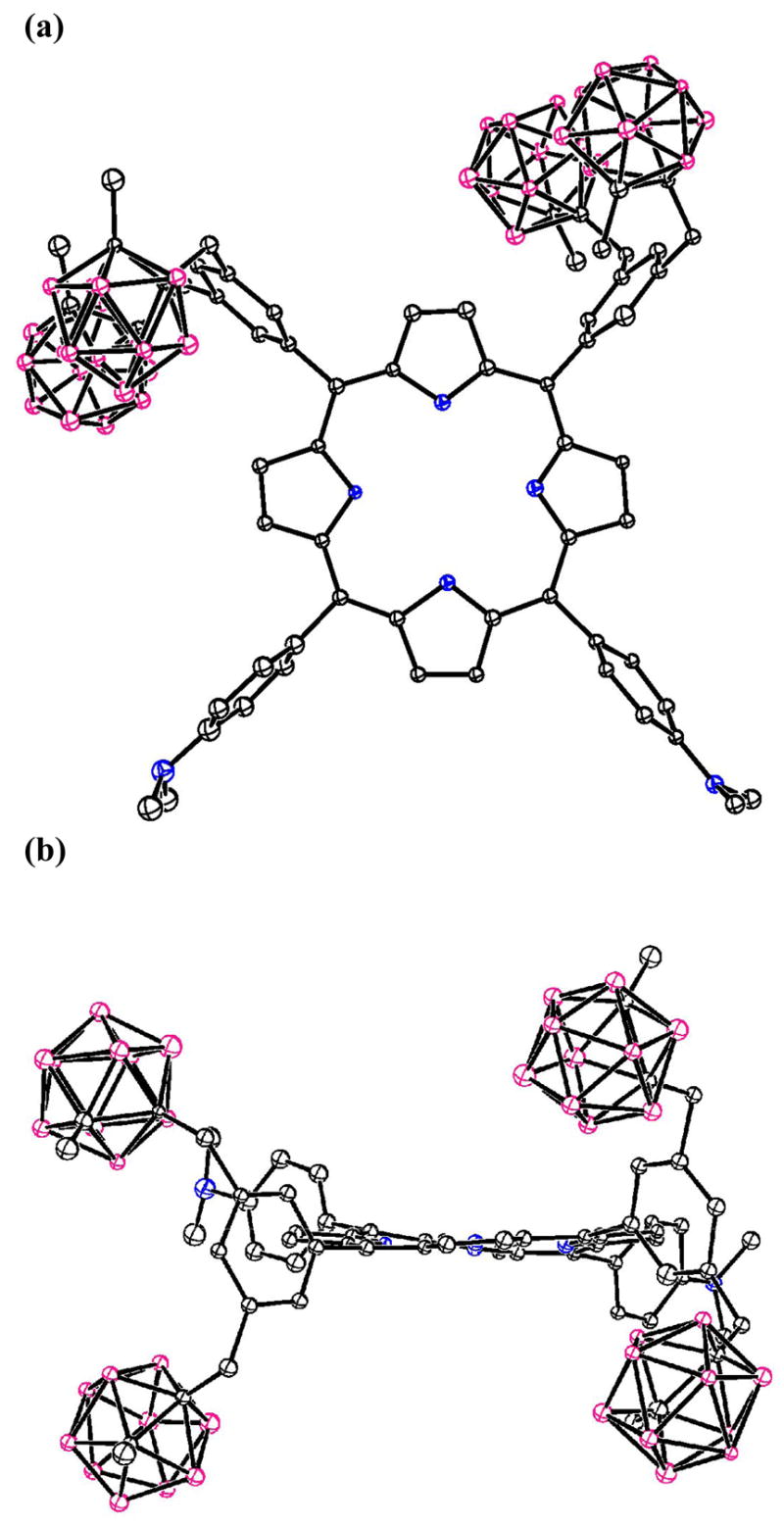
The molecular structure of porphyrin 6a with 20% spheres. (a) top view; (b) side view. Solvent molecules and H atoms are not shown.
2.3. Dark cytotoxicity
The dark toxicity of porphyrins 6c, 6d, 7c, 7d and 8d toward human carcinoma HEp2 cells was evaluated at concentrations up to 100 μM and the results obtained are shown in the Supplementary data. All compounds were found to be non-toxic to HEp2 cells in the dark under the conditions tested, up to 100 μM concentrations.
2.4. Cellular uptake
The time-dependent uptake of porphyrins 6c, 6d, 7c, 7d and 8d was evaluated in human glioblastoma T98G cells at a concentration of 10 μM over a period of 24 h and the results are shown in Figure 8. All porphyrins accumulated rapidly in the first 2 h, after which a plateau was reached for 6c, 6d, 7c and 7d, while 8d continued to steadily accumulate over time up to the 24 h period investigated. Porphyrin 6c bearing two quaternary ammonium groups on adjacent p-phenyl positions accumulated the most of this series compounds at short time points (< 4 h) while porphyrin 8d with three phosphonic acid groups, accumulated the most at longer time points (> 8 h). Porphyrin 7c containing only one quaternary ammonium group was the least taken-up by the T98G cells at all time points investigated. Similar uptake results were also obtained using human carcinoma HEp2 cells (results not shown).
Figure 8.
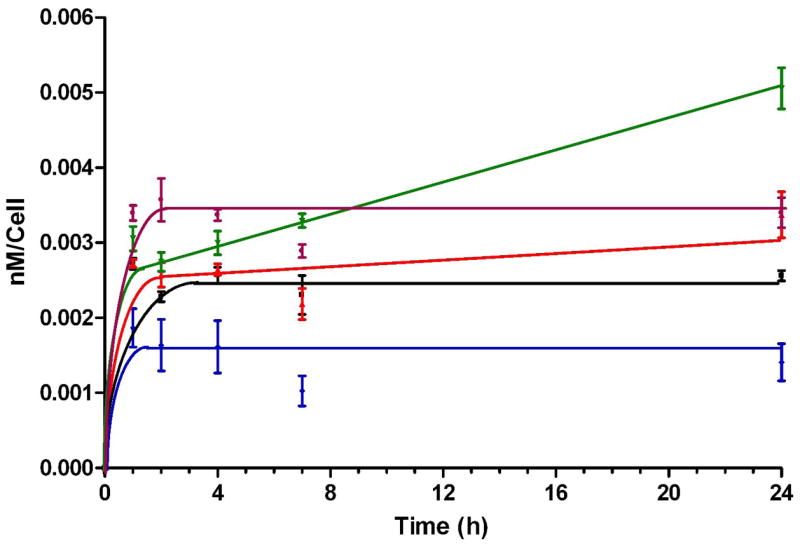
Time-dependent uptake of porphyrins 6c (purple), 6d (black), 7c (blue), 7d (red) and 8d (green) at 10 μM by T98G cells.
2.5. Intracellular localization
Fluorescence microscopy was used to investigate the preferential sites of intracellular localization of porphyrins 6c, 6d, 7c, 7d and 8d, using both HEp2 and T98G cells. The cells were exposed overnight to 10 μM of each porphyrin and then examined for intracellular fluorescence. Similar results were obtained with both cell lines; Figures 9b–13b show the fluorescence patterns observed in HEp2 cells. For the co-localization experiments the cells were incubated concurrently with porphyrin and one of the following organelle tracers for 30 min: MitoTracker Green 250 nM, LysoSensor Green, BODIPY FL C5-ceramide at 50 nM and 50 nM, DiOC6 5 μg/mL. The slides were washed three times with growth medium and new medium containing 50 mM HEPES pH 7.4 was added. The images were acquired using a Zeiss Axiovert 200M inverted fluorescence microscope fitted with standard FITC and Texas Red filter sets. The preferential site of intracellular localization for porphyrins 6c–6d, 7c–d, and 8d was found to be the lysosomes (Figures 9f–13f).
Figure 9.
