

Abstract
The linear pentadecapeptide antibiotic, gramicidin D, is a naturally occurring product of Bacillus brevis known to form ion channels in synthetic and natural membranes. The x-ray crystal structures of the right-handed double-stranded double-helical dimers (DSDHℛ) reported here agree with 15N-NMR and CD data on the functional gramicidin D channel in lipid bilayers. These structures demonstrate single-file ion transfer through the channels. The results also indicate that previous crystal structure reports of a left-handed double-stranded double-helical dimer in complex with Cs+ and K+ salts may be in error and that our evidence points to the DSDHℛ as the major conformer responsible for ion transport in membranes.
The ion-channel forming peptide antibiotic gramicidin A (gA; ref. 1), HCO-l-Val1-Gly2-l-Ala3-d-Leu4-l-Ala5-d-Val6-l-Val7-d-Val8-l-Trp9-d-Leu10-l-Trp11-d-Leu12-l-Trp13-d-Leu14-l-Trp15-NHCH2CH2OH, is selective for monovalent cations such as H+, Tl+, NH4+ and the alkali metals (2). Despite over two decades of study, the structure of the active gramicidin channel remains a source of controversy. Two principal models were originally proposed; an intertwined double-stranded double helix (DSDH)† proposed by Veatch et al. (3) and a head-to-head single-stranded helix (HHSH) proposed by Urry et al. (4). Five x-ray crystal structures of gramicidin have been reported, three uncomplexed forms crystallized from different length n-alcohols [methanol (5), ethanol (6), and n-propanol (7)] and two ion complexed forms [Cs+ (8) and K+ (9)] crystallized from methanol. All five structures are reported to be DSDHℒ dimers, with 5.6 residues/turn in the uncomplexed structures and 6.4 residues/turn in the cation complexed structures. Although the nature and number of hydrogen bonds differs between the uncomplexed structures and the Cs+ and K+ complexes, they all contain a common dimeric ribbon linked by 16 hydrogen bonds, and the d,l composition places all of the amino acid residues on one side of this ribbon. Because of steric crowding, the ribbon coils away from the side chain covered surface and forms a second chain of hydrogen bonds, thus forming a helical structure. Although there are 14 hydrogen bonds in the second set in both the complexed and uncomplexed structures, the registry is shifted by two residues in the complexed structures. This shifting makes the channel diameter larger and the channel length shorter to accommodate ions in the complexes. The biological relevance of these structures has been questioned primarily because the left-handed coils do not agree with the measured CD spectra of gramicidin in lipid bilayers.
In addition, two NMR determinations of gA have been reported; a solution structure in SDS micelles (10) and a solid-state structure in oriented lipid bilayers (11). Although there are small differences in the details of these structures, the folds are the same and are of the HHSHℛ type. It has been possible to reconcile the CD spectra and some of the NMR spectra on gA in lipid bilayers with the HHSHℛ dimer, and this model has been accepted as the consensus model responsible for ion transport by gA.
In this paper we report the crystal structures of the Cs+ complex of gramicidin crystallized from methanol and that of H3O+ bound gramicidin crystallized from glacial acetic acid. Both structures demonstrate a new fold, a right-handed double-stranded antiparallel double helix (DSDHℛ) form, that is consistent with most of the existing literature on gramicidin structure, ion conductance, and side-chain interactions including a previously published solution NMR structure of a Cs+ complexed gramicidin (12). More importantly, the structures demonstrate a remarkable similarity with the solid-state NMR data of gramicidin in oriented bilayers (11).
