

Cardiac contractility is regulated by a process termed excitation–contraction coupling (ECC) [1]. An increase in the body’s metabolic demands results in an increase in contractility. However, because every myocyte contracts with each beat, the heart cannot recruit additional myocytes to achieve this phenomenon. Hence, the myocyte has evolved numerous cellular mechanisms to regulate ECC. The β-adrenergic signaling pathway is a primary regulator of ECC [2].
Nitric oxide and cardiac function
Another key signaling pathway that regulates ECC is nitric oxide (NO) [3]. The heart is able to generate NO on a beat-per-beat basis via nitric oxide synthase (NOS) [4]. Cardiac myocytes constitutively express two NOS isoforms: endothelial NOS (NOS3) and neuronal NOS (NOS1). Both of these enzymes produce low levels of NO from the precursor L-arginine in a Ca2+/calmodulin-dependent manner. NOS1 and NOS3 differentially regulate contractility due to their differences in cellular localization [5,6]. Another isoform (inducible NOS, NOS2) is not normally expressed in healthy myocytes. However, its expression is induced during the inflammation that occurs in many cardiomyopathies (e.g., heart failure [7]). Once expressed, NOS2 produces much higher levels of NO (and related congeners) in a Ca2+-independent manner, leading to cardiac dysfunction [8–10].
Several studies, using a multitude of tools (various NO donors, specific and non-specific NOS inhibitors, cGMP analogs, transgenic and knockout mice, etc.) have examined the functional impact of NO and its signaling pathways and end targets. The results to date have yielded exciting information. However, the data generated have often been complex, leading to much controversy. Accordingly, NO has been found to be both a negative and positive inotropic agent. This dual effect, in part, has been found to be due to using diverse NO species and different NO concentrations, leading to S-nitrosylation or cGMP- or cAMP-dependent signaling [11–14]. These factors work in concert to exert positive or negative inotropic effects. For example, activation of the cGMP-dependent protein kinase through high [NO•] stimulation of guanylate cyclase will decrease the L-type Ca2+ current (ICa) [8,15]. Conversely, conditions that favor NO+ generation lead to nitrosylation of the L-type Ca2+ channel and an increase in ICa [15]. Nonetheless, inconsistencies have been observed for each effect. For example, studies have shown that cGMP is able to increase ICa [16], and nitrosylation can decrease ICa.[17]. These differences have been presumed to be due to different experimental protocols, NO donors, and/or animal species. In this issue of Nitric Oxide, Gonzalez et al. [18] have further addressed these factors by performing identical experimental protocols using the same preparation (isolated rat heart) and NO donor (SNAP, at different concentrations). It should be noted that additional factors, not studied by Gonzalez et al., also influence the functional response to NO, such as adrenergic state [19], site of NO production [20], and NOS isoforms [5].
Biphashic effect of the NO donor SNAP
In these well-designed experiments, Gonzalez et al. [18] have verified the biphasic effects of NO in an isolated rat heart preparation. That is, low concentrations of SNAP (0.1, 1, and 10 μM) exhibited a positive inotropic and lusitropic effect (increased left ventricular pressure development (LVPmax), contractility (dP/dtmax), and ventricular relaxation (dP/dtmin)). However, a high concentration of SNAP (100 μM) induced a negative inotropic and lusitropic effect in LVPmax, (dP/dt)max, and (dP/dt)min. These results demonstrate that the contractile effect is dependent on the concentration of NO generated.
These authors further investigated the signaling pathways activated by the different concentrations of SNAP/NO. The data suggest that whole-heart cGMP levels were increased only in the presence of the highest concentration of SNAP (100 μM). cGMP was confirmed as the signaling molecule for the functional effects of the high concentration of SNAP (100 μM) by using ODQ, an inhibitor of NO stimulation of guanylate cyclase. ODQ abolished the negative functional effects of high SNAP. However, ODQ did not abolish the positive inotropic effect of the low doses of SNAP, demonstrating that the effects of low SNAP are via the cGMP-independent pathway. Further experiments revealed that the cGMP-dependent protein kinase (PKG) was responsible for the cGMP-induced negative inotropic effects.