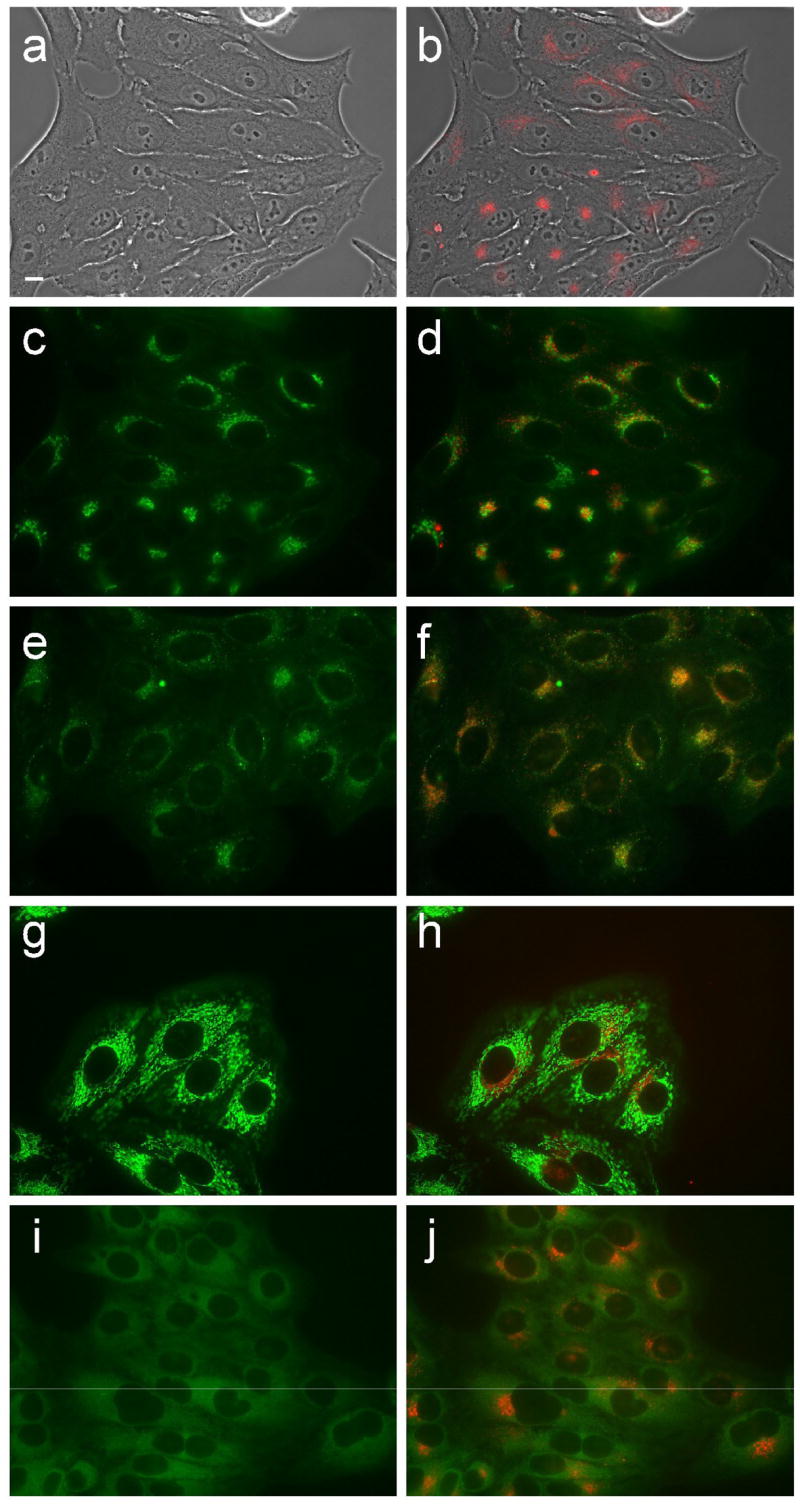
Intracellular localization of porphyrin 6c in HEp2 cells at 10 μM for 24 h. (a) phase contrast; (b) porphyrin fluorescence; (c) BODIPY FL- C5-Ceramide fluorescence (Golgi label); (d) overlay with porphyrin; (e) LysoSensor fluorescence (lysosome label); f: overlay; (g) MitoTracker fluorescence (mitochondria label); (h) overlay; (i) ERTracker Green FM fluorescence (ER label); (j) overlay. Scale bar: 10 μm.
Figure 13.
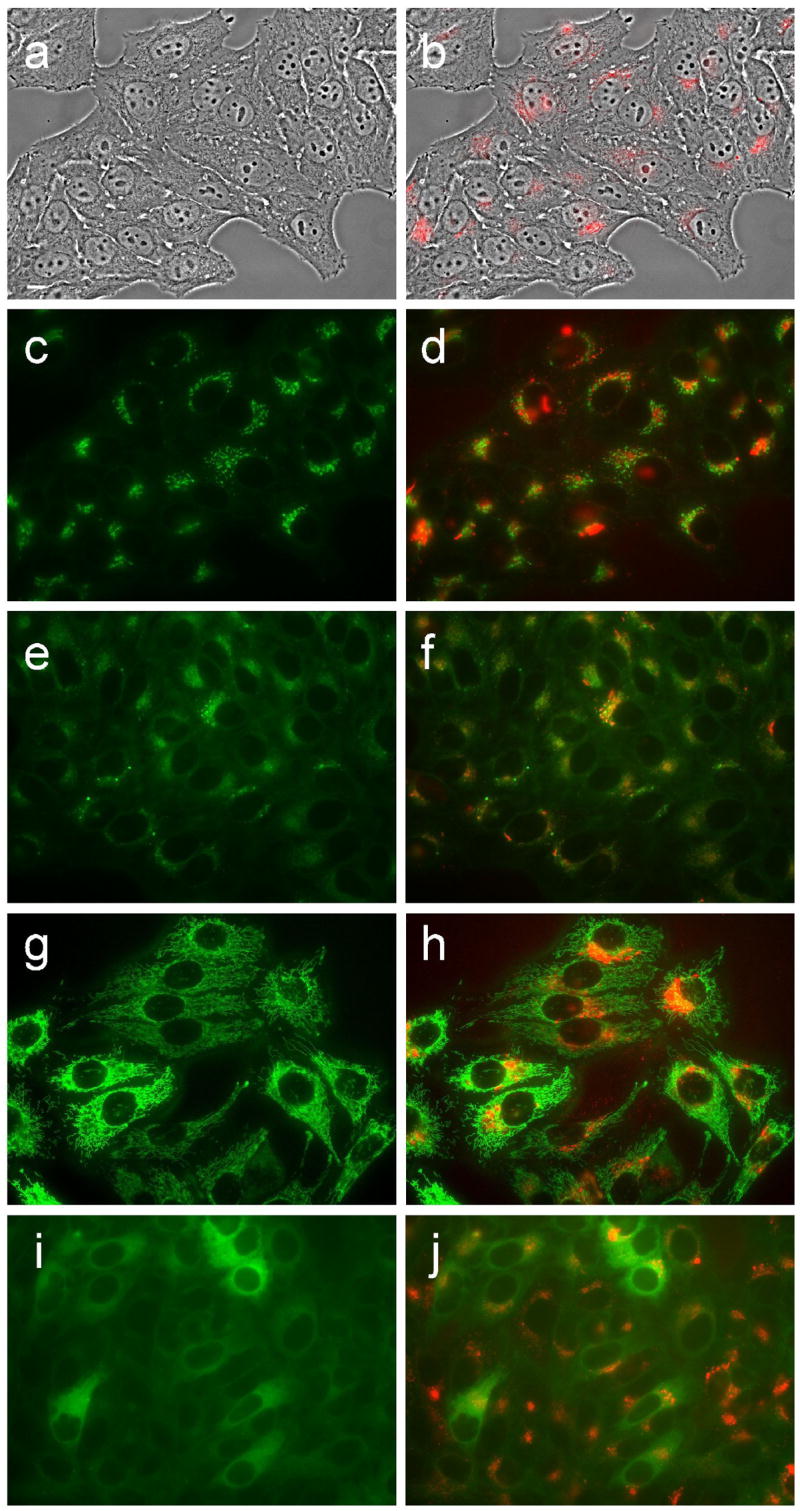
Intracellular localization of porphyrin 8d in HEp2 cells at 10 μM for 24 h. (a) phase contrast; (b) porphyrin fluorescence; (c) BODIPY FL- C5-Ceramide fluorescence (Golgi label); (d) overlay with porphyrin; (e) LysoSensor fluorescence (lysosome label); f: overlay; (g) MitoTracker fluorescence (mitochondria label); (h) overlay; (i) ERTracker Green FM fluorescence (ER label); (j) overlay. Scale bar: 10 μm.
3. Discussion
The new series of closo-carboranylporphyrins 5-8 was prepared in a single reaction using a MacDonald-type [2+2] condensation31 of either dimethylamino- or diethylphosphonate-dipyrromethane with dicarboranyl-benzaldehyde 3 (Scheme 1). We investigated various reaction conditions that led to the highest yields of the target porphyrins 5-8, bearing one to three water-solubilizing groups and two to six closo-carborane cages, including different acid catalysts, reagent concentrations, solvents, reaction times and temperatures.27,28 The optimal conditions involved the use of 10 mM dipyrromethane and benzaldehyde concentrations in dichloromethane and 2 equiv. of BF3•OEt2 for either 1 h (in the case of 4a + 3) or 4 min (for 4b + 3). The use of dipyrromethanes 4 in a [2+2] condensation rather than a mixed aldehyde condensation afforded higher yields of the target porphyrins containing at least one dipyrromethane motif, by minimizing the formation of H2OCP, which was always a side product in these reactions. When 5-(4-acetamidophenyl)dipyrromethane was used in place of 4a, lower yields of the targeted porphyrins were obtained due to the additional step required to cleave the acetamido group (ethanol/HCl at reflux for 24 h) and the isolation of a tetracarboranylcorrole as one of the main products. The condensation of 5-[bis(3,5-(2-methyl-o-carboran-1-yl)methyl)phenyl]dipyrromethane with phosphonate- or amino-substituted benzaldehydes also gave lower yields of the targeted porphyrins;16 in fact, no carborane-containing phosphonate-substituted porphyrins were obtained using this methodology. The acid-catalyzed scrambling was more significant in the [2+2] condensation of 4a with 3 probably as a result of the strong electron-donating effect of the dimethylamino group and the longer reaction time necessary to drive the reaction to completion; in this case the trans-porphyrin 5a was not among the major products of the reaction and H2OCP was the main product obtained in 8.5% yield. Under the above optimal conditions for the condensation of 4b and 3, porphyrin 7b bearing a single phosphonate ester was the major product obtained, followed by 8b and 6b; in this case H2OCP was only obtained in 2.1% yield. Although acid-catalyzed scrambling reactions are generally avoided in order to minimize challenging chromatographic separations, this methodology allowed us to isolate reasonable amounts of several interesting porphyrin targets for structure/in vitro biological activity relationships.