Structure Solution
For the Cs+ complexed structure, commercially available gramicidin D (Sigma) was crystallized by slow evaporation from methanolic solutions (30 mg/ml) containing 250 mM CsCl. Gramicidin D is a naturally occurring mixture composed of approximately 85% gA, and 10% and 5% of isomers having tyrosine (gC) and phenylalanine (gB), respectively, substituted for tryptophan at position 11. A large lenticular crystal suitable for data collection was mounted in a glass capillary with mother liquor, and data were collected to 1.4 Å on an R-Axis II image plate by using CuKα radiation while preserving the anomalous scattering from Cs+ (f" = 7.9 e− (Table 1)). The space group, P212121, and cell constants (a = 31.06 Å, b = 31.88 Å, c = 52.11 Å) are the same (<1% difference) as those reported by Wallace and Ravikumar (8) and yield two dimers/asymmetric unit. The structure was solved by molecular replacement with amore (13) by using a polyalanine model based on the hydrogen bonding scheme proposed by Wallace and Ravikumar (8). The best solution had a correlation coefficient of 58% and an R factor of 45.3% for the 1,542 data between 5.0 and 2.5 Å. Electron density maps based on these coordinates did not allow interpretation of amino acid side- chain positions, especially for Trp. Furthermore, this structure was demonstrated to have the incorrect hand based on Cs+ anomalous scattering phased by the protein atoms. Inversion of the atomic positions improved the electron density maps and allowed Trp, Leu, and Val side chains to be fit unambiguously. Despite the fact that both crystals were obtained from a gramicidin D mixture, there is no evidence of the other isomers in the electron density maps, as seen in the uncomplexed gramicidin dimers (7). It is possible the lower resolution of the complexed structures could obscure the subtle details of chemical heterogeneity at positions 1 and 11. The Cs+ anomalous scattering demonstrated that in the final refinement, the hand of the structure is correct. The refined structure bears no resemblance whatsoever to the hydrogen bonding scheme and left-handed structure proposed by Wallace and Ravikumar (8). We conclude that the structure proposed by Wallace and Ravikumar (8) is incorrect.
Table 1.
Crystallographic data regarding the data collection and structure refinement of the DSDHℛ complexes
| Cs+ | H+ | |
|---|---|---|
| Cell constants, Å | a = 31.06 | a = 20.58 |
| b = 31.88 | b = 27.90 | |
| c = 52.11 | c = 52.04 | |
| Space group | P212121 | P212121 |
| No. of reflections measured | 39,976 | 11,764 |
| No. of unique reflections | 17,776 | 3,416 |
| R merge, % | 5.5 | 5.0 |
| Completeness, % | 91.3 | 95.0 |
| No. of parameters | 5,644 | 1,299 |
| No. of restraints | 6,744 | 1,276 |
| RcrystF > 4σ F | 14.6 | 17.8 |
| All data | 15.6 | 19.1 |
| RfreeF > 4σ (F) | 19.5 | 23.8 |
| All data | 20.0 | 24.9 |
| rms deviation from ideal | ||
| Bond lengths (1-2), Å | 0.015 | 0.020 |
| Bond angles (1-3), Å | 0.035 | 0.040 |
The structure of the H+ complex was crystallized from a concentrated solution (50 mg/ml) of commercial gramicidin D (Sigma) in glacial acetic acid. Large rods appeared over several months. A fragment of one of these crystals was mounted in a glass capillary and data were collected to 1.7 Å in the same manner as the Cs+ complex. The cell constants are entirely new for gramicidin crystals (a = 20.58 Å, b = 27.90 Å, c = 52.04 Å) with space group P212121 and only one dimer/asymmetric unit. The structure was solved by molecular replacement with amore (13) by using the coordinates of the Cs+ complex. The best solution had a correlation coefficient of 71.4% and an R factor of 46% for 2,993 data between ∞ and 1.8 Å. Refinements for both the Cs+ complex and H+ derived structure were conducted with shelxl-97 (14) and the details are reported in Table 1. Both coordinate sets have been deposited in the Protein Data Bank with codes 1AV2 (Cs+) and 1BDW (H+).
