

Abstract
G protein-coupled receptors (GPCRs) convey extra cellular stimulation into dynamic intracellular action, leading to the regulation of cell migration and differentiation. T lymphocytes express Gαi2 and Gαi3, two members of the Gαi/o protein family, but whether these two Gαi proteins have distinguishable roles guiding T cell migration remains largely unknown due to a lack of member-specific inhibitors. This study details distinct Gαi2 and Gαi3 effects on chemokine receptor CXCR3-mediated signaling. Our data showed that Gαi2 was indispensable for T cell responses to three CXCR3 ligands: CXCL9, CXCL10, and CXCL11, as the lack of Gαi2 abolished CXCR3-stimulated migration and GTPγS incorporation. In sharp contrast, T cells isolated from Gαi3 knockout mice displayed a significant increase in both GTPγS incorporation and migration as compared to wild type T cells when stimulated with CXCR3 agonists. The increased GTPγS incorporation was blocked by Gαi3 protein in a dose-dependent manner. Gαi3-mediated blockade of Gαi2 activation did not result from Gαi3 activation, but instead, resulted from competition or steric hindrance of Gαi2 interaction with the CXCR3 receptor via the N-terminus of the second intracellular loop. A mutation in this domain abrogated not only Gαi2 activation induced by a CXCR3 agonist, but also the interaction of Gαi3 to the CXCR3 receptor. These findings reveal for the first time an interplay of Gαi proteins in transmitting GPCR signals. This interplay has heretofore been masked by the use of pertussis toxin, a broad inhibitor of the Gαi/o protein family.
Introduction
Heterotrimeric G proteins consist of α, β and γ subunits that couple to seven transmembrane receptors called G protein-coupled receptors (GPCRs). The G protein resides attached to the intracellular face of the plasma membrane in an inactive form consisting of the Gα subunit bound to GDP, a structure that is stabilized by interaction with the βγ dimer. Agonist binding to the receptor provokes a conformation change that facilitates the interaction of the Gα subunit to the receptor. Upon interaction with the receptor, Gα protein becomes activated, causing GDP exchange for GTP. This activation to the high energy Gα-GTP results in dissociation of the α and βγ subunits and propagation of downstream signaling by both the Gα-GTP and the βγ subunits. The GTP-binding proteins are classified by the signaling events they instigate, of which there are four major families: Gαi/o, Gαq/11, Gα12/13, and Gαs (1, 2). Members of the Gαi/o family, including Gαi1, Gαi2, and Gαi3, Gαo, Gαgust and Gαt, can all be irreversibly uncoupled from receptors by pertussis toxin (PTX) (3, 4). PTX catalyses ADP-ribosylation of a specific cysteine residue at position -4 from the C-terminus of the α subunits, and thus blocks downstream pathways by preventing receptor interaction (5). The bacterial toxin has proven to be an excellent tool in dissecting the essential role of Gαi proteins in chemotaxis, proliferation and differentiation of various cells (3, 4, 6, 7). However, it cannot distinguish unique properties of individual Gαi/o protein family members. Investigation of the details of GPCR and G protein interactions has drawn great attention in the past decade, due to the fact that the interactions dictate the specificity and amplitude of a GPCR-stimulated cellular response and GPCRs are targets for more than 30% of the current marketed drugs (8).
The primary PTX substrates evident in T lymphocytes are the 40-41 kDa αsubunits of the Gαi2 and Gαi3 proteins (9, 10). The importance of PTX-sensitive proteins in lymphocyte trafficking came initially from the unique phenotype of lck-PTX-transgenic mice that expressed PTX catalytic S1 subunit in thymocytes and peripheral T cells under the control of an lckpromoter (11). The transgenic mice had 2∼4 fold more mature CD4+ and CD8+ thymocytes in the thymus than did control mice and few or no T cells in the peripheral lymphoid tissues owing to a defect in thymic emigration (11). Thymocyte emigration appears to be controlled by either Gαi3 or Gαi2, since null mutation of either of them does not affect this process significantly (12, 13). In the periphery, however, their function differs considerably. Gαi2-deficient mice display impairment in lymph node development in several anatomic locations and in Peyer’s patch formation (12, 14). The mice develop inflammatory bowel disease due to lack of TGF-β responses in T cells that causes Th1-skewed hyperimmune responses in the colon (12, 15). In contrast, Gαi3-deficient mice develop no overt phenotype (16).
In the present study, we investigate unique properties of Gαi2 and Gαi3 in chemotaxis of activated T cells induced by CXCR3 ligands, since expression of the CXCR3 receptor has been associated with pathophysiology of Th1 type autoimmune diseases in both humans and experimental models (17-19). The receptor has three known ligands: monokine-induced IFN-γ(MIG)/CXCL9, IFN-γ-inducible 10 kDa protein (IP-10)/CXCL10, and IFN-γ-inducible T cell α chemoattractant (ITAC)/CXCL11. These three chemokines exhibited discernible biological effects in vivo and in vitro (20-24), but they all activated exclusively Gαi2 through the CXCR3 receptor, as indicated by a lack of CXCR3-mediated responses in Gαi2-/- T cells, regardless of the agonist employed. Interestingly, we found that Gαi3 was negatively involved in the CXCR3-mediated signaling. This translated to cellular responses, where a lack of Gαi3 significantly enhanced T cell migration toward CXCR3 agonists. Gαi3 appears to hinder Gαi2 activation via the N-terminus of the second intracellular loop. A mutation in this region abolished both activation of Gαi2 and the binding of Gαi3 to the receptor. This work begins to unravel an interplay between individual Gαi proteins that has heretofore been masked by the use of PTX.
Methods
Animals
Gαi2-knockout (KO), Gαi3-KO, and wild type (WT) control mice on the mixed 129Sv/C57BL/6 background were generated by gene targeting and backcrossed with C57BL/6 (B6) mice for six times as described (12, 25). Both female and male mice were used at 4-6 weeks of age unless otherwise indicated. The mice were housed in conventional cages at the animal facilities of the Massachusetts General Hospital in accordance with institutional guidelines.
T cell preparation
Single-cell suspensions prepared from lymph nodes and spleens were treated with a cocktail of rat anti-mouse monoclonal antibodies (Abs) against CD19, CD32, and CD16 followed by depletion of Ab-bound cells with BioMag goat anti-rat IgG (Polysciences Inc., PA) per the manufacturer’s instructions. This preparation isolated an ∼ 90% pure population of T cells. CXCR3 expression on these T cells was induced by stimulation for two days at 37°C and 5% CO2 with 2.0 μg/ml concanavalin A (ConA) in a complete RPMI medium (10% fetal calf serum, 2 mM L-glutamine, 100U/ml penicillin, 100μg/ml streptomycin, and 50 μM β-mercaptoethanol). The cells were continuously cultured for two more days in the medium with replenishment of 15% ConA supernatant, which gave rise to a maximal level of CXCR3 expression.
