

Abstract
 Anti-β-substituted γ,δ-unsaturated amino acids have been synthesized via a novel design of the Eschenmoser–Claisen rearrangement. The rearrangement gives good isolated yields and excellent diastereoselectivity due to (Z)-N,O-ketene acetal formation and the pseudochairlike conformations of the reaction intermediates. Upon reductive hydrolysis, important novel amino acids and amino alcohols were synthesized for the first time via this methodology.
Anti-β-substituted γ,δ-unsaturated amino acids have been synthesized via a novel design of the Eschenmoser–Claisen rearrangement. The rearrangement gives good isolated yields and excellent diastereoselectivity due to (Z)-N,O-ketene acetal formation and the pseudochairlike conformations of the reaction intermediates. Upon reductive hydrolysis, important novel amino acids and amino alcohols were synthesized for the first time via this methodology.
In the course of synthesis and conformational studies of biological active peptide ligands, a universal methodology is needed for the synthesis of nonproteinogenic amino acids with terminal unsaturation.1 The double bond has been a useful building block in organic synthesis due to its potential conversion to aldehydes, alcohols, halides, epoxides, amines, or carboxylic acids.2 It also can be used in peptide macro-cyclization via ring-closing metathesis.3 In addition, various β-side chain groups can be introduced in the synthesis so that the amino acid will provide biologically active functionalities in conformational constrained peptides.4 The Kazmaier–Claisen rearrangement has turned out to be very useful in the synthesis of this kind of amino acids and their applications in peptidomimetics.5 However, the Kazmaier strategy does not work well for racemic anti-β-substituted γ,δ-unsaturated amino acids. It is difficult to introduce an anti-β-substituent using the Z-allyl alcohol as a starting material, which is not always commercially available. Furthermore, the cis-oriented side chain can destabilize the chairlike transition state resulting in poor diastereoselectivities.6 The rearrangement product is a diastereomeric mixture that is usually hard to purify. Previous studies also have generated other ω-unsaturation by Ni(II) complex alkylation.7 However, attempts to prepare the anti product have failed due to epimerization. On the other hand, the Eschenmoser–Claisen rearrangement has been developed as a useful methodology for synthesis of γ,δ-unsaturated carboxylic amide derivatives.8 We envisioned that if an N,O-ketene acetal intermediate could be generated in its cis configuration, a chairlike transition state conformation would provide an anti-β-substituted product after rearrangement (Figure 1). During this study, we found that the Eschenmoser–Claisen rearrangement was a straightforward methodology for the synthesis of the desired amino acids. This methodology also can be used for the synthesis of amino alcohols which are common structural components in bioactive nature products and important synthetic building blocks in organic synthesis.9
Figure 1.
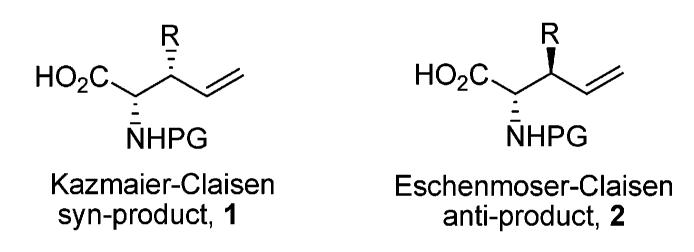
Syn- and anti-β-substituted γ,δ-unsaturated amino acids.