Structure of the Dimer
The two crystal structures contain nearly identical right-handed double-stranded double helices of gA with 7.2 residues/turn (Fig. 1). The hydrogen bonding pattern required to assemble such a dimer is substantially different from that found in any gramicidin structure or model previously reported. Gramicidin can be considered as two “zippers” of hydrogen bonds, one set, that is seen in all of the previous crystal structures, assembles the two-stranded antiparallel β-sheet and contains 16 hydrogen bonds. The second set arises from the coiling of gramicidin upon itself to relieve the steric crowding arising from having all side chains on the same side of the sheet. In the three similar, uncomplexed β5.6 DSDHℒ gramicidin dimers (5–7), this second set contains 14 hydrogen bonds. Although the two peptide strands are joined by 16 hydrogen bonds in both complexed and uncomplexed forms, the disposition of the strands relative to one another is opposite and hence the H-bonding pattern is different. These distinctly different double-stranded ribbons are coiled in opposite directions and stabilized by 14 additional hydrogen bonds in DSDHℒ but only by 10 additional hydrogen bonds in DSDHℛ. The DSDHℛ intradimer hydrogen bonding leaves six potential hydrogen bond sites in the backbone free. Four of these six sites are occupied by symmetry-related dimers above and below the central dimer, forming continuous tubes along the c-axis in both crystal structures. The amide nitrogen and carbonyl oxygens of Ala5 are the only sites on each backbone that do not hydrogen-bond to other sites on the backbone or a symmetry-related backbone. In all cases, this site is occupied by a solvent or counter ion.
Figure 1.

(a) Stereographic illustration of the structure of the Cs+ complexed DSDHℛ gramicidin dimer demonstrating the right-handed nature of the helix. (b) The H+ complexed DSDHℛ gramicidin dimer crystallized from glacial acetic acid.
The DSDHℛ Conformation in Lipid Membranes
NMR studies of 15N-labeled gA (including Trp side chains) in oriented lipid bilayers have been used to determine the orientation of the backbone N—H dipoles relative to the helix axis (15) and thus the structure. Three major multiresonance peaks have been attributed to N—H bonds making angles of 15°, 25°, and 39° with the direction of the magnetic field perpendicular to the bilayer normal. In a related study using 2H-labeled gramicidin, similar results also were obtained (16). The six crystallographically independent strands in the structures reported here provide information on the range of stable orientations of individual N—H bonds in the dimer. The precise angles made between the N—H bonds and the channel axis for all 16 backbone N—H groups and the 4 tryptophan N—H groups have been calculated for each of the six independent strands in the two structures. When these angles are displayed on a histogram (Fig. 2a) the profile shows a remarkable parallel with the observed 15N-NMR spectra. The x-ray observations not only match the distribution assigned to the three strongest peaks as calibrated by Nicholson et al. (15) but demonstrate the well-defined bimodal distribution reflected in the spectra and appear to account for many of the weaker peaks. A plot of the individual N—H angles (Fig. 2b) reveals a well defined pattern variation between the d (red) and l (blue) residues that accounts for the bimodal distribution. The d residues cluster around 35° and the l residues cluster near 15°. There is a clear structural reason for this behavior. The l-amino acids lie in a shallow conformation very similar to that of the HHSHℛ. However, the d-amino acids have a larger pitch than the l-amino acids that causes the DSDHℛ monomer to double in length compared with the HHSHℛ. Finally, the features at low field appear to be attributable to the N—H groups on the indole rings of the tryptophan residues. All 11 of the θ values of 40° or larger are angles made by N—H bonds in indole rings. Although the strength of these signals is weaker, their presence in the spectra suggests that the tryptophan orientations observed in the solid state are largely retained in the lipid environment.
Figure 2.