Cell membrane preparation
Cell membrane was prepared as described with some modifications (26). Briefly, T cells stimulated with or without ConA as above were collected, suspended in icecold hypotonic buffer (10mM Tris HCl, pH 7.6, 5mM EDTA, 3mM EGTA, 1X protease inhibitor cocktail, and 1mM PMSF), and Dounce homogenized. In some cases, the cells were treated with 10 ng/ml PTX for 1 hr at 37°C before suspension in hypotonic buffer and homogenization. Disrupted cells were centrifuged at 500 g at 4°C for 10 minutes to remove nuclei and unbroken cells. The remaining supernatant was spun at 32,000 g for 25 minutes at 4°C to pellet the cell membrane that was then suspended in membrane solubilization buffer (20mM glycine pH 7.2, 1mM MgCl2, 250mM sucrose, 1x protease inhibitor cocktail, and 1mM PMSF). The suspended cell membrane was left on ice for at least 2 hrs with periodic vortexing and then frozen until use.
GTPγS assay
Cell membrane equivalents of 20μg protein were first incubated with indicated concentrations of chemokines on ice for 1 hr to prime Gαi activation by the receptor (27). G protein activation was initiated by placing samples at room temperature (RT) for 2 hrs in the presence of 0.3 nM [35S] GTPγS (1250 Ci/mmol, Perkin Elmer, Boston, MA),) in a GTP-binding buffer (50mM Tris HCl, pH 7.4, 100mM NaCl, 1mM EGTA, and 5mM MgCl2) supplemented with fresh 0.1% BSA, 1mM DTT, and 2mM GDP (28). Activation of Gαi proteins at RT instead 37°C minimizes background radioactive incorporation (28). The concentrations used for each CXCR3 ligand were empirically determined based on the most robust migratory response and our preliminary tests. All chemokines were obtained from Peprotech Inc. (Rocky Hill, NJ). Where indicated, a myristolated Gαi3 protein or control Gαo protein (Calbiochem, San Diego, CA) at varying concentrations was preincubated with the cell membrane for 1 hr on ice to allow the added Gα protein to interact with the βγ subunit in the membrane prior to chemokine stimulation (27). To determine the Gαi protein in association with the receptor, 20μg/ml monoclonal Ab specific for Gαi2 (IgG2b, Chemicon, Temecula, CA) or an isotype-matched control Ab was added to the samples, along with the chemokine to block Gαi2 protein activation (29). The samples were then transferred to a 96-well plate and suctioned through a glass fiber filter by a TomTec cell harvester (Model MACH 3M, Hamden, CT). The filters were washed 5 times with a cold GTP-washing buffer (20mM Tris, pH 8.0, 100mM NaCl, and 25 mM MgCl2), dried, and radioactive count determined on a 1450 MicroBeta scintillation counter (Wallac, Turku, Finland). Percentages of specific GTPγS incorporation in activated T cell membranes were calculated as [(cpm of sample)-(cpm of background control)]/[(cpm of control without a stimulus)-(cpm of background control)] X 100, where background controls contained 1,000 times excess cold GTPγS relative to radioactive 35S-GTPγS.
Migration Assays
The Transwell system with 3 μm pore size polycarbonate membrane (Corning, Corning, NY) was used for all transmigration assays, since T cells are relatively small in size. This pore size significantly reduced the basal levels of T cell migration in our study. T cells at 3 × 106 cells/ml in Aim-V medium (Invitrogen Inc., Carlsbad, CA) were added to the upper chamber at 0.3 ml per well, with the indicated concentrations of chemokines in 0.6 ml Aim-V medium in the lower chamber. Freshly isolated T cells or T cells activated by ConA for 4 days were allowed to migrate for 4∼6 hrs at 37°C and 5% CO2. At the end of the migration, the cells in the lower chamber were collected and counted on a hemacytometer.
Western blot analysis
To examine Gαi2 expression in T cell membranes, T cells with or without stimulation were collected and lysed in cold nondenaturing lysis buffer (50mM Tris HCl, pH 7.5, 300mM NaCl, 5mM EDTA, 1% Triton X-100, 1mM PMSF, and 1x protease inhibitor cocktail), followed by centrifugation at 500 g for 10 minutes to remove nuclear fragments and unbroken cells. Crude cell membrane was pelleted from the supernatant by centrifugation at 32,000 g for 25 minutes. Protein samples of approximately 100 μg were separated on a 10% SDS-PAGE, transferred onto a nitrocellulose membrane, and immunoblotted with anti-Gαi2 monoclonal Ab (Chemicon, Temecula, CA). Gαi2 protein was visualized by incubation of the membrane with sheep anti-mouse Ab conjugated with horseradish peroxidase (HRP) (Amersham Biosciences, Piscataway, NJ) followed by reaction with SuperSignal West Pico ELC reagents (PIERCE, Rockford, IL) and exposure to Kodak film (Rochester, NY). For assessment of Gαi3 protein expression, Gαi2 protein was first depleted by anti-Gαi2 specific Ab owing to cross-reaction of anti-Gαi3 Ab with Gαi2 protein and similar molecular mass of these two G proteins. Briefly, T cell membranes prepared above were incubated for 1 hr on ice with anti-Gαi2 Ab-conjugated protein G-agarose beads that had previously been treated with 1 mg/ml Bovine serum albumin to block non-specific binding. The beads were removed by centrifuge and the depletion was repeated three times. The resultant Gαi2-depleted T cell membranes were subjected to western blotting analysis using anti-Gαi3 Ab (BIOMOL. International LP.) as detailed above. The same membranes were re-probed, after stripping, with anti-G protein pan β subunit Ab (Upstate, Lake Placid, NY) for equal protein loading controls.
Flow Cytometry
To determine chemokine receptor expression, T cells with or without stimulation with ConA or transfected Cos-7 cells were incubated with specific Abs followed by analysis on a FACSCalibur cytometer equipped with a Cellquest software (BD Bioscience, San Jose, CA). A murine CXCR3 specific Ab (clone 4c4) was a generous gift from Dr. Dominic Picarella, Millenium Pharmaceuticals) (30). Fluorescence-conjugated rat antimouse Abs specific for CCR7 or CXCR4 and mouse anti-human CXCR3 Ab were purchased from Biolegend (San Diego, CA), R&D systems (Minneapolis, MN) or BD Pharmingen (San Jose), respectively.