All new compounds were characterized by spectroscopic techniques (MS, 1H NMR, UV-Vis) and in addition, benzonitriles 1 and 2, benzaldehyde 3, dipyrromethane 4a and porphyrins 5b and 6a were also characterized by X-ray crystallography. It is interesting to note that while the porphyrin ring in porphyrin 5b, bearing two phosphonic esters on opposite p-phenyl positions is essentially flat, that of porphyrin 6a with two adjacent p-dimethylaminophenyl groups is moderately distorted from planarity, showing a twisted conformation. Such porphyrin distortions from planarity have been previously observed32 and seem to be associated with the electronic interactions between the water-solubilizing groups rather than with the 3,5-dicarboranylmethylphenyl groups.21,23 Upon quaternization of the dimethylamino groups of 6a, i.e. in 6c, a more pronounced distortion of the porphyrin is expected, as we have previously observed for a 5,10-di(4-trimethylamino)phenyl-15,20-diphenylporphyrin.19
As a consequence of the highly hydrophobic character of the closo-carborane cages, porphyrins 5b, 6a,b, 7a,b and 8b were completely insoluble in water. Two strategies are usually employed for conferring partial water-solubility to carboranylporphyrins,12 (1) the introduction of water-solubilizing groups (such as amino, hydroxyl or carboxylate), or (2) the deboronation of o-carborane cages to the corresponding anionic nido-carboranes. In the present study we synthesized, characterized and compared the in vitro biological properties of a new series of closo-carboranylporphyrins bearing either positively charged quaternary ammonium or negatively charged phosphonate water-solubilizing groups; these compounds could potentially interact with DNA33,34 and the phosphonate-substituted porphyrins could form multiple hydrogen bonds with biological substrates. Interestingly, porphyrin 5d bearing two phosphonic acid substituents on opposite p-phenyl positions showed very limited water-solubility, probably as a result of the formation of large aggregates and their subsequent precipitation in aqueous media.35 Porphyrins 5d, 6c, 6d, 7c, 7d and 8d showed similar absorption and emission spectra in DMSO, typical for meso-tetraphenylporphyrins, as shown in Figure 7. However, in HEPES buffer (20 mM, pH 7.4) containing 1% DMSO all porphyrins showed significant broadening of their emission bands and fluorescence quenching indicating aggregation (see Figure 7 and Supplementary data), as we have previously observed.36 Interestingly, porphyrins 5d, 6c, 7c and 7d displayed red-shifted (1–4 nm) emission bands while porphyrins 6d and 8d showed blue-shifted (3–5 nm) bands, suggesting the formation of different types of aggregates.
Figure 7.

Fluorescence emission spectra of porphyrins 5d (a), 6c (b), 6d (c), 7c (d), 7d (e) and 8d (f), at 1 × 10−6 M in DMSO (solid line), and in freshly prepared HEPES buffer (20 mM, pH 7.4) containing 1% DMSO (dashed line). Excitation at 420 nm.
Porphyrins 6c, 6d, 7c, 7d and 8d were all found to be non-toxic to HEp2 cells in the dark up to 100 μM concentrations (see Supplementary data). Higher concentrations were not investigated because the compounds precipitated. Our results are in agreement with previous studies reporting very low dark toxicities for both closo- and nido-carboranylporphyrins.21,23,34,36–42 The cellular uptake of this series of closo-carboranylporphyrins did not increase with their hydrophobic character. We determined the following order of increasing uptake at times < 8 h: 6c > 8d > 7d > 6d > 7c (Figure 8). At longer time periods porphyrin 8d, bearing three phosphonic acid groups, was the most accumulated within cells, and at 24 h twice the amount of this porphyrin was found within cells than 6d, containing two phosphonic acid groups on adjacent phenyl rings. Porphyrin 7d with only one phosphonic acid substituent and the highest percentage of boron by weight of all porphyrins showed slightly higher uptake than 6d at all time points investigated and delivered the highest amount of boron to T98G cells after 24 h (see Table 1 in Supplementary data). After 24 h the calculated amount of intracellular boron (in μg per billion cells or per gram of wet tissue) delivered by the porphyrins to T98G cells was between 92.6 ± 16.2 (for 7c) and 220.0 ± 35.0 (for 7d). Therefore all porphyrins are able to deliver therapeutic amounts of boron to human glioblastoma T98G cells. The differences in uptake observed for the phosphonate-containing porphyrins might be due to the unique aggregation behavior of these polyprotic compounds, as we have previously investigated.35 At physiologic pH the phosphonic acid groups mainly exist in their mono-anionic form (i.e. as PO3H−) and the resulting anionic porphyrins can aggregate as a result of both π-π interactions and hydrogen bond formation. It has been recently shown that negatively charged cobaltacarborane-porphyrins containing up to eight negative charges form large aggregates in aqueous solutions.36,43 On the other hand porphyrin 6c, bearing two quaternary ammonium groups accumulated within T98G cells about 2.5 times more than 7c with only one positive charge; in fact porphyrin 7c was the least taken up by cells of all the porphyrins studied and delivered the least amount of boron to T98G cells. The higher amphiphilicity of porphyrin 6c compared with 7c as a result of its more favorable lipophilic/hydrophilic balance as a consequence of its charge distribution might be responsible for the observed results. The uptake kinetics for this series of carboranylporphyrins is similar to that previously observed for both closo- and nido-carboranylporphyrins.21,36,37,39,44,45
The preferential sites of intracellular localization for porphyrins 6c, 6d, 7c, 7d and 8d were found to be the cell lysosomes, as evidenced by the punctate pattern observed by fluorescence microscopy and its overlay with the lysosome-specific probe Lysotracker Green. These results are in agreement with previous studies showing that carboranylporphyrins localize within cell lysosomes,21,36,39,45–48 maybe as a result of an endocytic mechanism of uptake.40,50
4. Conclusions
The syntheses of six new amphiphilic closo-carboranylporphyrins bearing either positively charged quaternary ammonium (6c, 7c) or negatively charged phosphonate (5d, 6d, 7d and 8d) groups is reported. The X-ray molecular structures of four intermediates (1, 2, 3 and 4a) and of two target porphyrins (5b and 6c) are discussed. All porphyrins were found to be non-toxic in the dark up to 100 μM concentrations and the extent of their cellular uptake did not increase with their hydrophobic character. The tetracarboranylporphyrin 6c, bearing two quaternary ammonium groups, was the most efficiently taken up by human glioma T98G cells at short times (up to 8 h), followed by dicarboranylporphyrin 8d bearing three phosphonic acid substituents. The hexacarboranylporphyrin 7d was found to deliver the highest amount of boron to T98G cells after 24 h. The preferential sites of subcellular localization for all porphyrins were found to be the cell lysosomes.
5. Experimental
5.1. Chemistry
All reactions were carried out under an argon atmosphere in dried solvents. Commercially available starting compounds were purchased from Sigma-Aldrich or from Katchem, Ltd. and used directly without further purification. Silica gel 60 (70–230 mesh, Merck) was used for column chromatography. Analytical thin-layer chromatography (TLC) was performed using Merck 60 F254 silica gel (precoated sheets, 0.2 mm thick). 1H-NMR spectra were obtained using a Bruker DPX 250 MHz spectrometer; chemical shifts are expressed in ppm relative to TMS (0 ppm). Electronic absorption spectra were measured on a Perkin Elmer Lambda 35 UV-Vis spectrophotometer. Low resolution mass spectra (MS) were obtained on a Bruker Proflex III MALDI-TOF mass spectrometer. High resolution mass spectra (HRMS) were obtained using ESI-TOF under negative mode on an Agilent Technologies Time-of-Flight LC/MS 6210; the isotope peaks were matched with calculated isotope patterns (see Supplementary data) and only the most abundant peaks are listed below for each porphyrin. Melting points were measured on a MELT-TEMP apparatus.
Bis(3,5-bromomethyl)benzonitrile (1)22
To a round-bottomed flask containing dichloromethane (460 mL) was added 3,5-dimethylbenzonitrile (5.00 g, 38 mmol), NBS (15.00 g, 84 mmol), and benzoyl peroxide (0.048 g, 0.20 mmol). The reaction mixture was heated to reflux while stirring under argon. Upon reaching reflux a white floodlight was positioned within four inches of the reaction vessel. After 1 h the reaction mixture was cooled to room temperature and 50 mL of distilled water was added to quench the reaction. The organic phase was washed once with water, once with brine, and then dried over Na2SO4, and the solvent removed under vacuum to yield a yellow/orange residue. The crude residue was dried onto silica and purified by silica column chromatography using ethyl acetate/hexane 1:4 for elution. The pure product was collected affording 4.47 g of the title compound as a white solid in 40 % yield. M. p. = 114–115 °C; MS (GC) m/z 289.8 [M+1]; 1H NMR (CDCl3) δ ppm: 4.50 (s, 4H, CH2), 7.65 (s, 2H, o-ArH), 7.68 (s, 1H, p-ArH).
Bis[3,5-(2-methyl-o-carboran-1-yl)methyl]benzonitrile (2)
n-Butyllithium (23.63 mL, 1.6 M in hexane) was added dropwise to a solution of 1-methyl-o-carborane (4.76 g, 30 mmol) in dry THF (450 mL) at a temperature between −5 and 0 °C, under argon. The mixture was stirred at this temperature for 1.5 h, and then compound 1 (4.37 g, 15.10 mmol) and LiI (0.628 g, 4.69 mmol) in THF (50 mL) were added. The final reaction mixture was allowed to warm to room temperature and after 20 h the reaction was quenched with water and the solvent removed under vacuum. The crude residue was dissolved in ethyl acetate and washed once with water, once with brine, dried over Na2SO4, and the solvent removed under vacuum. The yellow residue was dried onto silica and purified by silica column chromatography using ethyl acetate/hexane 1:4 for elution. The pure title compound was obtained as an off-white solid (2.70 g) in 51% yield (40 % conversion). Recovered 1.05 g of compound 1. M. p. = 235–237 °C; MS (ESI) m/z 443.24; 1H NMR (CDCl3) δ ppm 1.4–3.0 (br, 20H, BH), 2.22 (s, 6H, CH3), 3.58 (s, 4H, CH2), 7.36 (s, 1H, p-ArH), 7.56 (s, 2H, o-ArH).