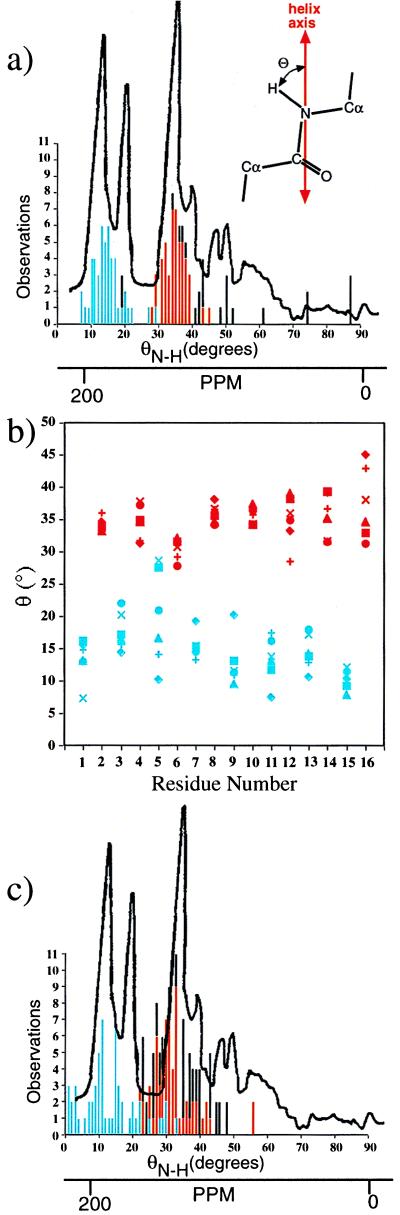
(a) Histogram of observed N—H dipole angles (θN—H) of the 16 backbone and 4 indole residues measured from the two crystal structures relative to the helix axis (120 observations) compared with the measured 15N-NMR pattern (adapted from ref. 15). The l-amino acids are recorded in blue, the d-amino acids in red, and Trp side chain in black. The θN—H scale was aligned with the ppm scale by using the calibration reported in ref. 15, 198 ppm ≈ 15°, 180 ppm ≈ 25°, and 146 ppm ≈ 39°. Although there is not a linear relationship between the two scales, the appropriate alignment using this calibration demonstrates the excellent agreement between the ensemble of N—H angles measured in the helices and the NMR data. The remarkable similarity between the two patterns strongly suggests that the DSDHℛ dimer is the conformation present in lipids where the spectra were recorded (15). (b) The observed variation in N—H dipole angles associated with the 16 backbone N—H bonds in the six strands of the gramicidin D in the DSDHℛ dimer. The characteristic difference in the angles of the N—H bonds of the d and l residues and the enhanced flexibility of l-Ala5 residue are noteworthy. (c) Comparison between the 15N-NMR spectrum described in a and the measured N—H angles in the two published HHSHℛ NMR structures (10, 11) (Protein Data Bank codes 1MAG and 1GRM). The measured crystallographic angles fit the 15N-NMR spectra better than the values calculated for the HHSHℛ models.
These results clearly demonstrate that the N—H conformations observed in the solid-state mimic the N—H conformations present in the lipid bilayer, where gramicidin is clearly active (2). Hence, the structure observed in the crystalline states and in the lipid bilayer may be the same. Because methanol stabilizes an inactive conformation of gramicidin (5), the cation must play a role in inverting the helical sense and hydrogen bonding in the more polar methanol environment. However, the new determination of gramicidin in a glacial acetic acid environment demonstrates that the cations are unnecessary if the environment is less polar. The dielectric constant of bulk glacial acetic acid is 5–6, very similar to that of the interior of lipid bilayers. This property of the solvent allows the handedness of the structure to invert and creates what must be the active, ion-conducting conformation as demonstrated by the similarity to the 15N-NMR results described above.
Since it was first postulated by Urry et al. in 1971 (4), a right-handed head-to-head single helical dimer (HHSHℛ) has been assumed to be the conformation of gramicidin responsible for ion transport in membranes (10, 11). The HHSHℛ model does not agree as well with the recorded data (Fig. 2c). The DSDHℛ dimer also would be compatible with the observed CD spectra in a lipid environment because it has the right-handed helical twist agreeing with the CD spectra of gA in long-chain lipids (17). The tryptophan side-chain orientations observed in the DSDHℛ dimer also exhibit unequivocal correlation with the biochemical data. Indole orientations determined for the Trp side chains are consistent with Raman scattering (18) which requires χ2 to be approximately ±90°. Furthermore, it has been shown that the extent of lipid interactions for each of the four Trp residues varies as follows: Trp9 > Trp11 > Trp13 > Trp15; however, all four Trp residues interact with the hydrophobic acyl chains of the lipid (19). The DSDHℛ dimer conformation is fully consistent with these observations having Trp15 exposed to water near the top and bottom of the dimer and Trp9 exposed only to lipid. The reported activity of covalently linked head-to-head dimers of gramicidin (20, 21) would appear to owe their activity to some aggregated state or conformation unrelated to the activity of native (or wild-type) gramicidin.