In vitro transcription and translation
Gαi2 and Gαi3 proteins were synthesized and 35S-labeled by the TNT® Quick Coupled in Vitro Transcription/Translation Systems per the manufacturer’s instructions (Promega, Madison, WI). In brief, 1 μg of a Gαi2 or Gαi3 plasmid was mixed with the rabbit reticulocyte lysate provided, 100μM myristic acid and [35S] methionine (1000 Ci/mmol) for 75 minutes at 30°C. The resulting product was verified by SDS-PAGE on the basis of the size and compared in the presence or absence of the specific plasmid.
Immunoprecipitation
To study CXCR3-associated Gαi proteins, Cos-7 cells were transfected with pcDNA plasmids containing either WT or mutated CXCR3 receptors or a control HA-tag· S1P1 (sphingosine 1-phosphate 1) receptor using LipoFECTamine according to the manufacturer’s instructions (Invitrogen). Three mutants were: 1) A R149N-CXCR3 that had asparagine (N) substitution for arginine (R) at position 149; 2) LLL-CXCR3 generated by replacement of alanines for leucines (L) at positions 332-334 in the carboxyl terminus; and ST (-)-CXCR3 constructed by substitution of all serines (S) and threonines (T) in the carboxyl terminus with alanines (24, 31, 32). The transfected cells were lysed and the cell membrane was prepared and suspended in a GPCR solubilization buffer (1mM MgCl2, 250mM sucrose, 1X protease inhibitor cocktail, and 1mM PMSF in PBS) as described (24, 31, 32). Cell membranes with equivalent of 60μg protein were incubated with 3μl of in vitro synthesized 35S-labeled Gαi2 or Gαi3 for 1 hr on ice to allow the added Gαi protein to interact with the βγ subunit in the membrane, after which the cell membrane was either left unstimulated or stimulated for 2 hrs on ice by indicated chemokines and activated at RT as described (33). The stimulated membranes were then cross-linked for 2 hrs on ice with 20μM dithiobis-sulfosuccinimidylpropionate (DTSSP) (PIERCE). The cross-linker was quenched by the addition of 50μM Tris pH 7.5 for 15 minutes on ice. To the chemokine-stimulated membranes, anti-CXCR3 Ab or control Ab was added, and the Ab-bound receptor was pulled down by pre-cleaned protein G-agarose beads (Invitrogen Inc.). For HA-S1P1-transfected cells, immunoprecipitation using anti-HA Ab was carried out in place of anti-CXCR3 Ab. The resultant beads were washed extensively with a cold GPCR washing buffer (1mM EGTA, 5mM MgCl2 and 0.1% BSA in PBS). Radioactivity of the resultant beads was quantified by scintillation counting on the 1450 MicroBeta scintillation counter to determine the amount of 35S-Gαi2 or 35S-Gαi3 in association with the CXCR3 receptor.
Statistical analysis
The Student’s two-tailed t test was used to analyze the significance of experimental groups compared to relevant controls.
RESULTS
Altered migration toward CXCR3 ligands of Gαi2- or Gαi3-deficient T cells
CXCR3 agonists induce chemotactic responses of activated T cells in vitro with a potency of CXCL11>CXCL10>CXCL9 in a PTX-sensitive manner (24, 34). These three chemokines also mediate distinct biological effects in vivo (20, 21, 23, 35). In an attempt to understand whether the varying responses evoked by these agonists resulted from activation of different Gαi proteins, we tested the role of Gαi2 and Gαi3 in the migration of activated T cells toward CXCL9, CXCL10 or CXCL11 (36). Stimulation of WT T cells with increasing concentration gradients of CXCR3 ligands elicited a weak chemotactic response to CXCL9 (data not shown), but vigorous responses to CXCL10 or CXCL11 in a dose-dependent manner (Figure 1A, B and E), mirroring previous investigations (37). However, migration toward these three individual chemokines was all reduced to control levels in cells lacking Gαi2 over a wide range of agonist concentrations tested (Figure 1A, B and E). The lack of chemotactic responses toward CXCR3 agonists was not due to aberrant cell viability or migration of these cells, because they migrated similarly as WT counterparts in response to CCL19 or CCL21 (data not shown), two ligands for the CCR7 receptor (Figure 1H). These results indicate that CXCR3-mediated migration depends predominantly on Gαi2, irrespective of the ligands.
FIGURE 1.



Chemotactic responses in the presence or absence of Gαi2 or Gαi3. A-E. Gαi2 and Gαi3 have opposing effects on CXCR3-stimulated chemotaxis. Activated T cells prepared from wild type (WT), Gαi2-/-, and Gαi3-/- mice were either left untreated (A-B, and E) or treated with 100 ng/ml PTX overnight (C and D), and then added to the upper chamber. CXCR3 agonists at indicated concentrations were added to the lower chamber in triplicate. After 4 hr incubation, the migrated cells in the lower chamber were collected and counted. Data are presented as mean percentages ± standard derivations (SD) of cell migration relative to cell input. One representative experiment in A-D and cumulative data from at least five independent experiments (n = 15) in E are shown. Chemokine concentrations used in E were the optimal concentrations for stimulating maximal migration toward CXCL9 (100ng/ml), CXCL10 (10ng/ml), CXCL11 (100ng/ml). Medium controls are designated as 100% and mean percentages ± SD of cell migration relative to the control are shown. * and **, statistical significance (p < 0.05) or (p < 0.01) respectively, in the presence vs. absence of a specific Gαi protein. F-H. Varying effects of Gαi2 and Gαi3 on T cell chemotaxis induced by different chemokines. The migration was assayed as above, except that non-stimulated T cells were used in place of activated T cells in chemotactic responses to CXCL12 and CCL19 in F and G. Data are presented as mean percentages ± SD of cell migration relative to cell input as A and B. One representative result of three independent experiments with each performed in triplicate.
Unexpectedly, the cell migration toward CXCL9 and CXCL11 was not attenuated, but instead, drastically increased in the absence compared to presence of Gαi3 (Figure 1A and E). The migration toward CXCL10 was also increased, albeit to a lesser extent (Figure 1B and E). But, such increased chemotaxis could not be induced by CCL21 (data not shown) or CCL19 in the cells, regardless of whether or not the cells were activated (Figure 1G and H). We also observed no augmented chemotactic response in Gαi3-/- cells stimulated with CXCL12, a ligand for another Gαi-dependent receptor CXCR4. In fact, CXCL12-provoked chemotaxis was diminished in a lack of either Gαi2 or Gαi3 (Figure 1F). The inflammatory chemokines MCP-1 and MIP-1α were unable to elicit a migratory response significantly given our T cell activation conditions (data not shown). Together, these data stress that the increased migration toward CXCR3 agonists in the absence of Gαi3 is receptor-specific.