Bis[3,5-(2-methyl-o-carboran-1-yl)methyl]benzaldehyde (3)
A solution of compound 2 (11.14 g, 25.14 mmol) in dry toluene (500 mL) was flushed with argon for 10 min. The reaction vessel was then cooled to 0 °C and DIBAL-H (1.5 M in toluene) (36.87 mL, 55.3 mmol) was slowly added dropwise while stirring under argon. The reaction was heated to 60–65 °C for 39 h, then cooled to 0 °C and a cold 10 % H2SO4 aqueous solution (228 mL) was slowly added to avoid foaming. Toluene (800 mL) was added and the reaction mixture was stirred for an additional 62 h under argon. The organic phase was washed once with Na2HCO3, once with water, once with brine, dried over Na2SO4 and the solvent removed under vacuum to yield a crude white solid. This solid was dissolved in dichloromethane, dried onto silica and purified by silica column chromatography using 1:1 hexane/dichloromethane for elution. Pure title compound was obtained as a white solid (7.07 g) in 62 % yield. The spectroscopic data for the title compound is in full agreement with that reported in the literature.21,23
5-(4-Dimethylamino)phenyldipyrromethane (4a) was prepared as described in the literature and its spectroscopic data are in full agreement with that previously reported.27
5-(4-Phosphonate diethylester)phenyldipyrromethane (4b)
A solution of compound 3 (0.047 g, 0.20 mmol) and freshly distilled pyrrole (0.80 mL, 11.5 mmol) was purged with nitrogen for 5 min. TFA (2 μL, 0.023 mmol) was added and the final solution was stirred at room temperature in a sealed reaction vessel for 20 min. The light green crude material was dried onto silica and purified by silica column chromatography using methanol/dichloromethane 1:33. The title compound was obtained (65 mg) in 86 % yield, as a yellow residue. 1H NMR (CDCl3) δ ppm: 1.36 (t, 6H, CH3, J = 7.0 Hz), 4.10 (m, 4H, CH2), 5.60 (s, 1H, CH), 6.01 (s, 2H, o-ArH), 6.18 (s, 2H, m-ArH), 6.78 (s, 2H, α-H), 7.35 (m, 2H, β-H), 7.62 (m, 2H, β-H), 9.56 (br, 2H, NH).
General Procedure for Porphyrin Synthesis
A solution of dipyrromethane 4 (1.12 mmol) and bis[3,5-(2-methyl-o-carboran-1-yl)methyl]benzaldehyde 3 (0.498 g, 1.12 mmol) in 112 mL of dichloromethane was purged with argon for 15 min. BF3•OEt2 (0.283 mL, 2.23 mmol) was added and the reaction was stirred in a sealed reaction vessel. After 1 h (for 4a) or 4 min (for 4b) DDQ (0.336 g, 1.48 mmol) and Et3N (0.31 mL, 2.23 mmol) were added and the reaction mixture stirred for 30 min. The mixture was then dried onto silica and filtered through a plug of silica gel using dichloromethane/hexane 2:1 for elution. The crude mixture was further purified using preparative silica TLC plates and dichloromethane/hexane 3:1 for elution to yield the porphyrins.
Deprotection
The diethyl ester porphyrins were dissolved in dichloromethane and purged with argon for 5 min. The reaction vessel was then sealed and bromotrimethylsilane was added by microliter syringe (16 equiv/phosphonate diester) while stirring at room temperature. The final mixture was stirred for 24 h. All solvents were removed under vacuum and the crude green residue was dissolved in a 1:4 H2O/THF solution and allowed to stir for 2 h. The organic solvent was removed under vacuum and the deprotected porphyrins were filtered and washed repeatedly with distilled water.
Quaternization
The dimethylamino porphyrins were dissolved in a 2:5 solution of CH2Cl2/CH3I and the resulting mixture allowed to stand without stirring in a sealed reaction vessel for 72 h at room temperature. The quaternized products precipitated out of solution and were filtered and washed repeatedly with dichloromethane. Porphyrins 6c and 7c were partially demethylated upon standing over a period of one week (for 6c) or one month (for 7c), thus reforming the starting porphyrins 6a and 7a, respectively.
5,15-Bis-(4-phosphonate diethyl ester)-10,20-bis[(3,5-(2-methyl-o-carboran-1-yl) methylphenyl]porphyrin (5b)
This porphyrin was obtained (31 mg, 3.6% yield) as a purple/red solid. M.p.: > 300 °C; MS (MALDI) m/z 1552.78 [M-CH3]+; 1H NMR (CD2Cl2) δ ppm: −2.80 (br, 2H, NH), 1.60 (t, 12H, CH3, J = 7.0 Hz), 1.3 – 3.4 (br, 40H, BH), 2.27 (s, 12H, CH3), 3.85 (s, 8H, CH2), 4.40 (m, 8H, CH2), 7.58 (s, 2H, ArH), 8.12 (s, 4H, ArH), 8.26 (m, 4H, ArH), 8.40 (m, 4H, ArH), 8.95 (m, 8H, β-H). UV-Vis (CH2Cl2) λmax: 419 nm (ε 331,000), 514 (15,400), 548 (6,000), 588 (4,800), 643 (2,800). Deprotection afforded the corresponding phosphonic acid porphyrin (5d) in 83% yield as a green solid. M.p.: > 300 °C; HRMS (ESI) m/z 1457.0262 [M+H]+, calculated for C60H88B40N4O6P2 1456.0195; 1H NMR (DMSO-d6) δ ppm: −2.92 (br, 2H, NH), 2.21 (s, 12H, CH3), 1.3 – 3.4 (br, 40H, BH), 3.92 (s, 8H, CH2), 7.63 (s, 2H, ArH), 8.15 (br, 8H, ArH), 8.29 (br, 4H, ArH), 8.85 (br, 8H, β-H). UV-Vis (DMSO) λmax: 420 nm (ε379,800), 515 (18,300), 550 (9,100), 589 (6,300), 645 (5,300).
5,10-Bis-(4-dimethylaminophenyl)-15,20-bis[3,5-(2-methyl-o-carboran-1-yl)methylphenyl] porphyrin (6a)
This porphyrin was obtained in 2.1% yield as a black solid. M.p.: > 300 °C, MS (MALDI) m/z 1380.90 [M+]; 1H NMR (CD2Cl2) δ ppm: −2.69 (s, 2H, NH), 1.25–3.1 (br, 40H, BH), 2.24 (s, 12H, CH3), 3.25 (s, 12H, CH3), 3.79 (s, 8H, CH2), 7.17 (br, 4H, ArH), 7.51 (s, 2H, ArH), 8.07–8.12 (m, 8H, ArH), 8.85 (br, 4H, β-H), 8.99 (s, 2H, β-H), 9.03 (br, 2H, β-H). UV-Vis (DMSO) λmax: 423 nm (ε 171,000), 523 (13,500), 568 (14,200), 595 (8,500), 657 (7,300). Quaternization of this porphyrin afforded the corresponding quaternary ammonium iodide salt (6c) in 83% yield as a purple/red solid. M.p.: > 300 °C, HRMS (ESI) m/z 706.1089 [M – 2I]2+, calculated for C66H102B40N6 706.1090; 1H NMR (acetone-d6) δ ppm: −2.79 (s, 2H, NH), 1.25–3.1 (br, 40H, BH), 2.37 (s, 12H, CH3), 4.07 (s, 8H, CH2), 4.29 (s, 18H, CH3), 7.86 (s, 2H, p-ArH), 8.22 (s, 4H, ArH), 8.55 (br, 4H, ArH), 8.65 (br, 4H, ArH), 8.88–8.99 (m, 8H, β-H). UV-Vis (DMSO) λmax: 419 nm (ε 406,100), 514 (11,800), 548 (6,300), 588 (4,500), 644 (3,200).
5,10-Bis-(4-phosphonic acid diethyl ester)-15,20-bis[(3,5-(2-methyl-o-carboran-1-yl) methylphenyl]porphyrin (6b)
This porphyrin was obtained in 2.6% yield as a purple solid. M.p. 282.8 °C; MS (MALDI) m/z 1552.78 [M-CH3]+; 1H NMR (CD2Cl2) δ ppm: −2.83 (br, 2H, NH), 1.55 (t, 12H, CH3, J = 7.0 Hz), 1.3 – 3.4 (br, 40H, BH), 2.27 (s, 12H, CH3), 3.85 (s, 8H, CH2), 4.39 (m, 8H, CH2), 7.58 (s, 2H, ArH), 8.13 (s, 4H, ArH), 8.25 (m, 4H, ArH), 8.41 (m, 4H, ArH), 8.97 (m, 8H, β-H). UV-Vis (CH2Cl2) λmax: 419 nm (ε 352,900), 514 (18,000), 549 (6,800), 589 (5,500), 643 (3,400). Deprotection afforded the corresponding phosphonic acid porphyrin (6d) in 78% yield as a green solid. M.p. >300 °C; HRMS (ESI) m/z 1457.0260 [M + H]+, calculated for C60H88B40N4O6P2 1456.0195; 1H NMR (d-DMSO) δ ppm: −2.92 (br, 2H, NH), 1.3 – 3.4 (br, 40H, BH), 2.27 (s, 12H, CH3), 3.97 (s, 8H, CH2), 7.72 (s, 2H, ArH), 8.08–8.16 (br, 8H, ArH), 8.32 (br, 4H, ArH), 8.84 (m, 8H, β-H). UV-Vis (DMSO) λmax: 419 nm (ε395,100), 514 (17,200), 549 (8,300), 589 (5,900), 645 (4,900).