The Channel and Ion Binding
The channel in which Cs+ binds is fairly uniform, averaging 4.6 Å in van der Waals diameter over the entire length of the dimer. No significant inward tilting of carbonyls to complex the bound ions and water molecules is detected. This finding contradicts the hypothesis that as ions pass through the channel, carbonyl oxygens tilt inward to form a coordination sphere around them (22, 23). Tian et al. (24) have presented evidence that the gramicidin dimer does not distort during ion passage. Our results are consistent with this model with little distortion in the backbone. The more uniform, slightly convex surface of the DSDHℛ dimer is better suited to membrane entry and lipid packing (Fig. 3 a and c) than is the HHSHℛ dimer (Fig. 3 b and d). The electrostatic potential of the outer surface of the DSDHℛ dimer is predominantly neutral and well suited for membrane insertion (Fig. 3). The channel lumen has a predominantly negative electrostatic surface potential suited to cation passage. In addition, these electrostatic surface calculations suggest a higher concentration of negative potentials in the Cs+ binding sites (Fig. 5). It appears that electrostatic interaction between the Cs+ ions and the channel wall, which is lined with backbone hydrogen bonds, orients π electrons in the amide bonds toward the center of the channel to easily interact with passing ions. Because electrostatic potentials are much stronger in low dielectric media (e.g., in the lipid bilayer), the interactions of the Trp side chains with the uniform channel may be creating local dipoles.
Figure 3.
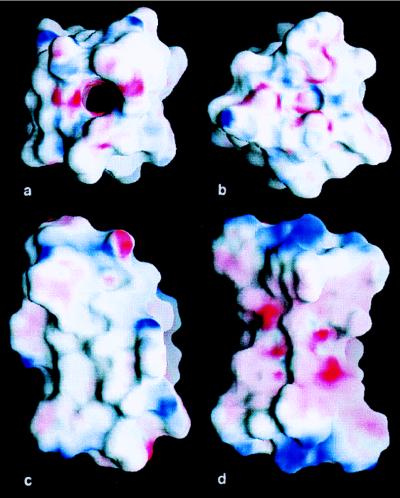
grasp maps of the electrostatic surfaces of the DSDHℛ and HHSHℛ (Protein Data Bank code 1MAG). View down the (a) DSDHℛ dimer and (b) the HHSHℛ dimer in an aqueous (ɛ = 80) environment. The colors run from +20 kT/e (blue) to −20 kT/e (red). The inner surface of the open DSDHℛ channel has a negative electrostatic charge required to attract and transport cations. In contrast, the HHSHℛ dimer does not have an open channel for ion passage. Exterior view of the (c) DSDHℛ dimer and (d) HHSHℛ dimer in a lipidic environment (ɛ = 2). The colors run from +40 kT/e (blue) to −40 kT/e (red). The exterior of the DSDHℛ dimer is largely neutral for membrane insertion, with partially charged atoms near the ends of the molecules. In contrast, at the same contours, the center of HHSHℛ dimer has a negative electrostatic surface in the centers of the molecule that would be embedded in the lipid bilayer.
Figure 5.