Gαi2 and CXCR3 expression in T cells
The opposing responses of Gαi2- and Gαi3-deficient T cells to CXCR3 ligands argue for Gαi3 antagonizing the function of Gαi2. To conclude this, we must rule out that the increased migration toward CXCR3 agonists in Gαi3-/- cells resulted from increased expression of Gαi2 that could potentially provide a compensatory mechanism to offset the lack of Gαi3 (38). Figure 2A clearly showed that Gαi2 expression was not increased in activated Gαi3-/- T cells compared to WT T cells (lanes 3 vs. 1, low panel), as was the case for Gαi3 expression in activated Gαi2-/- T cells (lanes 5 vs.1, low panel). Neither Gαi2 nor Gαi3 expression was increased in freshly isolated KO T cells compared to WT T cells (Figure 2A, top panel). We also detected comparable levels of the CXCR3 receptor on these three groups of T cells (Figure 2B). The expression of other receptors, including CCR7 on stimulated and unstimulated T cells and CXCR4 on resting T cells, was also comparable in these cells, in spite of some variations (Figure 2B). To exclude another possibility that the increased chemotactic response was ascribed to coupling of the CXCR3 receptor to other G protein family members in the absence of Gαi3, the cells were treated with 100 ng/ml PTX overnight before they were assayed for chemotaxis. The pretreatment ablated CXCR3-mediated chemotactic responses in both WT and Gαi3-deficient T cells (Figure 1C and D), confirming involvement of PTX-sensitive Gαi2 protein only in the increased migratory response in the absence of Gαi3. Our investigation thus concludes that Gαi3 participates as an inhibitor in CXCR3-stimulated signaling and null mutation of Gαi3 directly accounts for the increased migration of Gαi3-/- T cells toward CXCR3 ligands.
FIGURE 2.
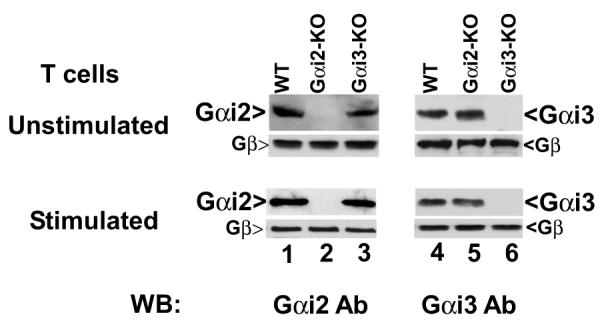

Gαi and chemokine receptor expression A. Gαi expression in T cells by immunoblotting analysis. Cell membranes of unstimulated (upper panel) or stimulated (lower panel) T cells prepared from indicated mice were analyzed by immunoblotting using anti-Gαi2 specific monoclonal Ab (left panel). Cell membranes with Gαi2 depletion were analyzed by anti-Gαi3 Ab (right panel). The blots were stripped and re-probed with anti-G protein pan β subunit Ab for equal protein loading controls. One representative result of four (left) or two (right) experiments performed is shown. B. Analysis of chemokine receptor expression on T cells. T cells prepared from indicated mice were either left unstimulated for staining CCR7 and CXCR4 receptors or stimulated with Con A for 4 days for measuring CCR7 and CXCR3 expression on a flow cytometer. The numbers within the panels indicate the percentages of positive cells on gated CD3+ T cell population. Representative results of four (CXCR3) and three (CXCR4 and CCR7) experiments performed are shown.
Altered GTPγS incorporation in the absence of Gαi2 or Gαi3
Our migration data show involvement of both Gαi2 and Gαi3 in CXCR3-mediated signaling, presumably with Gαi2 activating downstream effectors to drive T cell migration. Yet, Gαi3-mediated inhibition of Gαi2 activity in a receptor-specific fashion has never been explored. So, in the subsequent studies, we set out to determine whether Gαi3 directly blocked Gαi2 activation or diverted signals downstream of Gαi2. For downstream signaling disruption, we would expect Gαi3 activation to be an indispensable receptor-stimulated event in the G protein-signaling cascade, which can be readily measured by a GTPγS incorporation assay. All three CXCR3 chemokines at the concentrations optimal for cell migration were able to stimulate significant GTPγS incorporation in WT T cell membranes as previously reported (Figure 3A) (39). In contrast, T cells from Gαi2-/- mice failed to incorporate GTPγS significantly above controls following stimulation with CXCL9 or CXCL11 under similar conditions (Figure 3A). A small increase in GTPγS incorporation was seen in the cells stimulated with CXCL10, consistent with lesser Gαi3 inhibition in response to this chemokine (Figure 3A and Figure 1B). The lack of GTPγS incorporation in Gαi2-/- T cell membranes was not an incapability of these cells to incorporate the GTP analog, since adenosine elicited comparable GTPγS incorporation in T cells isolated from all these mice (Figure 3A) (40). Instead, the result argues strongly that Gαi3 cannot be activated by CXCR3 agonists, despite of its ability to inhibit Gαi2 function. Moreover, GTPγS incorporation in these T cells proportionally correlated with their migratory responses stimulated by CXCR3 agonists, increasing G protein activation in Gαi3-/- T cells and lacking such activation in Gαi2-/- T cells as compared to WT T cells. The increased GTPγS incorporation in Gαi3-/- cells was solely contributed by Gαi2 activation, as demonstrated by complete abrogation of the response following PTX treatment (Figure 3B). The findings not only indicate that Gαi2 is the predominant G protein activated by the CXCR3 receptor, but also suggest that CXCR3 receptor activation cannot elicit exchange of GDP for GTP in Gαi3 protein, arguing that Gαi3 interferes with Gαi2 signaling without being activated.
FIGURE 3.




CXCR3 agonist-stimulated G protein activation in the presence or absence of Gαi2 or Gαi3. A. CXCR3 agonist-stimulated GTPγS incorporation in activated T cells. Cell membranes equivalent to 20μg protein prepared from activated T cells with or without Gαi2 or Gαi3 were stimulated in triplicate with 100ng/ml CXCL9, 10ng/ml CXCL10, 100ng/ml CXCL11, or 10 μM adenosine in the presence of 0.3 nM 35S-GTPγS as described in materials and methods. Percentages ± SD of specific 35S-GTPγS incorporation relative to background controls are shown with unstimulated cell membranes designed as 100%. The result represents four separate experiments. * and ** are statistical significance (p < 0.05) or high significance (p < 0.01), respectively, in the presence vs. absence of a specific Gαi protein. B. Inhibition of G protein activation by PTX treatment. Activated T cells prepared from indicated mice were treated with 10ng/ml PTX for 1 hr before the cell membranes were prepared and assayed for GTPγS incorporation induced by either 10ng/ml CXCL10 or 100ng/ml CXCL11 as Figure 3A. Data represent three separate experiments with each sample in triplicate as Figure 3A. C. A blockade of G protein activation in Gαi3-/- T cell membrane by anti-Gαi2 Ab. G protein activation in cell membrane prepared from Gαi3-/- T cells was assayed in the presence of either 3 μg anti-Gαi2 monoclonal Ab or isotype-matched control Ab (Control Ab) during chemokine stimulation as in A. The data represent three independent experiments with each sample in triplicate as A. ** statistical significance (p < 0.01) in the presence vs. absence of a specific anti-Gαi2 Ab. D. Inhibition of Gαi2 activation in Gαi3-/- T cell membranes by exogenous Gαi3 protein. Cell membranes prepared from Gαi3-/- T cells were pre-incubated with indicated concentrations of in vitro synthesized Gαi3 or Gαo protein before stimulation of the membrane by 100 ng/ml CXCL11 and assaying G protein activation as in A. The data represent three separate experiments with each sample in triplicate. * and **, statistical significance (p < 0.05) or (p < 0.01), respectively, in the presence of Gαi3 vs. Gαo protein.