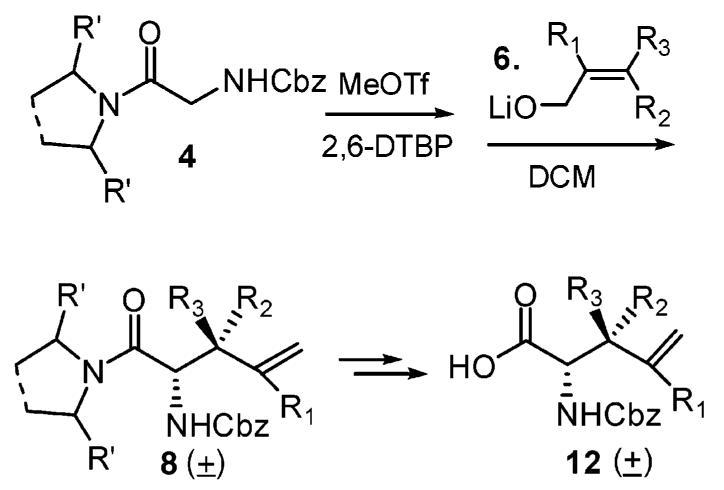
Glycine amide derivatives 4 were synthesized using the secondary amine pyrrolidine 3a and N,N-diisopropylamine 3b (Scheme 1), as we expected that a 2,5-diphenylpyrrolidine C2-symmetric auxiliary and other enantiopure secondary amines would provide an excellent enantioselective rearrangement later on.10 Various commercially available allyl alcohols were used successfully for the rearrangement as shown in Table 1. After Meerwein salt formation and lithium allyl oxide 6 addition, rearrangement provided products with different β-substituents upon warmup. It should be indicated that the β,β-dimethyl-γ,δ-unsaturated amino acid derivative 8a-2 could not be synthesized by direct alkylation using 3-bromo-3-methyl-1-butene because an SN2′ addition would happen instead.
Scheme 1.
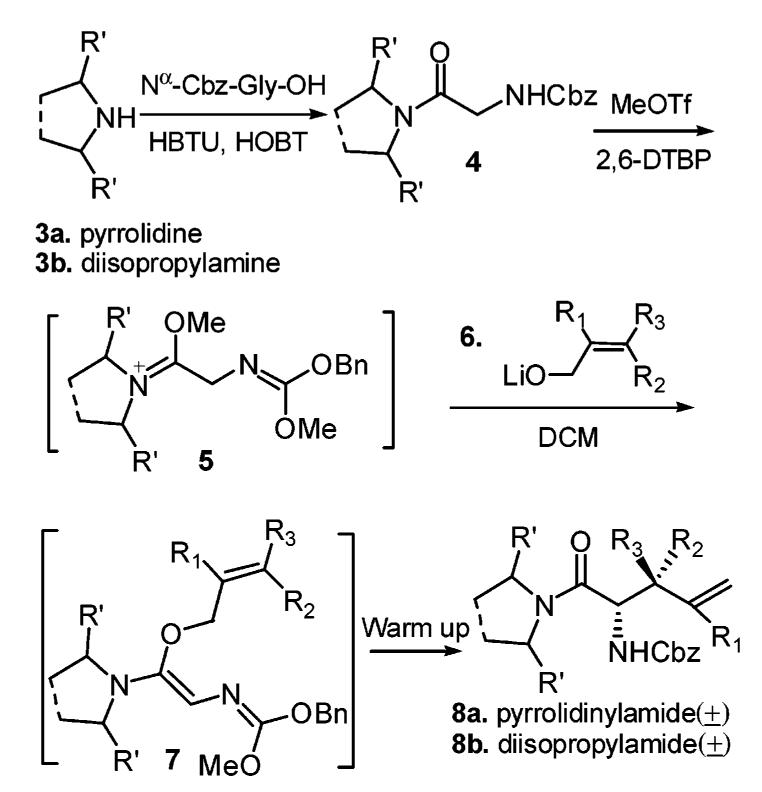
Meerwein Salt Formation and Eschenmoser–Claisen Rearrangement
Table 1.