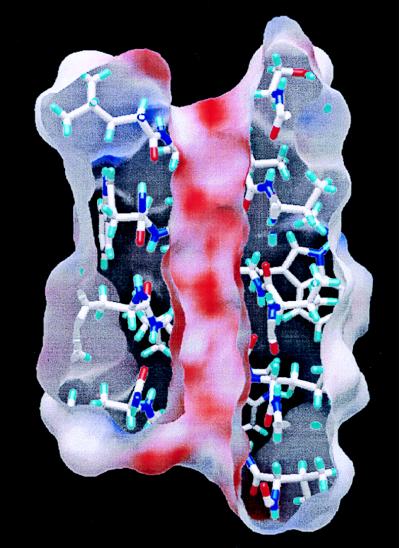
A slice through the DSDHℛ dimer reveals a largely negative internal electrostatic surface. The electrostatic surface is colored from +50 kT/e (blue) to −50 kT/e (red). The electrostatic surface spirals around the inside surface, following the carbonyl oxygens.
Each channel contains a single Cs+ ion that is distributed over three sites in the channel lumen. Each Cs+ has eight closest contacts on average with six interactions arising from nearby carbonyl oxygens with 3.6- to 4.5-Å distances and a pair of water molecules above and below the cation position at 2.8–3.1 Å (Fig. 4). The refined occupancies of the Cs+ ions sum to near unity with water molecules present between Cs+ sites and distributed throughout the channel when Cs+ is elsewhere in the channel. Although crystallographic refinement of atomic thermal parameters and their occupancies are highly correlated, the thermal parameters of the Cs+ ions closely match the average thermal parameters of the nearby backbone atoms, suggesting that the refined Cs+ occupancies are reasonable. There does not appear to be Cs+ present anywhere except in the channel lumen as confirmed by the anomalous scattering. These results are consistent with a nonpolar channel exterior that should not bind charged ions. Furthermore, there is no evidence of any anion binding in the channel lumen as previously reported (8). However, the water structure within the Cs+ bound channel contains 7.5 bound waters taking into account the 3-fold disorder. Furthermore, the structure obtained from acetic acid contains a chain of 7.5 disordered waters distributed over 15 sites, representing the equilibrium water structure inside the channel lumen. Tripathi and Hladky (25) report that as each ion passes through the gramicidin channel 7 waters are carried through the channel when the conducting ion is Na+, K+, or Cs.
Figure 4.

(a) A representative example of the interactions between Cs+ and gramicidin in the center of each channel. There are six closest interactions formed between carbonyls and the Cs+. (b) Similar interactions are formed at the ends of the channel but with poorer coordination.
The positions of the bound metal ions in gramicidin have also been studied extensively spectroscopically. Recent 13C-NMR investigations find that Na+ interacts most strongly with the carbonyl of Leu10 and two axial water molecules (26) and that transient interactions are made with the carbonyls of Val8 and Trp15. The Cs+ in the central binding site in the channel lumen is 3.7–3.8 Å from the carbonyl carbon of Leu10. Furthermore, the major coordinating atom, aside from the 2 axial water molecules, is the oxygen of Val8, suggesting interaction with the carbonyls of Val8 as well. In the peripheral binding sites, there is proximal interaction with the carbonyl of Trp15 (3.7–3.8Å).
Summary
The DSDHℛ structure reveals a 1:2 complex of Cs+:gA; demonstrates a mechanism of single file ion transfer through the gA lumen; satisfies NMR, CD, and Raman spectra for the conducting form of gA; and resolves previous disparities between solid-state and lipid solvent conformations of gA. These results suggest that, although the uncomplexed DSDHℒ appears to play a critical role in the initial insertion of gA into the membrane (27), the DSDHℛ dimer is the major conformer responsible for ion transfer and may constitute the “normal” channel. Although it may be too soon to rule out the possibility that the long cherished head-to-head helical dimer model is also a conducting species, these new structures and their agreement with CD and 15N-NMR spectra strongly suggest that the structural form described in this paper is the membrane transport form and that other existing data on gramicidin function should be re-examined in the light of these unexpected findings. It remains to be seen whether other conformations of complexed and unusual gA homo- and heterodimers contribute to the complex solution spectra and physiological and biological properties of gA. Of immediate interest will be the mechanics of interconversion between DSDHℒ to DSDHℛ in membrane lipids.