5-(4-Dimethylaminophenyl)-10,15,20-tris[3,5-(2-methyl-o-carboran-1-yl)methylphenyl] porphyrin (7a)
This porphyrin was obtained in 6.3% yield as a red/brown solid. M.p. >300 °C; MS (MALDI) m/z 1679.10 [M]+; 1H NMR (CD2Cl2) δ ppm: −2.73 (s, 2H, NH), 1.25–3.1 (br, 60H, BH), 2.27 (s, 18H, CH3), 3.29 (s, 6H, CH3), 3.85 (s, 12H, CH2), 7.20 (br, 2H, ArH), 7.57 (s, 3H, ArH), 8.12–8.16 (m, 8H, ArH), 8.90 (br, 6H, β-H), 9.10 (br, 2H, β-H). UV-Vis (DMSO) λmax: 418 nm (ε 253,900), 518 (14,300), 562 (11,000), 592 (8,100), 653 (6,300). Quaternization afforded the corresponding quaternary ammonium iodide salt (7c) in 92% yield as purple red solid. M.p. 186.3 °C; HRMS (ESI) m/z 1695.7502 [M + H – I]+, calculated for C71H122B60N5 1694.5730; 1H NMR (acetone-d6) δ ppm: −2.78 (s, 2H, NH), 1.25–3.1 (br, 60H, BH), 2.38 (s, 18H, CH3), 4.09 (s, 12H, CH2), 4.28 (s, 9H, CH3), 7.85 (s, 3H, ArH), 8.23 (s, 6H, ArH), 8.60 (s, 4H, ArH), 8.92 (m, 8H, β-H); UV-Vis (DMSO) λmax: 420 nm (ε 389,800), 514 (14,900), 548 (7,700), 588 (5,800), 644 (4,400).
5-(4-Phosphonic acid diethyl ester)-10,15,20-tris[(3,5-(2-methyl-o-carboran-1-yl)methyl phenyl]porphyrin (7b)
This porphyrin was obtained in 8.0% yield as a purple solid. M.p. 294.7 °C; MS (MALDI) m/z 1756.10 [M – CH3]+; 1H NMR (CD2Cl2) δ ppm: -2.80 (br, 2H, NH), 1.36 (t, 6H, CH3, J = 7.0 Hz), 1.3 – 3.4 (br, 60H, BH), 2.28 (s, 18H, CH3), 3.86 (s, 12H, CH3), 4.38 (m, 4H, CH2), 7.59 (s, 3H, ArH), 8.12 (s, 6H, ArH), 8.26 (m, 2H, ArH), 8.41 (m, 2H, ArH), 8.97 (m, 8H, β-H). UV-Vis (CH2Cl2) λmax: 419 nm (ε 359,100), 514 (21,900), 548 (11,000), 588 (7,400), 643 (3,500). Deprotection afforded the corresponding phosphonic acid porphyrin (7d) in 94% yield as a black solid. M.p. >300 °C; HRMS (ESI) m/z 1717.4781 [M+H]+, calculated for C68H115B60N4O3P 1716.4735; 1H NMR (DMSO-d6) δ ppm: −2.91 (s, 2H, NH), 2.24 (s, 18H, CH3), 1.3 – 3.4 (br, 60H, BH), 3.92 (s, 12H, CH2), 7.68 (br, 3H, ArH), 8.12 (br, 8H, ArH), 8.26 (br, 2H, ArH), 8.86 (br, 8H, β-H). UV-Vis (DMSO) λmax: 420 nm (ε 418,100), 515 (16,900), 549 (8,000), 589 (5,700), 645 (4,700).
5,10,15-Tris-(4-phosphonic acid diethyl ester)-20-bis[(3,5-(2-methyl-o-carboran-1-yl) methylphenyl]porphyrin (8b)
This porphyrin was obtained in 4.5% yield as a purple solid. M.p. 267 °C; MS (MALDI) m/z 1362.40 [M]+; 1H NMR (CD2Cl2) δ ppm: −2.83 (br, 2H, NH), 1.55 (t, 18H, CH3, J = 7.0 Hz), 1.3 – 3.4 (br, 20H, BH), 2.27 (s, 6H, CH3), 3.84 (s, 4H, CH2), 4.40 (m, 12H, CH3), 7.58 (s, 1H, ArH), 8.08 (s, 2H, ArH), 8.26 (m, 6H, ArH), 8.42 (m, 6H, ArH), 8.97 (m, 8H, β-H). UV-Vis (CH2Cl2) λmax: 419 nm (ε 385,000), 514 (16,300), 549 (6,700), 589 (5,000), 643 (3,000). Deprotection afforded the corresponding phosphonic acid porphyrin (8d) in quantitative yield as a blue-green solid. M.p.: > 300 °C; HRMS (ESI) m/z 1196.5739 [M+H]+, calculated for C52H61B20N4O9P3 1195.5655; 1H NMR (DMSO) δ ppm: −2.94 (br, 2H, NH), 1.3 – 3.4 (br, 20H, BH), 2.27 (s, 6H, CH3), 3.96 (br, 4H, CH2), 7.72 (s, 1H, ArH), 8.08–8.16 (m, 8H, ArH), 8.31–8.34 (m, 6H, ArH), 8.84 (br, 8H, β-H). UV-Vis (DMSO) λmax: 420 nm (ε331,700), 515 (14,600), 549 (7,100), 589 (5,100), 645 (4,200).
5.2. Molecular Structures
Crystal structures were determined using data collected at low temperature on a Nonius KappaCCD diffractometer equipped with MoKα radiation (λ=0.71073 Å) and a graphite monochromator. Refinement was by full-matrix least squares, and all nonhydrogen atoms were treated anisotropically with H atoms included, except as described below. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications no. CCDC 663420–663423, CCDC 663925 and CCDC 644633. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK, (fax: +44- (0)1223–336033 or e-mail: deposit @ccdc.cam.ac.uk).
Crystal data for 1: colorless, C9H7Br2N, Mr=289.0, monoclinic space group P21/c, a=7.574(2), b=4.4039(10), c=28.315(8) Å, β=96.475(8)°, V=938.4(4) Å3, Z=4, ρcalcd=2.045 gcm−3, μ=8.579 mm−1, T=90K, 8408 data collected, R=0.021 (F2>2σ), Rw=0.050 (all F2) for 2279 unique data having θ<28.2° and 110 refined parameters. Absorption corrections by multi-scan method.
Crystal data for 2: colorless, C15H33B20N, Mr=443.6, monoclinic space group P21/n, a=17.526(2), b=7.8927(10), c=20.262(3) Å, β=109.743(6)°, V=2638.0(6) Å3, Z=4, ρcalcd=1.117 gcm−3, μ=0.052 mm−1, T=150K, 21359 data collected, R=0.071 (F2>2σ), Rw=0.202 (all F2) for 5089 unique data having θ<27.1° and 328 refined parameters.
Crystal data for 3: colorless, C15H34B20O, Mr=446.6, orthorhombic space group P212121, a=7.8923(10), b=12.211(2), c=27.548(5) Å, V=2654.9(7) Å3, Z=4, ρcalcd=1.117 gcm−3, μ=0.054 mm−1, T=110K, 22125 data collected, R=0.050 (F2>2σ), Rw=0.143 (all F2) for 4549 unique data having θ<30.5° and 327 refined parameters.
Crystal data for 4a: tan, C17H19N3, Mr=265.4, monoclinic space group P21/n, a=8.0907(10), b=8.735(2), c=20.075(4) Å, β=96.093(12)°, V=1410.7(5) Å3, Z=4, ρcalcd=1.249 gcm−3, μ=0.075 mm−1, T=110K, 22814 data collected, R=0.045 (F2>2σ), Rw=0.121 (all F2) for 5080 unique data having θ<32.5° and 189 refined parameters.
Crystal data for 5b: brown, C68H104B40N4O6P2. 3 C7H8, Mr=1844.3, monoclinic space group C2/c, a=25.249(7), b=22.017(6), c=19.894(5) Å, β=105.52(2)°, V=10656(5) Å3, Z=4, ρcalcd=1.150 gcm−3, μ=0.092 mm−1, T=110K, 48441 data collected, R=0.112 (F2>2σ), Rw=0.343 (all F2) for 7630 unique data having θ<23.3 and 569 refined parameters. All toluene solvent molecules are disordered, and were treated in half-populated sites with isotropic C atoms and a common Uiso for each molecule. Solvent H atoms were not included.
Crystal data for 6a: Black, C64H96B40N6. 4 CHCl3, Mr=1859.3, triclinic space group P-1, a=11.3640(12), b=18.753(2), c=23.905(4) Å, α=85.692(5), β=86.915(5), γ=73.912(6)°, V=4878.1(11) Å3, Z=2, ρcalcd=1.266 gcm−3, μ=0.384 mm−1, T=106K, 11914 data collected, R=0.171 (F2>2σ), Rw=0.457 (all F2) for 7736 unique data having θ<20.0° and 573 refined parameters. Due to the very weak scattering power of the crystals even at low temperature, it was not possible to do full anisotropic refinement. Only the Cl atoms were refined anisotropically, while the lighter atoms were treated as isotropic.