The superoxide radical is needed to react with NO to form nitrosylating agents (i.e., NO+, peroxynitrite, etc. [21]). The authors used tempol, a superoxide scavenger, to examine if the functional effects of low-dose SNAP occur via redox-mediated modifications such as S-nitrosylation or S-glutathiolation. Tempol did not have any effect on the functional response to high-dose SNAP, which is consistent with high SNAP being cGMP-dependent. However, tempol did inhibit the functional effects of low-dose SNAP (1 μM). Although tempol had no effect on the high -dose SNAP, it should be noted that ODQ induced a further increase in (dP/dt)max in the presence of low-dose SNAP (1 μM). Intriguingly, high-dose SNAP in the presence of ODQ elicited a positive inotropic effect. These data suggest an intricate balance between the cGMP and nitrosylation signaling pathways that warrants further investigation. An interesting experiment would be to repeat the ODQ/tempol experiments with 10 μM SNAP. This concentration of SNAP seemed to yield a variable increase in total cGMP (although not significant), with a corresponding lower increase in (dP/dt)max. These experiments may better reveal the balance between the cGMP and nitrosylation signaling pathways. For example, would tempol induce a negative inotropic effect with 10 μM SNAP? Because cGMP signaling is compartmentalized [22], a change in whole cell cGMP may not have been observed, but there could have been a sufficient increase in local cGMP levels to elicit a negative inotropic effect. Because ODQ + 1 μM SNAP induced a larger positive inotropic effect than 1 μM SNAP alone, this suggests that there is compartmentalized cGMP signaling that is superseded by nitrosylation. Hence, ODQ should elicit a much larger positive inotropic effect with 10 μM SNAP than with 1 μM SNAP.
A possible determinant of which pathway (cGMP or nitrosylation) dominates could be the ECC proteins that are modified. The authors did not specifically examine which protein(s) was phosphorylated by PKG, but speculated that troponin I (TnI) is involved (Fig. 1, left panel). PKG phosphorylation of TnI will desensitize the myofilaments to Ca2+, which could indeed explain the negative inotropic effect[23]. Also speculated was PKG phosphorylation of Cav1.2 which would limit Ca2+ influx [24] and also lead to a decrease in contractility (Fig. 1, left panel). However, the authors also observed a slower rate of relaxation, (dP/dt)min, which cannot be explained by phosphorylation of TnI or Cav1.2. The authors did not report the effect of PKG inhibition on relaxation; therefore, we do not know if this effect is also via PKG or some other cGMP-mediated pathway and further work needs to be done to determine the ECC protein(s) involved. Previous studies have also shown that ECC proteins can be nitrosylated (or at least modified by the cGMP-independent pathway) such as RyR (Fig. 1, right panel). S-nitrosylation of RyR will increase its open probability [25], allowing more release of Ca2+ from the SR to increase contractility. However, this increase in RyR open probability alone will not be sufficient to maintain the increase in contractility [26]. In addition, modification in RyR cannot be responsible for the increased lusitropy. Thus, there must be another primary target of nitrosylation, which could be the SERCA/phospholamban complex (Fig. 1, right panel). An increase in SERCA activity would increase the rate of relaxation [1], as well as increase the SR Ca2+ load, and, along with increased RyR open probability, would increase contractility. It has been demonstrated that SERCA1 (not the cardiac isoform) can undergo S-glutathiolation to increase its activity[27].
Fig. 1.
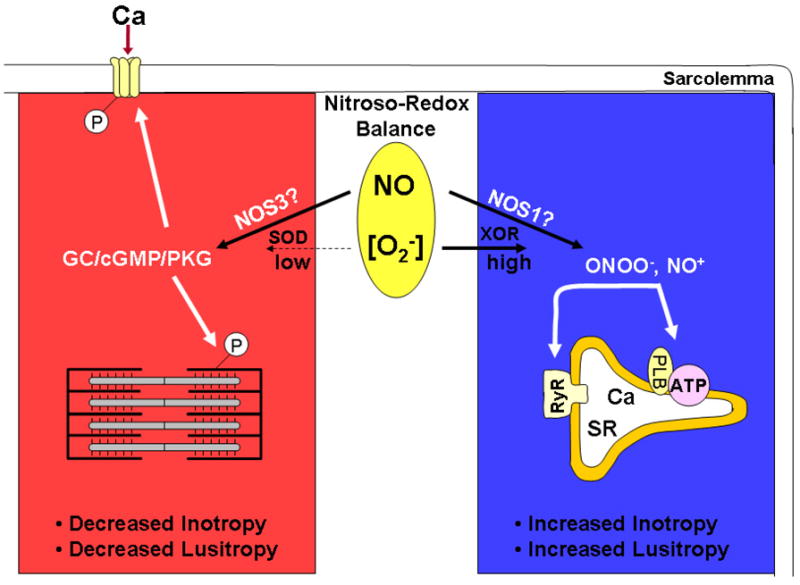
NO signaling pathways in cardiac myocytes. O2−,superoxide radical; SOD; superoxide dismutase; GC, guanylate cyclase; P, phosphorylation; XOR, xanthine oxidoreductase, ONOO−, peroxynitrite; NO+, nitrosonium ion; SR, sarcoplasmic reticulum; ATP, SR Ca-ATPase; PLB, phospholamban; RyR, SR Ca release channel (see text for other abbreviations).