In addition, the results reported here raise serious doubts as to the accuracy of the previously published crystallographic structures of the Cs+ (8) and K+ (9) complexes of gramicidin. Chemically, the presence of anions within the channels of both the Cs+ and K+ structures is anomalous because the single-valency cation selectivity of gramicidin is well established (2). Similarly, there appears to be no reason for Cs+ to bind external to the channel as found in the previously published model. More troubling are the crystallographic anomalies that appear to be present. Careful examination of the figures in ref. 8 reveals that several amino acids appear to have the wrong chirality. Given the necessity for the Cα—Cβ bond to project away from the helix axis, incorrect gramicidin acid chirality severely distorts the local geometry at these sites. In the K+ (9) structure the data were truncated at 2.5 Å despite the claimed presence of diffraction data to 1.8 Å, several Trp side chains were omitted from the refinement and many of the backbone torsion angles refined to highly unusual conformations. Such observations are considered hallmarks of incorrect structures (28). Therefore, the improved resolution of the structures reported here, 1.4 Å and 1.7 Å, coupled with the absence of unusual structural and crystallographic features, and confirmation of the hand by Cs+ anomalous scattering, strongly demonstrate the correctness and accuracy of the new structures. Unequivocal resolution of the sources of the anomalous features of the previously reported C+ (8) and K+ (9) complexes must await release of the diffraction data. It should be noted that the crystal structures of uncomplexed gramicidin for which atomic coordinates have been deposited in the Protein Data Bank, refined at 0.86, 1.13, and 1.20 Å resolution, exhibit no structural anomalies.
Table 2.
Average and rmsd values for the φ, ψ angles of the l- and d-amino acids in the three DSDHℒ dimers and the newly determined DSDHℛ dimers
| Structure (Type) | 〈l〉 | 〈d〉 | |
|---|---|---|---|
| Methanol (x-ray) | (DSDHℒ ββ5.6) | −156 ± 7/116 ± 19 | 98 ± 21/−138 ± 8 |
| Ethanol (x-ray) | (DSDHℒ ββ5.6) | −152 ± 8/109 ± 22 | 101 ± 19/−140 ± 8 |
| n-Propanol (x-ray) | (DSDHℒ ββ5.6) | −153 ± 8/109 ± 21 | 101 ± 20/−141 ± 8 |
| Arseniev (NMR) | (HHSHℛ β6.3) | −118 ± 18/133 ± 24 | 128 ± 22/−119 ± 19 |
| Cross (NMR) | (HHSHℛ β6.3) | −116 ± 6/134 ± 13 | 132 ± 17/−124 ± 7 |
| Burkhart (x-ray) | (DSDHℛ ββ7.2) | −110 ± 11/140 ± 8 | 148 ± 6/−136 ± 8 |
Acknowledgments
This research was supported by Grant GM-32812 from the National Institutes of Health.
ABBREVIATIONS
- DSDH
double-stranded double helix
- HHSH
head-to-head single-stranded helix
- gA
gramicidin A
Footnotes
Data depostion: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID codes 1AV2 and 1BDW).
These can be either the left handed (i.e., DSDHℒ or HHSHℒ) or right-handed (i.e., DSDHℛ or HHSHℛ).
