

Abstract
Microglial activation is a hallmark of brain abscess. The continual release of proinflammatory mediators by microglia following bacterial challenge may contribute, in part, to the destruction of surrounding normal tissue characteristic of brain abscess. Therefore, attenuating chronic microglial activation during the course of CNS bacterial infections may have therapeutic benefits. The purpose of this study was to evaluate the ability of the natural peroxisome proliferator-activated receptor (PPAR)-γ agonist 15-deoxy-Δ12,14- prostaglandin J2 (15d-PGJ2) to modulate microglial activation in response to Staphylococcus aureus, one of the main etiologic agents of brain abscess in humans. 15d-PGJ2 was a potent inhibitor of proinflammatory cytokine (IL-1β, TNF-α, IL-12 p40) and CC chemokine (MIP-1β, MCP-1) production in primary microglia, but had no effect upon the expression of select CXC chemokines (MIP-2, KC). 15d-PGJ2 also selectively inhibited the S. aureus-dependent increase in microglial TLR2, CD14, MHC class II, and CD40 expression, whereas it had no effect on the co-stimulatory molecules CD80 and CD86. Microarray analysis revealed additional inflammatory mediators modulated by 15d-PGJ2 in primary microglia following S. aureus exposure, the majority of which were chemokines. These results suggest that suppressing microglial activation through the use of 15d-PGJ2 may lead to the sparing of damage to normal brain parenchyma that often results from brain abscess.
Keywords: chemokines, cytokines, 15d-PGJ2, microglia, PPAR-γ, Staphylococcus aureus
Brain abscesses develop in response to a parenchymal infection with pyogenic bacteria, beginning as a localized area of cerebritis and evolving into a suppurative lesion surrounded by a well-vascularized fibrotic capsule. Brain abscess remains a significant medical problem despite recent advances made in detection and therapy (Mathisen and Johnson 1997; Townsend and Scheld 1998). In addition, the emergence of multi-drug resistant strains of bacteria has become a confounding factor that is magnified the inability of many antibiotics to reach high therapeutic levels in brain tissue.
We have established a mouse experimental brain abscess model using Staphylococcus aureus where lesion sites are greatly exaggerated compared to the localized nature of bacterial growth, reminiscent of an overactive immune response (Kielian et al. 2001a, 2001b; Kielian and Hickey 2002; Baldwin and Kielian 2004). This phenomenon is also observed in human brain abscess, where lesions can encompass a large portion of brain tissue, often spreading well beyond the initial focus of infection. In addition to peripheral immune cell infiltrates, microglial activation is a hallmark of brain abscess development (Flaris and Hickey 1992; Kielian and Hickey 2002; Baldwin and Kielian 2004). Microglia are the resident mononuclear phagocytes of the CNS parenchyma and participate in innate immune responses (Aloisi 2001; Hanisch 2002). As such, microglia are uniquely poised to provide an initial line of defense against invading microorganisms into the CNS prior to leukocyte infiltration. Our group has recently reported that exposure of microglia to S. aureus leads to the elaboration of a wide array of proinflammatory mediators and enhanced expression of surface receptors that play a pivotal role in bacterial recognition and antigen presentation (Kielian et al. 2001b, 2002). Based on these findings, we have proposed that chronic microglial activation may contribute, in part, to the destruction of normal CNS parenchyma characteristic of brain abscess through the excessive production of proinflammatory mediators. Evidence to support this concept is provided by our recent studies demonstrating the prolonged expression of TNF-α, IL-1β, and MIP-2/CXCL2, factors produced by S. aureus-stimulated microglia ( Kielian et al. 2002), concomitant with continued macrophage/microglial activation in mice with S. aureus-induced brain abscess (Baldwin and Kielian 2004). Additional evidence demonstrating that microglia contribute to the exacerbation of CNS disease is provided by studies in Alzheimer’s disease and experimental autoimmune encephalomyelitis (EAE), the animal model for multiple sclerosis, where activated microglia have been shown to secrete a wide array of inflammatory cytokines and cytotoxic agents, including TNF-α, IL-1β, and nitric oxide (NO; Meda et al. 1995; Renno et al. 1995; Sheng et al. 1995; Benveniste 1997; Tran et al. 1997; Lue et al. 2001; Griffin and Mrak 2002). Therefore, attenuating microglial activation subsequent to effective bacterial neutralization in the brain may result in less damage to normal parenchyma and improvements in cognitive and neurological functions, all pathological changes associated with brain abscess.