Results of Eschenmoser–Claisen Rearrangement
| entry | allylation agent | alcohol equiv. |
reaction temp °C |
reaction time h |
anti/syn | yield (%)a |
|---|---|---|---|---|---|---|
| 1 | 4 | −35–rt | 3.5 | N/A | 8a-1, 75 | |
| 2 | 2.2 | −35–35 | 4 | N/A | 8a-2, 44 | |
| 3 | 2.2 2.2 |
−35–rt | 2 4 |
9.6:1 8.0:1 |
8a-3, 61 8b-3, 56 |
|
| 4 | 4 | −35–rt | 6 | N/A | 8a-4, 72 | |
| 5 | 4 2.2 |
−35–rt | 4 17 |
16.8:1 20:1 |
8a-5, 74 8b-5, 77 |
Isolated yields. Compounds a are from pyrrolidinylamide, and compounds b are from diisopropylamide.
Crotyl alcohol is a trans/cis mixture (19:1) from Sigma-Aldrich.
The Eschenmoser–Claisen rearrangement results are summarized in Table 1. Various amounts of MeOTf were investigated and 2.2 equiv was found to be the best. Mechanistically, two equivs of methyl triflate was consumed in intermediate 5 (Scheme 1). In practice, a minimum of 2.2 equiv of the alcohol was needed to get good yields. Larger amounts of alcohol usually gave higher yields. Stoichiometric amounts of 2,6-di-tert-butyl-pyridine were used to scavenge triflic acid formed during the Meerwein salt formation.11
The diastereomeric ratios were determined via proton NMR spectra of the crude samples. We were delighted to see that the reaction generated good to excellent diastereoselectivities. N,N-Diisopropylamine (Scheme 1, 4b) provided a little better result in entry 5 (Table 1). These good diastereoselectivities can be explained via unique (Z)-N,O-ketene acetal formation and an excellent chairlike transition state in the rearrangement.12 Unlike the Kazmaier–Claisen intermediate where the enolate oxygen stays preferentially cis to the glycyl nitrogen due to chelation control, the Eschenmoser–Claisen intermediate could adopt two possible configurations (Figure 2) due to the lack of the enolate oxygen. Presumably, the thermodynamically more stable intermediate 9 is the predominant configuration.13 It should be noted that we tried to quench intermediate 9 using several saturated alcohols. However, this N,O-ketene acetal intermediate was unstable and hydrolyzed easily even in neutral aqueous solution.
Figure 2.
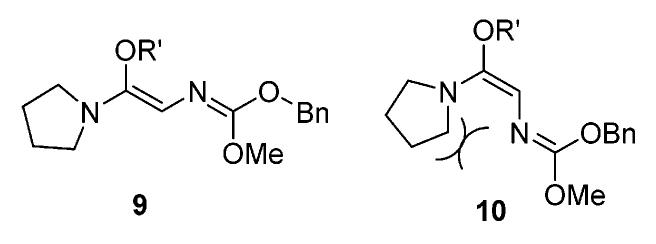
(Z)- and (E)-N,O-ketene acetal reaction intermediates.
The hydrolysis of these rearranged amides was tricky because of the presence of a terminal double bond and the N-Cbz protecting group. There are many amide bond hydrolysis methods reported.14 However, basic conditions cleaved the Cbz group and caused epimerization. Strong acidic conditions also removed the Cbz group and led to partial hydrolysis of the terminal double bond. A reduction–hydrolysis approach was convenient in this case, and among many reducing agents, lithium trimethoxyaluminum hydride15 and sodium aluminum hydride16 worked well.
Epimerization was observed during workup because amino aldehydes are notorious for racemerization under either basic or acidic conditions.17 The presence of a β-substituent and a γ-double bond makes the α-proton even more labile. The side reaction was initiated upon aldehyde generation during the hydrolysis of the reduced complex. To minimize epimerization, we developed in situ modified Lindgren oxidation18 at low temperature (Scheme 2). In this way, the aldehyde was oxidized simultaneously to the carboxylic acid. Several amino alcohols were obtained from further reduction of the amino aldehydes that were isolated at low temperature by extraction (Scheme 2). Little epimerization was observed during the workup process. The diastereomerically pure amino alcohols were obtained after flash column chromatography purification.
Scheme 2.