References
- 1. Hotchkiss R D. Adv Enzymol. 1944;4:153–199. [Google Scholar]
- 2.Mueller P, Rudin D O. Biochem Biophys Res Commun. 1965;26:398–404. doi: 10.1016/0006-291x(67)90559-1. [DOI] [PubMed] [Google Scholar]
- 3.Veatch W R, Fossel E T, Blout E R. Biochemistry. 1974;13:5249–5256. doi: 10.1021/bi00723a001. [DOI] [PubMed] [Google Scholar]
- 4.Urry D W, Goodall M C, Glickson J D, Mayers D F. Proc, Natl Acad Sci USA. 1971;68:672–676. doi: 10.1073/pnas.68.8.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langs D A, Smith G D, Courseille C, Précigoux G, Hospital M. Proc Natl Acad Sci USA. 1992;88:5345–5349. doi: 10.1073/pnas.88.12.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langs D A. Science. 1988;241:188–191. doi: 10.1126/science.2455345. [DOI] [PubMed] [Google Scholar]
- 7.Burkhart, B. M., Gassman, R. M., Langs, D. A., Pangborn, W. A. & Duax, W. L. (1998) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 8.Wallace B A, Ravikumar K. Science. 1988;241:182–187. doi: 10.1126/science.2455344. [DOI] [PubMed] [Google Scholar]
- 9.Doyle D A, Wallace B A. J Mol Biol. 1997;266:963–977. doi: 10.1006/jmbi.1996.0837. [DOI] [PubMed] [Google Scholar]
- 10.Arseniev A S, Barsukov I L, Bystrov V F, Lomize A L, Ovchinnikov Y A. FEBS Lett. 1985;186:168–174. doi: 10.1016/0014-5793(85)80702-x. [DOI] [PubMed] [Google Scholar]
- 11.Ketchem R R, Hu W, Cross T A. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 12.Nekrasov A N, Stepanov A V, Timofeev V P. FEBS Lett. 1995;371:35–38. doi: 10.1016/0014-5793(95)00800-o. [DOI] [PubMed] [Google Scholar]
- 13.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 14.Sheldrick G M. shelxl-97: A Program for the Refinement of Crystal Structures. Göttingen, Germany: Univ. of Göttingen; 1997. [Google Scholar]
- 15.Nicholson L K, Moll F, Mixon T E, LoGrasso P V, Lay J C, Cross T A. Biochemistry. 1987;26:6621–6626. doi: 10.1021/bi00395a009. [DOI] [PubMed] [Google Scholar]
- 16.Datema K P, Pauls K P, Bloom M. Biochemistry. 1986;25:3796–3803. doi: 10.1021/bi00361a010. [DOI] [PubMed] [Google Scholar]
- 17.Killian J A, Prasad K U, Hains D, Urry D W. Biochemistry. 1988;27:4848–4855. doi: 10.1021/bi00413a040. [DOI] [PubMed] [Google Scholar]
- 18.Hu W, Cross T A. Biochemistry. 1995;34:14147–14155. doi: 10.1021/bi00043a020. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi H, Nemoto Y, Harada I. Biochemistry. 1990;29:1572–1579. doi: 10.1021/bi00458a031. [DOI] [PubMed] [Google Scholar]
- 20.Urry D W, Goodall M C, Glickson J D, Mayers D F. Proc Natl Acad Sci USA. 1971;68:1907–1911. doi: 10.1073/pnas.68.8.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stankovic C J, Heinemann S H, Delfino J M, Sigworth F J, Schreiber S L. Science. 1989;244:813–817. doi: 10.1126/science.2471263. [DOI] [PubMed] [Google Scholar]
- 22.Smith R, Thomas D E, Atkins A R, Separovic F, Cornell B A. Biochim Biophys Acta. 1990;1026:161–166. doi: 10.1016/0005-2736(90)90059-w. [DOI] [PubMed] [Google Scholar]
- 23.Separovic F, Gehrmann J, Milne T, Cornell B A, Lin S Y, Smith R. Biophys J. 1994;67:1495–1500. doi: 10.1016/S0006-3495(94)80623-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian F T, Lee K-C, Hu W, Cross T A. Biochemistry. 1996;35:11959–11966. doi: 10.1021/bi961170k. [DOI] [PubMed] [Google Scholar]
- 25.Tripathi S, Hladky S B. Biophys J. 1998;74:2912–2917. doi: 10.1016/S0006-3495(98)77998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woolf T B, Roux B. Biophys J. 1997;72:1930–1945. doi: 10.1016/S0006-3495(97)78839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Connell A M, Koeppe R W, II, Andersen O S. Science. 1990;250:1256–1259. doi: 10.1126/science.1700867. [DOI] [PubMed] [Google Scholar]
- 28.Kleywegt G J, Brünger A T. Structure. 1996;4:897–904. doi: 10.1016/s0969-2126(96)00097-4. [DOI] [PubMed] [Google Scholar]