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily of proteins that regulate the expression of genes involved in reproduction, metabolism, development, and immune responses (Desvergne and Wahli 1999; Straus and Glass 2001; Kielian and Drew 2003). Three PPAR subtypes exist, α, δ (also referred to as β), and γ, that exhibit differential tissue distributions and ligand specificities (Desvergne and Wahli 1999; Straus and Glass 2001). Cells of the monocyte/macrophage lineage were recently shown to express PPAR-γ, suggesting a role for this receptor in the function of these cells (Ricote et al. 1998a). Subsequently, select PPAR-γ ligands including the naturally occurring prostaglandin metabolite 15-deoxy-Δ12,14- prostaglandin J2 (15d-PGJ2), were demonstrated to inhibit the production of a variety of proinflammatory molecules by monocytes/macrophages and microglia (Jiang et al. 1998; Ricote et al. 1998a; Petrova et al. 1999; Bernardo et al. 2000, 2003; Drew and Chavis 2001; Kielian and Drew 2003). However, to date, the effects of 15d-PGJ2 on microglial activation have only been examined in response to the gram-negative cell wall product lipopolysaccharide (LPS) or a combination of TNF-α and IFN-γ (Petrova et al. 1999; Bernardo et al. 2000, 2003; Drew and Chavis 2001). The effects of 15d-PGJ2 on microglial activation in response to the gram-positive bacterium S. aureus have not yet been investigated. This study demonstrates that 15d-PGJ2 is a potent and selective inhibitor of microglial activation in response to S. aureus, suggesting that it may exert beneficial effects during the chronic stage of brain abscess by minimizing damage to normal brain parenchyma that is a common sequelae of disease.
Materials and methods
Reagents and isolation of primary microglia
Primary microglia were isolated from C57BL/6 pups (1–8 days of age) as previously described (Esen et al. 2004). Briefly, mixed glial cell cultures were resuspended in complete Dulbecco’s modified Eagle’s medium (DMEM, 4.5 g/L glucose, Biowhittaker, Walkersville, MD, USA) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 200 mM L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin and 0.25 μg/mL fungizone (P/S/F mix, Biowhittaker), OPI medium supplement (oxalacetic acid, pyruvate, insulin, Sigma, St Louis, MO, USA), and 0.5 ng/mL recombinant mouse granulocyte–macrophage colony-stimulating factor (GM–CSF; BD Pharmingen, San Diego, CA, USA) and maintained in a humidified atmosphere of 95% air/5% CO2. When cultures reached confluency (1–2 weeks), flasks were shaken overnight at 50 g at 37°C to recover microglia. The purity of microglial cultures was evaluated by immunohistochemical staining using antibodies against CD11b (BD Pharmingen) and glial fibrillary acidic protein (GFAP; Dako Corp., Carpenteria, CA, USA) to identify microglia and astrocytes, respectively. The purity of primary microglia cultures was routinely greater than 90%, with astrocytes representing the major cell contaminant. However, we have recently demonstrated that astrocytes are sensitive to the inhibitory effects of 15d-PGJ2 following S. aureus exposure at levels equivalent to those used for primary microglia (Phulwani and Kielian, manuscript in preparation).
The murine N9 microglial cell line was kindly provided by Dr Ricciardi-Castagnoli (University of Milano Bicocca, Milano, Italy) and maintained in Isovec’s minimal defined medium (IMDM, Biowhittaker) supplemented with 5% FBS (0.2 μm filtered), 50 μM β-mercaptoethanol, 200 mM L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and fungizone. Heat-inactivated S. aureus (strain RN6390, kindly provided by Dr Ambrose Cheung, Dart-mouth Medical School) was prepared as previously described (Kielian et al. 2002). 15d-PGJ2 was purchased from Cayman Chemical (Ann Arbor, MI, USA). All reagents and culture media were verified to have endotoxin levels < 0.03 EU/mL as determined by Limulus amebocyte lysate assay (LAL; Associates of Cape Cod, Falmouth, MA, USA).
ELISA
Quantitation of cytokine and chemokine levels in microglial conditioned medium was performed using standard sandwich enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions (OptEIA mouse TNF-α, IL-1β, IL-12, MCP-1/CCL2, and MIP-1β /CCL4, BD Pharmingen; DuoSet mouse MIP-2/CXCL2 and KC/CXCL2, R & D Systems, Minneapolis, MN, USA). The level of sensitivity for all ELISAs was 15.6 pg/mL.