Relevance to endogenous NO production
The divergent contractile effects of NOS1 and NOS3 were hypothesized to be due to their different localization [5]. Differential effects of NO on contractility may be more complicated than merely different NOS isoforms. As suggested by Gonzalez et al. [18], these effects could be due to which specific signaling pathway is activated by each NOS isoform.
There may be discrete local redox environments enveloping each NOS isoform. Studies have shown that within cardiac myocytes NOS1 colocalizes with xanthine oxidoreductase [28], a producer of superoxide radicals and important for the generation of nitrosylating agents. Hence, the signaling pathway activated via NOS1 may be through the formation of peroxynitrite leading to S-nitrosylation. Previous studies have shown that NOS1 signaling leads to positive inotropic and lusitropic effects [5,6], which is consistent with Gonzalez et al. [18] (i.e., nitrosylation leads to positive inotropic and lusitropic effects). Thus, NOS1 may predominately signal via nitrosylating agents leading to increased contractility (Fig. 1, right panel).
The local redox environment for NOS3 may be different from that for NOS1. NOS3 colocalizes with superoxide dismutase [29], a scavenger of superoxide radicals, which will lead to enhanced activation of guanylate cyclase and cGMP production. Further evidence supporting the activation of the cGMP pathway primarily by NOS3 is the observation that the cGMP-specific phosphodiesterase (PDE5) is associated with NOS3 [30]. We have demonstrated that NOS3 signaling leads to negative inotropic effects via inhibition of ICa [31], which is consistent with Gonzalez et al. [18] (i.e., cGMP leads to negative inotropic effects). Thus, NOS3 may predominately signal via cGMP, leading to decreased contractility (Fig. 1, left panel).
A potentially important factor in determining which signaling pathway (cGMP or nitrosylating) will be dominant is the balance between NO and reactive oxygen species (ROS) known as the nitroso-redox balance (Fig. 1, middle circle). This critical balance is altered during heart failure [32] and may have profound effects on NOS1/NOS3 signaling. Much higher levels of NO (via NOS2) and superoxide (via NADPH, xanthine oxidoreductase, mitochondria) are generated during many cardiomyopathies. Altered reactive nitrogen species (RNS) and ROS production could disrupt the spatially localized redox environment encircling NOS1/NOS3. This, in turn, could change which signaling pathway is activated by each NOS isoform. For example, after myocardial infarction NOS1 is translocated from the SR to the caveolae, which depresses the response to β-adrenergic stimulation[33]. This NOS1-mediated decrease in the β-AR response has also been observed in normal myocytes [34]. Also, in patients with right ventricular hypertrophy, cGMP levels increased by PDE5 inhibition increased (not decreased) contractility [35]. In addition, pathophysiological levels of peroxynitrite have been shown to lead to decreased (not increased) cardiac inotropy by modulation of RyR and phospholamban [9,36]. Thus, the isoform-specific production of NO may not be as important as which signaling pathway is activated and/or dominant.
In summary, NO is an important modulator of cardiac contractility, resulting in either positive or negative inotropy. Many factors have been found to be responsible for this NO-induced biphasic effect, including NOS isoforms and the concentration of NO. In their study, Gonzalez et al. have nicely demonstrated that the contractile effects of NO may be more complex and dependent on an intricate balance between the cGMP and nitrosylating signaling pathways.