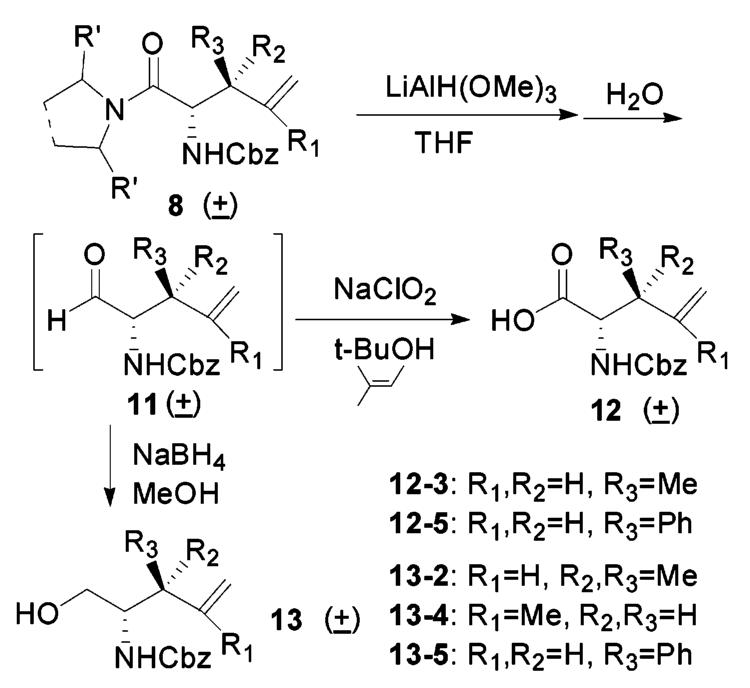
Reductive Hydrolysis, Amino Acid, and Amino Alcohol Generation
The lack of epimerization using the modified reductive hydrolysis and aldehyde oxidation was demonstrated using an 1H NMR spectra comparison between the epimerized and the diastereomerically pure product 12-5 (Figure 3). The downfield minor diastereomeric peaks from in situ oxidation are almost undetectable, indicating minimum epimerization. The broad peak at 4.63 ppm was likely from a rotamer of the product. This was proven by NOE experiments (Figure 4).19 Irradiation of the peak at 4.63 ppm resulted in the inversion of its exchange peak at 4.78 ppm. The size of the exchange peak increased as the NOE mixing time was increased.
Figure 3.
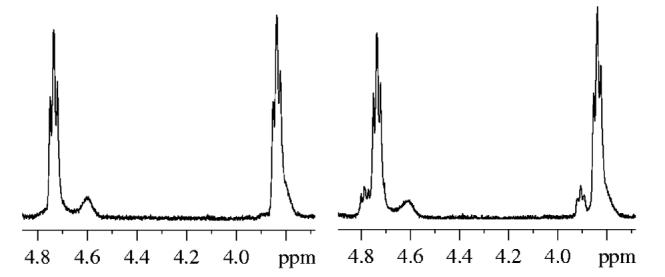
1H NMR spectra of 12-5 from different oxidation approaches. Left: in situ oxidation. Right: oxidation after workup.
Figure 4.
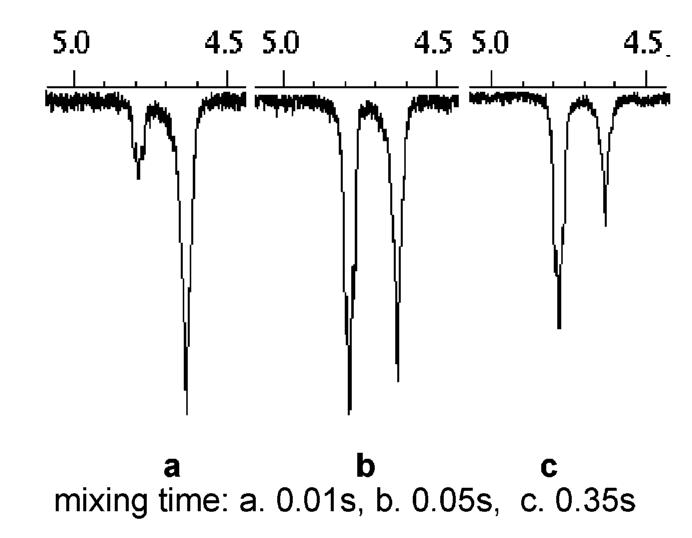
Rotational interexchange study of 12-5 by NOE.