Inhibition of CXCR3-mediated Gαi2 activation by Gαi3
The consistent and significant increases in GTPγS incorporation in Gαi3 -/- T cell membranes and in the migration of these T cells argue strongly for inhibition of Gαi2 activation by Gαi3. Moreover, inhibition of Gαi2 activation without a concomitant rise in Gαi3 activation raises an intriguing possibility that Gαi3 may interact with the receptor after ligand stimulation, but remain inactive and continue its association with the receptor, by which it blocks Gαi2 interaction with the receptor. To address this, we first used an Ab directed against the C-terminus of the Gαi2 protein to block the receptor-G protein interaction in the GTPγS incorporation assay. As shown in Figure 3C, pretreatment with Gαi2-specific Ab but not control Ab completely abolished CXCL10- and CXCL11-stimulated GTPγS incorporation in Gαi3-/- T cell membranes. A specific and complete blockade of G protein activation by anti-Gαi2 Ab further rules out activation of other G proteins by the CXCR3 receptor. Secondly, in vitro synthesized Gαi3 protein was added to the GTPγS incorporation assay prior stimulation with CXCL11 to block Gαi2 activation. Cell membranes prepared from activated Gαi3 -/- T cells were used in the assay to ensure no competition from endogenous Gαi3. As shown in Figure 3D, the addition of 50 nM Gαi3 resulted in a significant decrease in GTPγS incorporation of CXCL11-stimulated membranes, with 100 nM exogenous Gαi3 protein leading to complete inhibition of Gαi2 activation. Under similar conditions, however, addition of Gαo, another member of the Gαi/o family, did not affect Gαi2 activation, indicating a specific blockage of CXCR3 activation by Gαi3 protein.
Association of Gαi3 with the CXCR3 receptor
We next determined whether Gαi3 could physically interact with the CXCR3 receptor following stimulation with CXCR3 ligands. Co-immunoprecipitation (IP) of an activated G protein with its receptor has rarely been documented owing to transient interactions between the activated receptor and a G protein. Since Gαi3 is not activated by the CXCR3 receptor, Gαi3 would be retained by the receptor after stimulation. To test this possibility, Cos-7 cells were transiently transfected with constructs encoding the CXCR3 receptor or its variants that have been shown to alter migration and GTPγS incorporation (24). Cell surface expression of these receptors was compared by flow cytometry (Figure 4A). The results showed comparable levels of the ST(-)-CXCR3 variant and WT CXCR3 and slightly but similarly decreased levels of the DNY-CXCR3 and LLL-CXCR3 variants on the cells. To evaluate the effects of these mutations on Gαi3 binding, cell membrane fractions prepared from these transfected cells were incubated with in vitro synthesized 35S-labeled Gαi2 or Gαi3 and stimulated with or without CXCL11, as the chemokine induced the most robust response among the three CXCR3 agonists. Immunoprecipitation of the receptor using anti-CXCR3 Ab revealed Gαi3 interacting with WT CXCR3 receptor after stimulation with CXCL11, with the presence of 35S-labeled Gαi3 only in CXCL11-stimulated but not in non-stimulated immunoprecipitates (Figure 4B). Co-immunoprecipitation of Gαi3 with the C-terminal mutants LLL-CXCR3 and ST(-)-CXCR3 was also evident in CXCL11-stimulated cell membranes. However, the interaction between Gαi3 and the receptor apparently did not occur when the G protein-binding motif DRY was mutated to DNY (R149N mutant) in the N-terminal portion of the second intracellular loop (Figure 4B). The finding that this motif is required for interaction between Gαi3 and the CXCR3 receptor recapitulates its critical role in proper migratory responses of the CXCR3 receptor and other chemokine receptors (24, 41). Therefore, “incomplete activated” Gαi3 may prevent Gαi2-mediated signaling via the DRY site.
FIGURE 4.
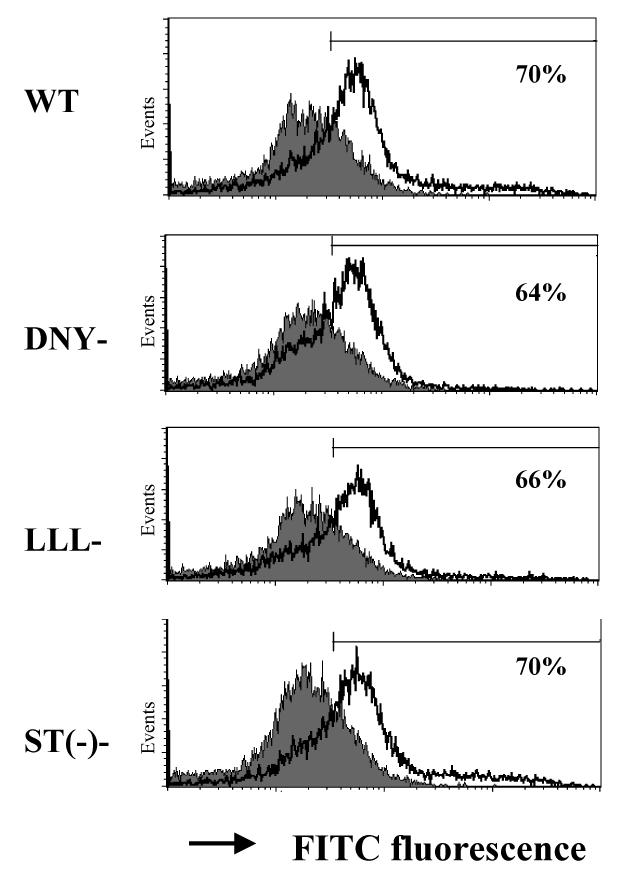


Similar structural requirement for Gαi3 binding to the CXCR3 receptor and for Gαi2 activation. A. Analysis of CXCR3 expression in transfected Cos-7 cells. Cos-7 cells transfected with WT or mutated CXCR3 receptors were stained with anti-CXCR3 Ab conjugated with Alexa and analyzed by flow cytometry. Percentages of CXCR3+ cells on gated viable cell populations are indicated within the panels, where the filled profiles are negative controls obtained by isotype-matched control Ab conjugated with Alexa. One representative result of three experiments performed is shown. B. Interaction of Gαi3 with the CXCR receptor at the G protein-binding domain. Cell membranes prepared from Cos-7 cells transfected with constructs harboring either WT or CXCR3 receptor variants were incubated with 35S-labeled Gαi2 or 35S-labeled Gαi3 protein for 2 hrs and then stimulated with 100ng/ml CXCL11. The associated Gαi proteins were immunoprecipitated by anti-CXCR3 Ab and quantified by scintillation counting on a 1450 MicroBeta scintillation counter (left panel). Association of 35S-Gαi2 with the S1P1 receptor in unstimulated cell membrane was also shown as a 35S-Gαi2 binding control (right panel). Data represent three separate experiments with two samples per assay (n = 6). **, statistical significance (p < 0.01) obtained by comparing mutated CXCR3 receptors with the WT one. C. Requirement of the G protein-binding motif for Gαi2 activation. Cell membranes prepared from Cos-7 cells transfected with indicated constructs as in A were stimulated with 100ng/ml CXCL11 or 10 μM adenosine in the presence of 0.3 nM 35S-GTPγS and the resultant 35S-GTPγS incorporation was measured as Figure 3A. The data represent three separate experiments with each sample in triplicate (n = 9). **, statistical significance (p < 0.01) obtained by comparing mutated CXCR3 receptors with the WT receptor.