Acknowledgments
M.T.Z. is supported by the National Institutes of Health (R01 HL079283).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM, Ziolo MT. When is cAMP not cAMP? Effects of compartmentalization. Circ Res. 2001;89:373–375. [PubMed] [Google Scholar]
- 3.Hare JM. Nitric oxide and excitation-contraction coupling. J Mol Cell Cardiol. 2003;35:719–729. doi: 10.1016/s0022-2828(03)00143-3. [DOI] [PubMed] [Google Scholar]
- 4.Pinsky DJ, Patton S, Mesaros S, Brovkovych V, Kubaszewski E, Grunfeld S, Malinski T. Mechanical transduction of nitric oxide synthesis in the beating heart. Circ Res. 1997;81:372–379. doi: 10.1161/01.res.81.3.372. [DOI] [PubMed] [Google Scholar]
- 5.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 6.Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322–1329. doi: 10.1161/01.RES.0000078171.52542.9E. [DOI] [PubMed] [Google Scholar]
- 7.Ziolo MT, Maier LS, Piacentino V, 3rd, Bossuyt J, Houser SR, Bers DM. Myocyte nitric oxide synthase 2 contributes to blunted beta-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation. 2004;109:1886–1891. doi: 10.1161/01.CIR.0000124231.98250.A8. [DOI] [PubMed] [Google Scholar]
- 8.Ziolo MT, Harshbarger CH, Roycroft KE, Smith JM, Romano FD, Sondgeroth KL, Wahler GM. Myocytes isolated from rejecting transplanted rat hearts exhibit a nitric oxide-mediated reduction in the calcium current. J Mol Cell Cardiol. 2001;33:1691–1699. doi: 10.1006/jmcc.2001.1420. [DOI] [PubMed] [Google Scholar]
- 9.Ziolo MT, Katoh H, Bers DM. Expression of inducible nitric oxide synthase depresses beta-adrenergic-stimulated calcium release from the sarcoplasmic reticulum in intact ventricular myocytes. Circulation. 2001;104:2961–2966. doi: 10.1161/hc4901.100379. [DOI] [PubMed] [Google Scholar]
- 10.Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 11.Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- 12.Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. 1999;84:1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 14.Chesnais JM, Fischmeister R, Mery PF. Positive and negative inotropic effects of NO donors in atrial and ventricular fibres of the frog heart. J Physiol. 1999;518:449–461. doi: 10.1111/j.1469-7793.1999.0449p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes: Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono K, Trautwein W. Potentiation by cyclic GMP of beta-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. J Physiol. 1991;443:387–404. doi: 10.1113/jphysiol.1991.sp018839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez DR, Fernandez IC, Ordenes PP, Treuer AV, Eller G, Boric MP. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Niric Oxide. 2008 doi: 10.1016/j.niox.2007.09.086. in press. [DOI] [PubMed] [Google Scholar]
- 19.Ziolo MT, Katoh H, Bers DM. Positive and negative effects of nitric oxide on Ca(2+) sparks: influence of beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H2295–H2303. doi: 10.1152/ajpheart.2001.281.6.H2295. [DOI] [PubMed] [Google Scholar]
- 20.Katori T, Donzelli S, Tocchetti CG, Miranda KM, Cormaci G, Thomas DD, Ketner EA, Lee MJ, Mancardi D, Wink DA, Kass DA, Paolocci N. Peroxynitrite and myocardial contractility: in vivo versus in vitro effects. Free Radic Biol Med. 2006;41:1606–1618. doi: 10.1016/j.freeradbiomed.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 21.Paolocci N, Ekelund UE, Isoda T, Ozaki M, Vandegaer K, Georgakopoulos D, Harrison RW, Kass DA, Hare JM. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: potential role for nitrosylation. Am J Physiol Heart Circ Physiol. 2000;279:H1982–8. doi: 10.1152/ajpheart.2000.279.4.H1982. [DOI] [PubMed] [Google Scholar]
- 22.Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziolo MT, Lewandowski SJ, Smith JM, Romano FD, Wahler GM. Inhibition of cyclic GMP hydrolysis with zaprinast reduces basal and cyclic AMP-elevated L-type calcium current in guinea-pig ventricular myocytes. Br J Pharmacol. 2003;138:986–994. doi: 10.1038/sj.bjp.0705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 26.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 27.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 28.Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci USA. 2004;101:15944–15948. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brahmajothi MV, Campbell DL. Heterogeneous basal expres. Res. 1999;85:575–587. doi: 10.1161/01.res.85.7.575. [DOI] [PubMed] [Google Scholar]
- 30.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Kohr MJ, Wheeler DG, Ziolo MT. Endothelial Nitric Oxide Synthase Decreases {beta}-adrenergic Responsiveness via Inhibition of the L-type Ca2+ Current. Am J Physiol Heart Circ Physiol. 2008 doi: 10.1152/ajpheart.01249.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 34.Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011–3016. doi: 10.1161/01.cir.0000019516.31040.2d. [DOI] [PubMed] [Google Scholar]
- 35.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 36.Kohr MJ, Wang H, Wheeler DG, Velayutham M, Zweier JL, Ziolo MT. Targeting of phospholamban by peroxynitrite decreases {beta}-adrenergic stimulation in cardiomyocytes. Cardiovasc Res. 2008;77:353–361. doi: 10.1093/cvr/cvm018. [DOI] [PMC free article] [PubMed] [Google Scholar]