The relative stereochemistry of 12-3 was confirmed by comparing its 13C NMR chemical shift to the minor product synthesized from an ester enolate Claisen rearrangement.1 A single-crystal X-ray structure of compound 8b-5 also was obtained (Figure 5). An anti relationship of the α-amino group and the β-phenyl group was unambiguously shown in this structure.
Figure 5.
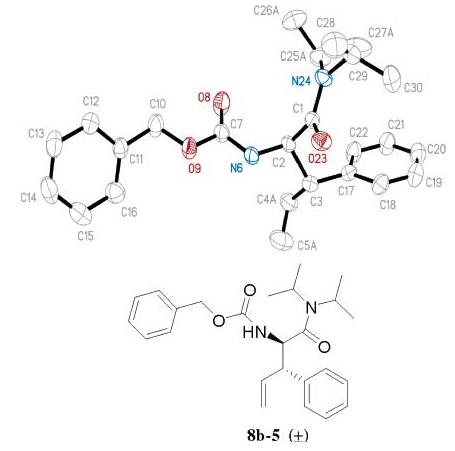
X-ray structure of product 8b-5 with an anti-β-substituent.
In summary, we have developed a convenient methodology for the synthesis of anti-β-substituted γ,δ-unsaturated amino acids for the first time using a Meerwein Eschenmoser–Claisen rearrangement. This [3,3]-sigmatropic rearrangement gives excellent diastereoselectivities. Both the amino acid and the amino alcohol can be easily purified and are ready for scale-up. The enantiomerically pure synthesis of these novel compounds using enantiopure C2-symmetric 2,5-diphenylpyrrolidine is currently under investigation.
Supplementary Material
Acknowledgment
This work was supported by U.S. Public Health Service grants DK 17420 and the National Institute of Drug Abuse, DA 06284 and DA 13449. We also thank Prof. Richard Glass (the University of Arizona) for useful discussions regarding the rearrangement reaction mechanisms.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic characterization (1H NMR, 13C NMR, HRMS, IR) of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Grubbs RH, Miller SJ, Blackwell HE. U.S. patent 5811515 1998; (b) Bartlett PA, Barstow JF. J. Org. Chem. 1982;47:3933. [Google Scholar]; (c) Gu X, Ying J, Min B, Cain JP, Davis P, et al. Biopolymers. 2005;80:151. doi: 10.1002/bip.20208. [DOI] [PubMed] [Google Scholar]
- 2.Rutjes TPJT, Wolf LB, Schoemaker HE. J. Chem. Soc., Perkin Trans. 1. 2000;24:4197. [Google Scholar]
- 3.(a) Fernandez MM, Diez A, Rubiralta M, Montenegra E, Casamitjana N. J. Org. Chem. 2002;67:7587. doi: 10.1021/jo025999i. [DOI] [PubMed] [Google Scholar]; (b) Hoffmann T, Harald L, Waibel R, Gmeiner P. Angew. Chem., Int. Ed. 2001;40:3361. doi: 10.1002/1521-3773(20010917)40:18<3361::aid-anie3361>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hruby VJ, Balse PM. Curr. Med. Chem. 2000;7:945. doi: 10.2174/0929867003374499. [DOI] [PubMed] [Google Scholar]; (b) Hruby VJ, Li G, Haskell-Luevano C, Shenderovich M. Biopolymers. 1997;43:219. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kazmaier U. Angew. Chem., Int. Ed. 1994;33:998. [Google Scholar]; (b) Kazmaier U, Krebs A. Angew. Chem., Int. Ed. 1995;34:2012. [Google Scholar]; (c) Kazmaier U, Mues H, Krebs A. Chem. Eur. J. 2002;8:1850. doi: 10.1002/1521-3765(20020415)8:8<1850::AID-CHEM1850>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; (d) Kazmaier U, Maier S. J. Org. Chem. 1999;64:4574. doi: 10.1021/jo9904821. [DOI] [PubMed] [Google Scholar]; (e) Qiu W, Gu X, Soloshonok VA, Carducci MD, Hruby VJ. Tetrahedron Lett. 2001;42:145. [Google Scholar]