In parallel experiments, immunoprecipitation of the CXCR3 receptor did not pull down Gαi2, as expected, presumably due to its rapid dissociation from the CXCR3 receptor upon activation. The 35S-Gαi2 could interact well with the S1P1 receptor, another PTX-sensitive GPCR (Figure 4B, right panel), ruling out that undetectable 35S-Gαi2 in CXCR3 immunoprecipitates resulted from a lack of binding function of the protein. Furthermore, significant GTPγS incorporation induced by CXCL11 was seen in the cell membranes prepared from Cos-7 cells transfected with WT CXCR3 receptor and all variants, except for the R149N variant that was totally ineffective (Figure 4C). The inability of the R149N mutant to activate Gαi2 and to interact with Gαi3 suggests that Gαi3 may compete for or sterically interfere with Gαi2 binding to the CXCR3 receptor directly or indirectly via the DRY domain, by which it controls the amplitude of a migratory response elicited by the receptor.
DISCUSSION
Our results clearly show that a deletion of Gαi3 augments Gαi2-mediated migratory responses toward CXCR3 ligands, whereas deletion of Gαi2 ablates the response. The Gαi3-mediated inhibition of CXCR3 signaling is not reliant on the exchange of GDP for GTP by Gαi3, but it does require ligand-stimulated interaction between Gαi3 and the CXCR3 receptor. Presumably, a conformational change of the receptor caused by ligand binding provides conditions that are optimal for Gαi3 binding to the receptor. However, the receptor-bound Gαi3 appears unable to undergo a conformational change required for dissociation of GDP and/or binding to GTP, thus preventing it from full activation and leaving the receptor. By occupancy of the receptor, Gαi3 blocks Gαi2 activation. It has been shown that the DRY motif at the N-terminus of the second intracellular loop is key for proper G protein recognition and interaction (42). Our data suggest that this motif may be also essential for the blockade induced by Gαi3, which requires further investigation to be concluded. Substitution of arginine (R)149 amino residue to asparagine (N) ablates not only activation of Gαi2 but also binding of Gαi3 to the receptor. Under similar conditions, receptors carrying mutations at the C-terminus can activate Gαi2 and preserve Gαi3 interaction similarly to the WT CXCR3 receptor (Figure 4B). Lack of Gαi3 binding to and Gαi2 activation by the R149N mutant cannot be ascribed to the levels of receptor expression on the cells. Although the R149N-CXCR3 mutant was expressed on the cells at levels slightly lower than the WT CXCR3 receptor, its expression was comparable to the LLL-CXCR3 mutant (figure 4A) (24). The latter could activate Gαi2 and bind with Gαi3 as the WT CXCR3 receptor (Figure 4B and 4C). Murine Gαi2 and Gαi3 are 83% identical in amino residue sequence. This structural similarity may be the basis for their competition for the same binding domain of the CXCR3 receptor or for Gαi3 interference with Gαi2 interaction of the receptor. A subtle difference in the receptor conformation induced by a specific agonist is likely to determine whether the activated receptor can interact and activate a given Gαi protein or interact with a Gαi protein without inducing its full activation. Therefore, function of a given Gαi protein as an activator or inhibitor is receptor specific and the interplay between these two G proteins may be essential in the control of the amplitude of a GPCR-induced signal. To the best of our knowledge, this is the first description of a G protein interacting with a receptor after ligand binding for the sole purpose of diminishing the other G protein interaction with the receptor. This finding gives novel insight into regulation of GPCRs’ signaling by heterotrimeric G proteins.
GPCRs initiate a signaling cascade through activation of heterotrimeric G proteins following stimulation by an extracellular agonist (43). To meet the complexity and versatility of cell migration in the body, many chemokine receptors have evolved to recognize more than one chemokine, such as the CXCR3 and CCR7 receptors, or one chemokine receptor can be activated by multiple chemokines like CCL5 (RANTES), CCL3 (MIP-1α), MCP-2 and MCP-3 (44). Our finding that a migratory response is tied proportionally to the amplitude of G protein activation indicates that stoichiometry of G protein activation is directly linked to the propensity of a cell to migrate. The precise activation level of the Gαi2 protein stimulated by ligation of the CXCR3 receptor is determined by the affinity of the ligand as well as the interplay of Gαi2 and Gαi3 with the receptor. In accordance with CXCR3 signaling dependent of Gαi2 and inhibition of the signaling by Gαi3, our in vivo study showed that similar to T cells from mice lacking the CXCR3 receptor, Gαi2-deficient T cells failed to elicit an acute graft-vs.-host defense (GVHD) reaction after transfused into full MHC-mismatched Balb/c SB-17 SCID (severe combined immunodeficiency) mice (Jin et al., manuscript in preparation) (30, 45). In contrast, transfer of Gαi3-deficient T cells into the SCID mice stimulated an aggravating GVHD response compared to WT T cells. Thus, an interplay among different heterotrimeric G proteins is likely to have a role to play in vivo, introducing another level of complexity in the control of cell migration systemically. The complex regulation of GPCR signaling at multiple levels is crucial in temporal and spatial regulation of cell migration in the body.