5.3. Absorption and Emission Spectra
The absorption spectra were measured on a Perkin Elmer Lambda 35 UV-vis spectrometer with 10 mm path length quartz cuvettes equipped with a photodiode detector, in the 300–700 nm wavelength range with 0.1 nm accuracy. Emission spectra were obtained on a Perkin Elmer LS55 luminescence spectrophotometer equipped with a Xenon lamp, in the 500–700 nm wavelength range with 1 nm accuracy. Stock solutions of all porphyrins in DMSO at 1 × 10−4 M were prepared. The aqueous solutions were prepared by adding 100 μL of each porphyrin DMSO stock solution into 10 mL of HEPES (N-2-hydroxyethylpiperazine-N′-2-ethansulfonic acid) buffer, 20 mM, pH 7.4. All measurements were performed within 2 h of solution preparation.
5.4. Cell Culture
HEp2 and T98G cells were acquired from the ATCC and maintained in a 50:50 mix of Advanced MEM:DMEM supplemented with 5% FBS (all from Invitrogen). The cultures were passaged twice weekly to maintain sub confluent stocks. Each porphyrin was prepared as a 10 mM stock in DMSO and diluted to a working concentration of 10 μM in two stages. First, equal volumes of 10 mM stock and Cremophor EL (Fluka) were mixed in a microfuge tube and sonicated using a bath sonicator for 10 min. This mixture was then diluted with DMSO to a final porphyrin concentration of 1 mM. This was then diluted by dropwise addition of medium to give a final concentration of 10 μM. The medium-containing porphyrin was then sonicated for 30 min.
5.4.1. Dark cytotoxicity
HEp2 cells were plated as described above and allowed 36 – 48 h to attach. The cells were then exposed to various concentrations of porphyrin up to 100 μM and incubated overnight. The loading medium was supplemented with CellTitre Blue (Promega) as per manufacturer’s instructions. Cell viability was measured by reading the fluorescence at 520/584nm using a BMG FLUOstar plate reader. The signal was normalized to 100% viable (untreated cells) and 0% viable (cells treated with 0.2% saponin (Sigma).
5.4.2. Time-dependent cellular uptake
T98G cells were plated at 20000 per well in a Costar 96 well plate and allowed to grow 36 h. Porphyrin stocks were prepared as above to 2x stocks. The porphyrin was then diluted in medium to final working concentrations. The cells were exposed to 10 μM porphyrin for 0, 1, 2, 4, 8, and 24 h. The uptake was stopped by removing the loading medium and by washing the cells with PBS. The cells were solubilized by the addition of 100 μl of 0.25% Triton X-100 (Calbiochem) in PBS. To determine compound concentration, fluorescence emission was read at 410/650 nm (excitation/emission) using a BMG FLUOstar plate reader. The cell numbers were quantified using CyQuant reagent (Molecular Probes).
5.4.3. Intracellular localization
HEp2 cells were plated on LabTek II two chamber coverslips and incubated 36 h. The cells were then exposed to 10 μM of each porphyrin for 24 h. For co-localization experiments using organelle tracers, cells were incubated concurrently with compound the following morning for 30 min with the following organelle tracers: Mitochondria: MitoTracker Green (Molecular Probes) 250 nM; Lysosomes: LysoSensor Green (Molecular Probes) 50 nM; Endoplasmic reticulum: ERTracker Green FM (Molecular Probes) 2 μg/ml (for 1 h); Golgi: BODIPY FL- C5-Ceramide (Molecular Probes) 1 mM. The slides were washed three times with growth medium and fed medium containing 50 mM HEPES pH 7.4. Fluorescent microscopy was performed using a Zeiss Labovert 200M inverted fluorescent microscope fitted with a standard FITC filter set (Ex/Em 470nm/540nm) for organelle tracers and standard Texas Red filter set (560nm/600 nm LP) set for compound detection (Chroma). The images were acquired with a Zeiss Axiocam MRM CCD camera fitted to the microscope.
Supplementary Material
Supplementary data
Dark cytotoxicity, fluorescence and HRMS data for porphyrins is available.
Figure 10.
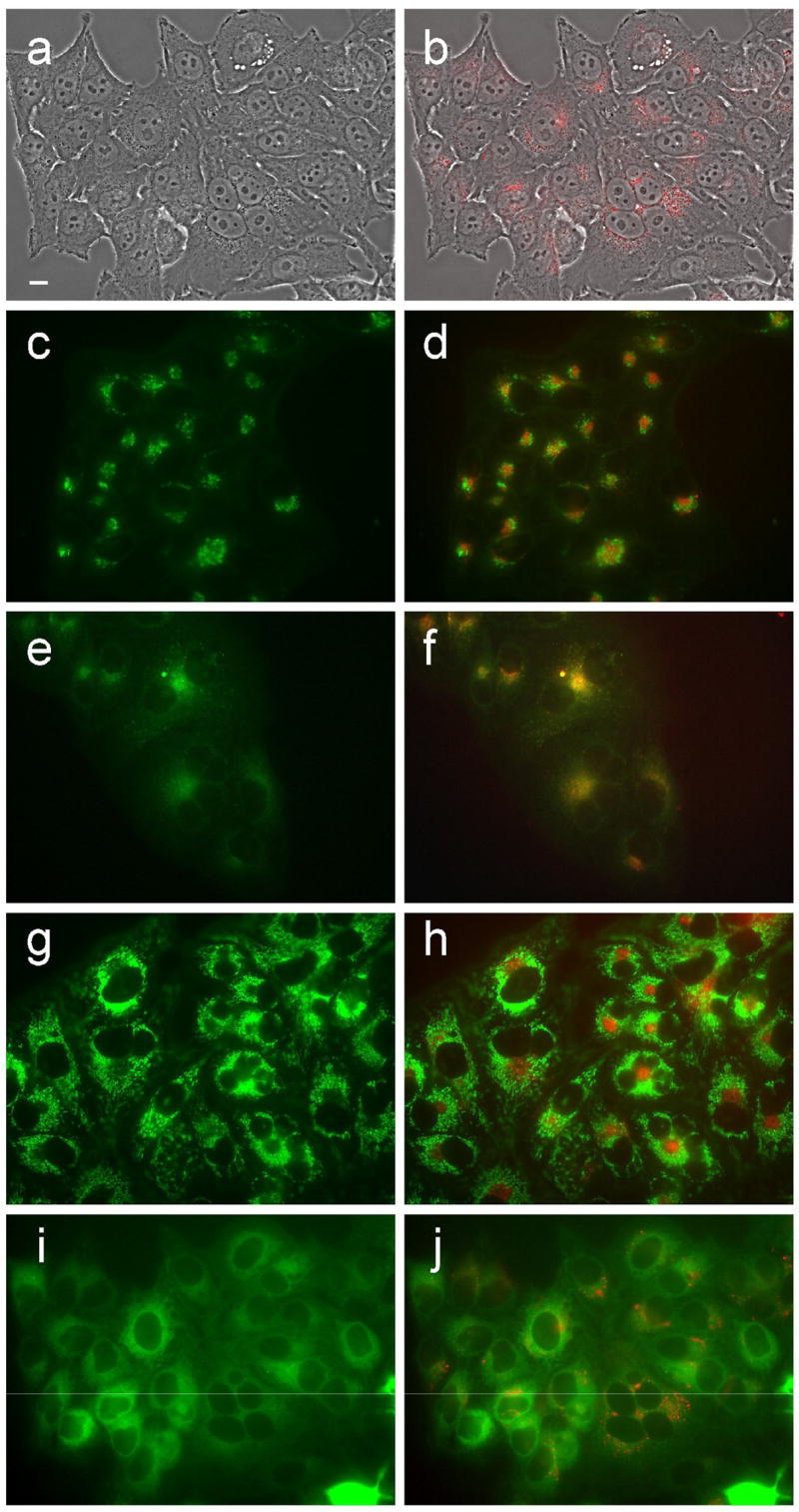
Intracellular localization of porphyrin 6d in HEp2 cells at 10 μM for 24 h. (a) phase contrast; (b) porphyrin fluorescence; (c) BODIPY FL- C5-Ceramide fluorescence (Golgi label); (d) overlay with porphyrin; (e) LysoSensor fluorescence (lysosome label); f: overlay; (g) MitoTracker fluorescence (mitochondria label); (h) overlay; (i) ERTracker Green FM fluorescence (ER label); (j) overlay. Scale bar: 10 μm.
Figure 11.