- 6.Kazmaier U. J. Org. Chem. 1996;61:3694. doi: 10.1021/jo960014g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gu X, Scott C, Ying J, Tang X, Hruby VJ. Tetrahedron Lett. 2003;44:5863. [Google Scholar]; (b) Gu X, Ndungu JM, Qiu W, Ying J, Carducci MD, Wooden H, Hruby VJ. Tetrahedron. 2004;60:8233. [Google Scholar]
- 8.(a) Coates B, Montgomery D, Sterenson PJ. Tetrahedron Lett. 1991;32:4199. [Google Scholar]; (b) Coats B, Montgomery D, Sterenson PJ. Tetrahedron Lett. 1994;50:4025. [Google Scholar]
- 9.Bergmeier SC. Tetrahedron. 2000;56:2561. [Google Scholar]
- 10.(a) He S, Kozmin SA, Rawal VH. J. Am. Chem. Soc. 2000;122:190. [Google Scholar]; (b) Qian X, Moris-Varas F, Fitzgerald MC, Wong C. Bioorg. Med. Chem. 1996;4:2055. doi: 10.1016/s0968-0896(96)00218-0. [DOI] [PubMed] [Google Scholar]
- 11.Resendes R, Nelson JM, Fischer A, Jakle F, Bartole A, Lough AJ, Manners I. J. Am. Chem. Soc. 2001;123:2116. doi: 10.1021/ja002750e. [DOI] [PubMed] [Google Scholar]
- 12.Wipf P. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Pergamon; Oxford, NY: 1991. p. 827. and references therein. [Google Scholar]
- 13.(a) Welch JT, Eswarakrishman S. J. Org. Chem. 1985;50:5909. [Google Scholar]; (b) Bartlett P, Hahne WF. J. Org. Chem. 1979;44:882. [Google Scholar]; (c) Ziegler FE. Acc. Chem. Res. 1977;10:227. [Google Scholar]
- 14.(a) Groves JT, Dias RM. J. Am. Chem. Soc. 1979;101:1033. [Google Scholar]; (b) Hub DH, Jeong JS, Lee HB, Ryu H, Kim YG. Tetrahedron. 2002;58:9925. [Google Scholar]; (c) Gassmart PC, Hodgson PKG, Balchunis RJ. J. Am. Chem. Soc. 1976;98:1275. [Google Scholar]; (d) Moghaddam FM, Ghaffarzadeh M. Synth. Commun. 2001;31:317. [Google Scholar]
- 15.Brown HC, Weissman PM. J. Am. Chem. Soc. 1965;87:5614. [Google Scholar]
- 16.Zakharkin LI, Maslin DN, Gavriljinko VV. Tetrahedron. 1969;25:5555. [Google Scholar]
- 17.(a) Ito JJ, Golebiowshi A. Chem. Rev. 1989;89:149. [Google Scholar]; (b) Rittle KE, Homnick CF, Ponticelle GS, Evans BE. J. Org. Chem. 1982;47:3016. [Google Scholar]
- 18.Kraus GA, Bruce Roth B. J. Org. Chem. 1980;45:4825. [Google Scholar]
- 19.Hatano T, Hemingwei RW. J. Chem. Soc., Perkin Trans. 2. 1997:1035. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.