Determination of receptors linked to Gαi proteins has been greatly aided by the use of PTX. PTX has also allowed insight into downstream effectors that are either partially or fully involved in propagating a signal by Gαi proteins. While PTX has provided extensive and important details about the agonist and signal transduction mechanisms, the specifics about individual Gαi/o family members has gone largely unresolved because PTX blocks receptor interaction by several Gαi/o family members. With the advent of the genetic manipulation of mice, we can tease out the specifics of Gαi1, Gαi2, and Gαi3 in a migratory response stimulated by a specific receptor. Studies of B cell migration in Gαi2-/- mice suggest an irreplaceable role of Gαi2 in chemotactic responses to chemokines CXCL12 and CXCL13 (14). On the other hand, Gαi2 and Gαi3 are redundant in thymic emigration as indicated by no significant defects in thymic exportation in Gαi2- and Gαi3-deficient mice (12, 13). Although the proportions and the numbers of CD4 and CD8 single positive thymocytes were increased in Gαi2-/- mice as lck-PTX-transgenic mice, the increases were not caused by a defect in thymic egress, rather by an accelerated transition from the double positive to single positive thymocytes as shown by our previous investigation (12, 46). In support, nearly normal numbers and percentages of CD4+ and CD8+ T cells were seen in the spleen of Gαi2-/- mice (11-13). Likewise, mature thymocytes in Gαi3-/- mice populated lymphoid tissues normally in the periphery and there was no aberrant accumulation of single positive thymocytes in the mice (13). The redundancy of Gαi2 and Gαi3 in thymic egress was also consistent with our recent study showing that Gαi2-KO and Gαi3-KO T cells migrated indistinguishably from WT T cells in response to an increasing concentration of sphingosine-1-phosphate (S1P), a lipid mediator controlling T cell egress (our unpublished data) (47).
How the redundancy works mechanistically is not fully understood at present. Association with the same subunit of Gβγ may be one of the common mechanisms whereby Gαi2 and Gαi3 can compensate for the absence of one to another. Hwang et al. have shown that functional silence of either Gαi2 or Gαi3 by specific siRNA has little effect on the migration of macrophages toward an increasing concentration gradient of C5a or C3a (48). Deletion of Gβ2, however, ablated C5a- or C3a-provoked migration, suggesting that both Gαi2 and Gαi3 can transduce the migration signal as long as the Gβ2 subunit is presented (48). On the contrary, the CXCR4 receptor requires both Gαi2 and Gαi3 for a full response. Absence of either gene impaired T cell migration induced by CXCL12 (Figure 1F), although decreased expression of the CXCR4 receptor on these cells may also be partially involved. We should point out that a slight increase or decrease (10∼15%) in the Gαi2 expression levels in Gαi3-/- T cells or Gαi3 in Gαi2-/- T cells was observed in some mice (Figure 2A). However, the levels of Gαi protein expression were not correlated with migratory changes for any of the studied chemokines. In particular, the slight decrease in Gαi2 expression in Gαi3-KO T cells was discordant with the increased activity for the CXCR3 receptor. Similarly, although the levels of Gαi2 and Gαi3 expression were slightly decreased in activated as compared to non-activated T cells, the decrease affects little their chemotactic response to CCL19 stimulation (Figure 1 G and H). This is probably due to the following two reasons. First, Gαi2 or Gαi3 is a lot more abundant than any individual chemokine receptor in a cell. Secondly, constant recycling of Gαi proteins from an active to inactive status further increases the size of a Gαi protein pool that is already far in excess of any individual chemokine receptor. Therefore, distinct function of Gαi2 and Gαi3 described in this study is unlikely due to a slightly altered level of Gαi2 or Gαi3 protein. Nevertheless, this does not exclude that the absolute levels of Gαi2 and Gαi3 can affect activation of a given GPCR, when the relative levels of Gαi2 and Gαi3 differ drastically. Our data thus argue that Gαi2 and Gαi3 proteins may play overlapping, distinct, antagonizing and additive roles depending on a specific receptor. The Gαi2 and Gαi3-null mutation mice provide a unique system to explore fundamental signaling differences of a specific GPCR coupling to Gαi2, Gαi3 or both and potential interplay between these Gαi proteins.
Acknowledgments
We thank members in Dr. Wu’s group for stimulating discussion, Mrs. Lu Zhang for technical assistance and animal husbandry, Dr. Dominic Picarella, Millenium Pharmaceuticals, for the murine CXCR3 specific Ab (clone 4c4), and Dr. Irene Kochevar for encouragement. This work is supported by the National Institutes of Health grants AI050822 and AI070785, Research Scholar Grant RSG-01-178-01-MGO from the American Cancer Society, and a pilot grant (to M.X.W.) from National Institutes of Health Project Grant DK43351 that funds the Center for the Study of Inflammatory Bowel Disease at the Massachusetts General Hospital, and by the Intramural Research Program of the NIH, NIEHS (to L.B) and DK074449 (to A. D. L).B.T. is supported in part by a NIH T32 training grant (AR07098-31) from the Department of Dermatology, Harvard Medical School.