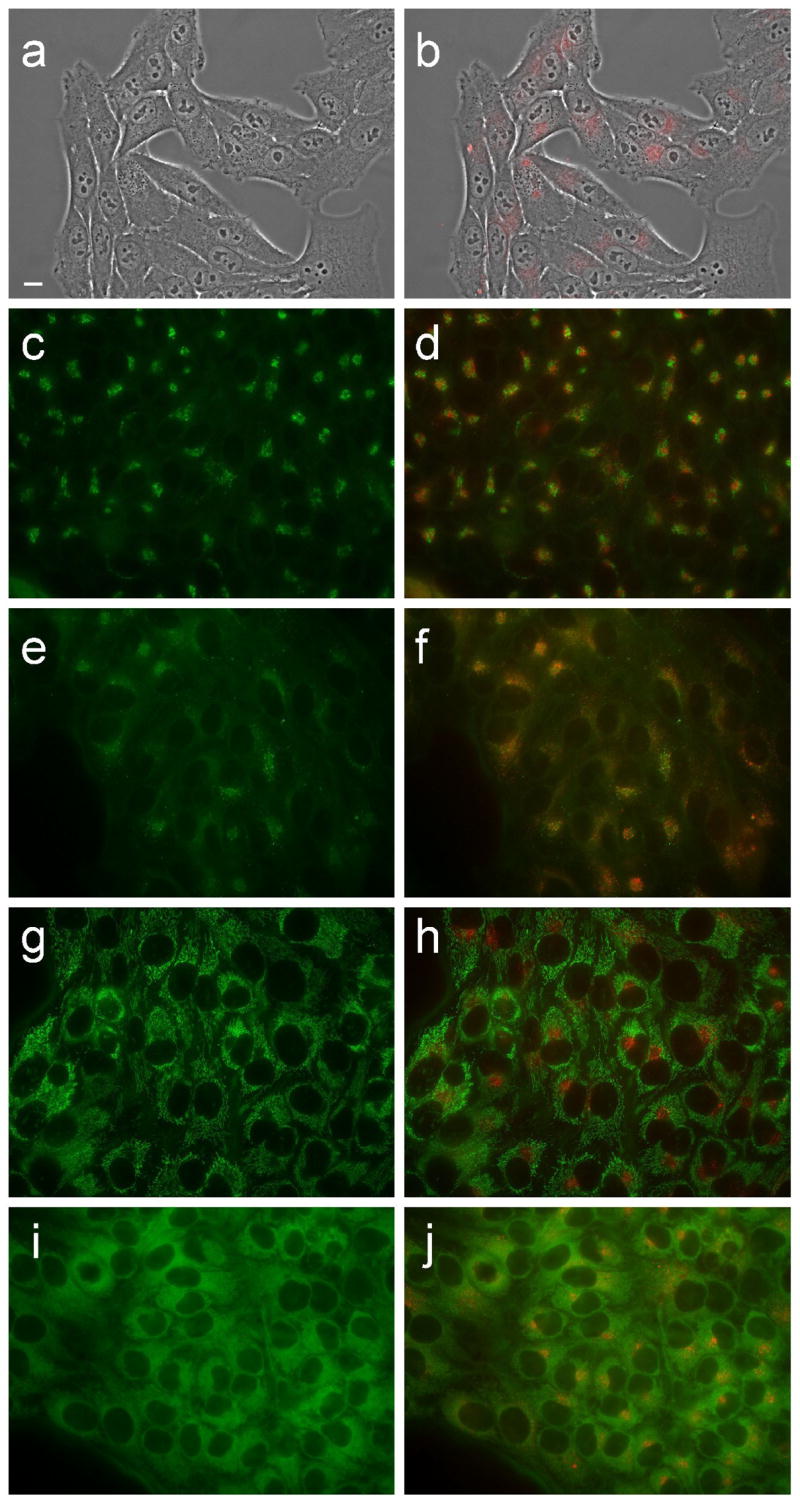
Intracellular localization of porphyrin 7c in HEp2 cells at 10 μM for 24 h. (a) phase contrast; (b) porphyrin fluorescence; (c) BODIPY FL- C5-Ceramide fluorescence (Golgi label); (d) overlay with porphyrin; (e) LysoSensor fluorescence (lysosome label); f: overlay; (g) MitoTracker fluorescence (mitochondria label); (h) overlay; (i) ERTracker Green FM fluorescence (ER label); (j) overlay. Scale bar: 10 μm.
Figure 12.
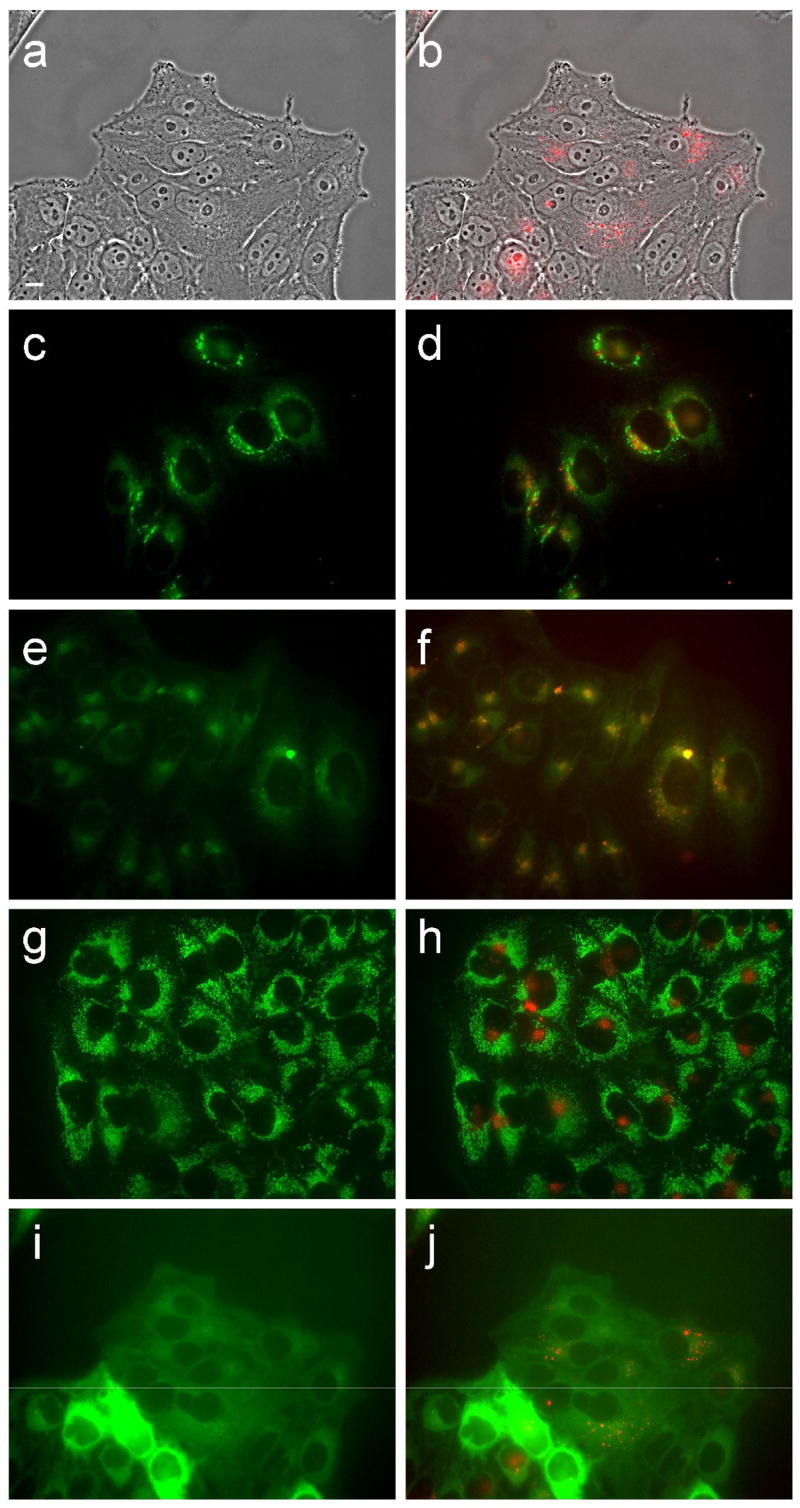
Intracellular localization of porphyrin 7d in HEp2 cells at 10 μM for 24 h. (a) phase contrast; (b) porphyrin fluorescence; (c) BODIPY FL- C5-Ceramide fluorescence (Golgi label); (d) overlay with porphyrin; (e) LysoSensor fluorescence (lysosome label); f: overlay; (g) MitoTracker fluorescence (mitochondria label); (h) overlay; (i) ERTracker Green FM fluorescence (ER label); (j) overlay. Scale bar: 10 μm.
Acknowledgments
The research described was supported by the National Institutes of Health, grant number R01 CA098902.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Barth RF, Soloway AH, Goodman JH, Gahbauer RA, Gupta N, Blue TE, Yang W, Tjarks W. Neurosurg. 1999;44:433–451. doi: 10.1097/00006123-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Barth RF. J Neuro-Oncol. 2003;62:1–5. doi: 10.1007/BF02699929. [DOI] [PubMed] [Google Scholar]
- 3.Barth RF, Coderre JA, Vicente MGH, Blue TE. Clin Cancer Res. 2005;11:3987–4002. doi: 10.1158/1078-0432.CCR-05-0035. [DOI] [PubMed] [Google Scholar]
- 4.Barth RF, Joensuu H. Radiother Oncol. 2007;82:119–122. doi: 10.1016/j.radonc.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa Y, Pooh K, Kobayashi T, Kageji T, Uyama S, Matsumura A, Kumada H. J Neuro-Oncol. 2003;62:87–99. doi: 10.1007/BF02699936. [DOI] [PubMed] [Google Scholar]
- 6.Diaz AZ. J Neuro-Oncol. 2003;62:101–109. doi: 10.1007/BF02699937. [DOI] [PubMed] [Google Scholar]
- 7.Busse PM, Harling OK, Palmer MR, Kiger WS, III, Kaplan J, Kaplan I, Chuang CF, Goorley JT, Riley KJ, Newton TH, Santa Cruz GA, Lu X-Q, Zamenhof RG. J Neuro-Oncol. 2003;62:111–121. doi: 10.1007/BF02699938. [DOI] [PubMed] [Google Scholar]
- 8.Joensuu H, Kankaanranta L, Seppälä T, Auterinen I, Kallio M, Kulvik M, Laakso J, Vähätalo J, Kortesniemi M, Kotiluoto P, Serén T, Karila J, Brander A, Järviluoma E, Ryynänen P, Paetau A, Ruokonen I, Minn H, Tenhunen M, Jääskeläinen J, Färkkilä M, Savolainen S. J Neuro-Oncol. 2003;62:123–134. doi: 10.1007/BF02699939. [DOI] [PubMed] [Google Scholar]
- 9.Capala J, Stenstam BH, Sköld K, af Rosenschöld PM, Giusti V, Persson C, Wallin E, Brun A, Franzen L, Carlsson J, Salford L, Ceberg C, Persson B, Pellettieri L, Henriksson R. J Neuro-Oncol. 2003;62:135–144. doi: 10.1007/BF02699940. [DOI] [PubMed] [Google Scholar]
- 10.Soloway AH, Tjarks W, Barnum BA, Rong FG, Barth RF, Codogni IM, Wilson JG. Chem Rev. 1998;98:1515–1562. doi: 10.1021/cr980493e. [DOI] [PubMed] [Google Scholar]
- 11.Bregadze VI, Sivaev IB, Glazun SA. Curr Med Chem, Anti-Cancer Agents. 2006;6:75–109. doi: 10.2174/187152006776119180. [DOI] [PubMed] [Google Scholar]
- 12.Renner MW, Miura M, Easson MW, Vicente MGH. Curr Med Chem, Anti-Cancer Agents. 2006;6:145–157. doi: 10.2174/187152006776119135. [DOI] [PubMed] [Google Scholar]
- 13.Ratajski M, Osterloh J, Gabel D. Curr Med Chem, Anti-Cancer Agents. 2006;6:159–166. doi: 10.2174/187152006776119126. [DOI] [PubMed] [Google Scholar]
- 14.Fairchild RG, Bond VP. Int J Rad Oncol Biol Phys. 1985;11:831–840. doi: 10.1016/0360-3016(85)90318-9. [DOI] [PubMed] [Google Scholar]
- 15.Gabel D, Foster S, Fairchild RG. Radiat Res. 1987;111:14–25. [PubMed] [Google Scholar]
- 16.This work was partially presented at the 12th International Congress on Neutron Capture Therapy for Cancer; Takamatzu (Japan). October 2006. [Google Scholar]; Vicente MGH, Easson MW. Proc 12th ICNCT for Cancer. 2006:231–233. [Google Scholar]
- 17.Mettath S, Munson BR, Pandey RK. Bioconj Chem. 1999;10:94–102. doi: 10.1021/bc9800872. [DOI] [PubMed] [Google Scholar]
- 18.Bustamante C, Gurrieri S, Pasternack RF, Purrello R, Rizzarelli E. Biopolymers. 1994;34:1099–1104. doi: 10.1002/bip.360340813. [DOI] [PubMed] [Google Scholar]
- 19.Kessel D, Luguya R, Vicente MGH. Photochem Photobiol. 2003;78:431–435. doi: 10.1562/0031-8655(2003)078<0431:lapeot>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Sibrian-Vazquez M, Nesterova IV, Jensen TJ, Vicente MGH. Bioconj Chem. accepted for publication. [Google Scholar]
- 21.Gottumukkala V, Luguya R, Fronczek FR, Vicente MGH. Bioorg Med Chem. 2005;13:1633–1640. doi: 10.1016/j.bmc.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 22.Bodwell GJ, Bridson JN, Houghton TJ, Yarlagadda B. Tetrahedron Lett. 1997;38:7475–7478. [Google Scholar]
- 23.Vicente MGH, Wickramasinghe A, Nurco DJ, Wang HJH, Nawrocky MM, Makar MS, Miura M. Bioorg Med Chem. 2003;11:3101–3108. doi: 10.1016/s0968-0896(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 24.McIlroy SP, Cló E, Nikolajsev L, Frederiksen PK, Nielsen CB, Mikkelsen KV, Gothelf KV, Ogilby PR. J Org Chem. 2005;70:1134–1146. doi: 10.1021/jo0482099. [DOI] [PubMed] [Google Scholar]
- 25.Lee CH, Lindsey JS. Tetrahedron. 1994;50:11427–11440. doi: 10.1016/j.tet.2008.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Littler BJ, Miller MA, Hung CH, Wagner RW, O’Shea DF, Boyle PD, Lindsey JS. J Org Chem. 1999;64:1391–1396. [Google Scholar]
- 27.Durantini EN. Molecules. 2001;6:533–539. [Google Scholar]
- 28.Littler BJ, Ciringh Y, Lindsey JS. J Org Chem. 1999;64:2864–2872. doi: 10.1021/jo982452o. [DOI] [PubMed] [Google Scholar]
- 29.Smith KM, Vicente MGH. Houben-Weyl Science of Synthesis. In: Weinreb SM, editor. Georg Thieme Verlag. Vol. 17. Stuttgart; 2004. pp. 1081–1235. [Google Scholar]
- 30.Muthukumaran K, Loewe RS, Ambroise A, Tamaru S, Li Q, Mathur G, Bocian DF, Misra V, Lindsey JS. J Org Chem. 2004;69:1444–1452. doi: 10.1021/jo034945l. [DOI] [PubMed] [Google Scholar]
- 31.Arsenault GP, Bullock E, MacDonald SF. J Am Chem Soc. 1960;82:4384–4389. [Google Scholar]
- 32.Senge MO. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego: 2000. pp. 239–347. [Google Scholar]
- 33.Lauceri R, Purrello R, Shetty SJ, Vicente MGH. J Am Chem Soc. 2001;123:5835–5836. doi: 10.1021/ja015581m. [DOI] [PubMed] [Google Scholar]
- 34.Vicente MGH, Nurco DJ, Shetty SJ, Osterloh J, Ventre E, Hegde V, Deutsch WA. J Photochem Photobiol B:Biol. 2002;68:123–132. doi: 10.1016/s1011-1344(02)00383-4. [DOI] [PubMed] [Google Scholar]
- 35.De Napoli M, Nardis S, Paolesse R, Vicente MGH, Lauceri R, Purrello R. J Am Chem Soc. 2004;126:5934–5935. doi: 10.1021/ja0494757. [DOI] [PubMed] [Google Scholar]
- 36.Hao E, Sibrian-Vazquez M, Serem W, Garno JC, Fronczek FR, Vicente MGH. Chem Eur J. 2007;13:9035–9042. doi: 10.1002/chem.200700752. [DOI] [PubMed] [Google Scholar]
- 37.Woodburn K, Phadke AS, Morgan AR. Bioorg Med Chem Lett. 1993;3:2017–2022. [Google Scholar]
- 38.Hill JS, Kahl SB, Stylli SS, Nakamura Y, Koo MS, Kaye AH. Proc Natl Acad Sci USA. 1995;92:12126–12130. doi: 10.1073/pnas.92.26.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicente MGH, Edwards BF, Shetty SJ, Hou Y, Boggan JE. Bioorg Med Chem. 2002;10:481–492. doi: 10.1016/s0968-0896(01)00300-5. [DOI] [PubMed] [Google Scholar]
- 40.Miura M, Micca PL, Fisher CD, Gordon CR, Heinrichs JC, Slatkin DN. Br J Radiol. 1998;71:773–781. doi: 10.1259/bjr.71.847.9771389. [DOI] [PubMed] [Google Scholar]
- 41.Ol’shevskaya VA, Zaitsev AV, Luzgina VN, Kondratieva TT, Ivanov OG, Kononova EG, Petrovskii PV, Mironov AF, Kalinin VN, Hofmann J, Shtil AA. Bioorg Med Chem. 2006;14:109–120. doi: 10.1016/j.bmc.2005.07.067. [DOI] [PubMed] [Google Scholar]
- 42.Ol’shevskaya VA, Nikitina RG, Zaitsev AV, Luzgina VN, Kononova EG, Morozova TG, Drozhzhina VV, Ivanov OG, Kaplan MA, Kalinin VN, Shtil AA. Org Biomol Chem. 2006;4:3815–3821. doi: 10.1039/b607766h. [DOI] [PubMed] [Google Scholar]
- 43.Kubat P, Lang K, Cigler P, Kozisek M, Matejicek P, Janda P, Zelinger Z, Prochazka K, Kral V. J Phys Chem B. 2007;111:4539–4546. doi: 10.1021/jp066494p. [DOI] [PubMed] [Google Scholar]
- 44.Matsumura A, Shibata Y, Yamamoto T, Yoshida F, Isobe T, Nakai K, Hayakawa Y, Kiriya M, Shimojo N, Ono K, Sakata I, Nakajima S, Okumura M, Nose T. Cancer Lett. 1999;141:203–209. doi: 10.1016/s0304-3835(99)00105-6. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen T, Brownell GL, Holden SA, Teicher BA. Biochem Pharma. 1993;45:147–155. doi: 10.1016/0006-2952(93)90387-c. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen T, Brownell GL, Holden SA, Kahl S, Miura M, Teicher BA. Radiat Res. 1993;133:33–40. [PubMed] [Google Scholar]
- 47.Callahan DE, Forte TM, Afzal SMJ, Deen DF, Kahl SB, Bjornstad KA, Bauer WF, Blakely EA. Int J Rad Oncol Biol Phys. 1999;45:761–771. doi: 10.1016/s0360-3016(99)00172-8. [DOI] [PubMed] [Google Scholar]
- 48.Hao E, Jensen TJ, Courtney BH, Vicente MGH. Bioconj Chem. 2005;16:1495–1502. doi: 10.1021/bc0502098. [DOI] [PubMed] [Google Scholar]
- 49.Osterloh J, Vicente MGH. J Porphyrins and Phthalocyanines. 2002;6:305–324. [Google Scholar]
- 50.Novick S, Laster B, Quastel MR. Int J Biochem Cell Biol. 2006;38:1374–1381. doi: 10.1016/j.biocel.2006.01.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Dark cytotoxicity, fluorescence and HRMS data for porphyrins is available.