Reference List
- 1.Wilkie TM, Gilbert DJ, Olsen AS, Chen XN, Amatruda TT, Korenberg JR, Trask BJ, de Jong P, Reed RR, Simon MI. Nat.Genet. 1992;1:85–91. doi: 10.1038/ng0592-85. [DOI] [PubMed] [Google Scholar]
- 2.Fields TA, Casey PJ. Biochem.J. 1997;321(Pt 3):561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaslow HR, Burns DL. FASEB J. 1992;6:2684–2690. doi: 10.1096/fasebj.6.9.1612292. [DOI] [PubMed] [Google Scholar]
- 4.Kehrl JH. Immunity. 1998;8:1–10. doi: 10.1016/s1074-7613(00)80453-7. [DOI] [PubMed] [Google Scholar]
- 5.Sunyer T, Monastirsky B, Codina J, Birnbaumer L. Mol.Endocrinol. 1989;3:1115–1124. doi: 10.1210/mend-3-7-1115. [DOI] [PubMed] [Google Scholar]
- 6.Hamm HE. Proc.Natl.Acad.Sci.U.S.A. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamm HE, Gilchrist A. Curr.Opin.Cell Biol. 1996;8:189–196. doi: 10.1016/s0955-0674(96)80065-2. [DOI] [PubMed] [Google Scholar]
- 8.Wise A, Gearing K, Rees S. Drug Discov.Today. 2002;7:235–246. doi: 10.1016/s1359-6446(01)02131-6. [DOI] [PubMed] [Google Scholar]
- 9.Beals CR, Wilson CB, Perlmutter RM. Proc.Natl.Acad.Sci.U.S.A. 1987;84:7886–7890. doi: 10.1073/pnas.84.22.7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SY, Ang SL, Bloch DB, Bloch KD, Kawahara Y, Tolman C, Lee R, Seidman JG, Neer EJ. Proc.Natl.Acad.Sci.U.S.A. 1988;85:4153–4157. doi: 10.1073/pnas.85.12.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaffin KE, Perlmutter RM. Eur.J.Immunol. 1991;21:2565–2573. doi: 10.1002/eji.1830211038. [DOI] [PubMed] [Google Scholar]
- 12.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Nat.Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 13.Jiang M, Spicher K, Boulay G, Martin-Requero A, Dye CA, Rudolph U, Birnbaumer L. Methods Enzymol. 2002;344:277–298. doi: 10.1016/s0076-6879(02)44721-0. [DOI] [PubMed] [Google Scholar]
- 14.Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. Immunity. 2005;22:343–354. doi: 10.1016/j.immuni.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Wu JY, Jin Y, Edwards RA, Zhang Y, Finegold MJ, Wu MX. J.Immunol. 2005;174:6122–6128. doi: 10.4049/jimmunol.174.10.6122. [DOI] [PubMed] [Google Scholar]
- 16.Huang TT, Zong Y, Dalwadi H, Chung C, Miceli MC, Spicher K, Birnbaumer L, Braun J, Aranda R. Int.Immunol. 2003;15:1359–1367. doi: 10.1093/intimm/dxg135. [DOI] [PubMed] [Google Scholar]
- 17.Balashov KE, Rottman JB, Weiner HL, Hancock WW. Proc.Natl.Acad.Sci.U.S.A. 1999;96:6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Callahan MK, Huang D, Ransohoff RM. Curr.Top.Dev.Biol. 2005;68:149–181. doi: 10.1016/S0070-2153(05)68006-4. [DOI] [PubMed] [Google Scholar]
- 19.Papadakis KA. Curr.Allergy Asthma Rep. 2004;4:83–89. doi: 10.1007/s11882-004-0048-7. [DOI] [PubMed] [Google Scholar]
- 20.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. J.Exp.Med. 2001;193:975–980. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, Luster AD. Immunity. 2000;12:483–494. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Schlossman SF, Edwards RA, Ou CN, Gu J, Wu MX. Proc.Natl.Acad Sci U.S.A. 2002;99:878–883. doi: 10.1073/pnas.022326699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiting D, Hsieh G, Yun JJ, Banerji A, Yao W, Fishbein MC, Belperio J, Strieter RM, Bonavida B, Ardehali A. J.Immunol. 2004;172:7417–7424. doi: 10.4049/jimmunol.172.12.7417. [DOI] [PubMed] [Google Scholar]
- 24.Colvin RA, Campanella GS, Sun J, Luster AD. J.Biol.Chem. 2004;279:30219–30227. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- 25.Hornquist CE, Lu X, Rogers-Fani PM, Rudolph U, Shappell S, Birnbaumer L, Harriman GR. J.Immunol. 1997;158:1068–1077. [PubMed] [Google Scholar]
- 26.Fukushima N, Kimura Y, Chun J. Proc.Natl.Acad.Sci.U.S.A. 1998;95:6151–6156. doi: 10.1073/pnas.95.11.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sklar LA, Vilven J, Lynam E, Neldon D, Bennett TA, Prossnitz E. Biotechniques. 2000;28:976–5. doi: 10.2144/00285rr03. [DOI] [PubMed] [Google Scholar]
- 28.Harrison C, Traynor JR. Life Sci. 2003;74:489–508. doi: 10.1016/j.lfs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Spiegel AM, Shenker A, Weinstein LS. Endocr.Rev. 1992;13:536–565. doi: 10.1210/edrv-13-3-536. [DOI] [PubMed] [Google Scholar]
- 30.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C. J.Exp.Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wess J. Pharmacol.Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- 32.Prossnitz ER, Schreiber RE, Bokoch GM, Ye RD. J.Biol.Chem. 1995;270:10686–10694. doi: 10.1074/jbc.270.18.10686. [DOI] [PubMed] [Google Scholar]
- 33.Sklar LA, Vilven J, Lynam E, Neldon D, Bennett TA, Prossnitz E. Biotechniques. 2000;28:976–5. doi: 10.2144/00285rr03. [DOI] [PubMed] [Google Scholar]
- 34.Smit MJ, Verdijk P, van der Raaij-Helmer EM, Navis M, Hensbergen PJ, Leurs R, Tensen CP. Blood. 2003;102:1959–1965. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z, Kaptanoglu L, Haddad W, Ivancic D, Alnadjim Z, Hurst S, Tishler D, Luster AD, Barrett TA, Fryer J. J.Immunol. 2002;168:3205–3212. doi: 10.4049/jimmunol.168.7.3205. [DOI] [PubMed] [Google Scholar]
- 36.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. J.Exp.Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu B, Humbles A, Bota D, Gerard C, Moser B, Soler D, Luster AD, Gerard NP. Eur.J.Immunol. 1999;29:3804–3812. doi: 10.1002/(SICI)1521-4141(199911)29:11<3804::AID-IMMU3804>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 38.Haunerland NH, Spener F. Prog.Lipid Res. 2004;43:328–349. doi: 10.1016/j.plipres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Cox MA, Jenh CH, Gonsiorek W, Fine J, Narula SK, Zavodny PJ, Hipkin RW. Mol.Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- 40.Palmer TM, Gettys TW, Stiles GL. J.Biol.Chem. 1995;270:16895–16902. doi: 10.1074/jbc.270.28.16895. [DOI] [PubMed] [Google Scholar]
- 41.Arai H, Charo IF. J.Biol.Chem. 1996;271:21814–21819. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- 42.Wess J. Pharmacol.Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- 43.Roberts DJ, Waelbroeck M. Biochem.Pharmacol. 2004;68:799–806. doi: 10.1016/j.bcp.2004.05.044. [DOI] [PubMed] [Google Scholar]
- 44.Gonsiorek W, Zavodny P, Hipkin RW. J.Immunol.Methods. 2003;273:15–27. doi: 10.1016/s0022-1759(02)00415-5. [DOI] [PubMed] [Google Scholar]
- 45.Duffner U, Lu B, Hildebrandt GC, Teshima T, Williams DL, Reddy P, Ordemann R, Clouthier SG, Lowler K, Liu C, Gerard C, Cooke KR, Ferrara JL. Exp.Hematol. 2003;31:897–902. doi: 10.1016/s0301-472x(03)00198-x. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Finegold MJ, Jin Y, Wu MX. Int.Immunol. 2005;17:233–243. doi: 10.1093/intimm/dxh204. [DOI] [PubMed] [Google Scholar]
- 47.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 48.Hwang JI, Fraser ID, Choi S, Qin XF, Simon MI. Proc.Natl.Acad.Sci.U.S.A. 2004;101:488–493. doi: 10.1073/pnas.0307549100. [DOI] [PMC free article] [PubMed] [Google Scholar]