Cell viability assays
The effects of 15d-PGJ2 on microglial cell viability were evaluated using a standard MTT assay based upon the mitochondrial conversion of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) into formazan crystals as previously described (Esen et al. 2004). Results are reported as the raw OD570 values (mean ± SEM).
Flow cytometric analysis
To examine the effect of 15d-PGJ2 on microglial surface marker expression following S. aureus stimulation, one-color flow cytometric analysis was utilized. Primary antibodies included TLR2 and TLR6 (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA) and CD14, CD40, CD80, CD86, and MHC Class II (all from BD PharMingen). Cells were then incubated with the appropriate biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA, USA), and detected using a streptavidin–Alexa 488 conjugate (Molecular Probes, Eugene, OR, USA). Cells were analyzed using a FACSCalibur cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) with compensation set based on the staining of cells with secondary antibodies alone.
Protein extraction and western blotting
Protein extracts were prepared from primary microglia cultured in 6-well plates by lysing cells in 200 μL of RIPA buffer [1% NP-40, 0.1% sodium dodecyl sulfate (SDS) in phosphate-buffered saline (PBS), pH 7.4] supplemented with a Complete™ protease inhibitor cocktail tablet (Roche, Indianapolis, IN, USA). Lysates were allowed to incubate on ice for 30 min followed by centrifugation at 21 000 g for 15 min at 4°C to pellet debris. Calculation of total protein in microglial extracts was determined by using a standard protein assay (bicinchoninic acid protein assay reagent, BCA; Pierce, Rockford, IL, USA).
PPAR-γ expression was evaluated by Western blot analysis. Microglial protein extracts (10 μg of total protein) were electrophoresed on 4–10% Tris–HCl Ready Gels (Bio-Rad, Hercules, CA, USA) and transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore, Bedford, MA, USA) using a semi-dry transfer apparatus (Bio-Rad). Blots were probed using a rabbit anti-mouse PPAR-γ antibody specific for PPAR-γ2 (Affinity Bioreagents, Golden, CO, USA) followed by a donkey anti-rabbit IgG–HRP conjugate (Jackson Immunoresearch, West Grove, PA, USA). Duplicative blots were probed with a rabbit anti-actin polyclonal antibody (A-5060; Sigma) to verify uniformity in gel loading. Blots were developed using the ChemiGlow West substrate (Alpha Innotech, San Leandro, CA, USA) and visualized by exposure to X-ray film (Kodak Biomax, Rochester, NY, USA).
Analysis of the effects of 15d-PGJ2 on microglial inflammatory molecule expression by microarray analysis
The effects of 15d-PGJ2 on gene expression in S. aureus-stimulated primary microglia were examined by microarray analysis as previously described (Kielian et al. 2004). Briefly, samples were evaluated for changes in inflammatory gene expression using a custom-designed oligonucleotide microarray synthesized by MWG Biotech (Highpoint, NC, USA). This microarray consisted of 285 genes including apoptotic and neurotrophic factors, pattern recognition receptors, cytokines, chemokines, and their receptors. The complete list of genes can be found at http://www.uams.edu/kielianlab. Microarray printing onto glass slides, probe labeling, hybridization, and data acquisition were performed by the UAMS Microarray Core Facility.
The production of labeled target riboprobes from 5 μg DNA-free total mRNA isolated from primary microglia were generated with the 3DNA Array 350 kit from Genisphere (Hatfield, PA, USA). Hybridization was performed on the Discovery Hybridization Station (Ventana Medical Systems, Tucson, AZ, USA). Slides were removed from the hybridization station, dried, and scanned using a Perkin Elmer ScanArray 5000 (Perkin Elmer Life and Analytical Sciences, Boston, MA, USA). Quantitation of signal intensities was performed as described below.
Microarray data interpretation and statistical analysis
Microarray expression data was Lowess transformed in ScanArray Express (Perkin Elmer). The normalized data was formatted and filtered in Microsoft Excel and subsequently imported to Decision- Site Browser (Spotfire®, Somerville, MA, USA) where it was log2-transformed. Each experimental group (methyl acetate, S. aureus only, and S. aureus + 15d-PGJ2) consisted of three biological replicates. A biological replicate from each group was directly compared with one replicate from each of the other two groups using a loop design, resulting in three such loops. RNA from each of the pairings was used to make four technical replicates (top and bottom arrays on each slide and a dye swap). From the biological and technical replicates, 12 intensity value pairs were measured per combination of experimental treatments and the ratios of these pairs calculated.
For a given comparison of biological replicates in different experimental groups, a one-sample t-test was performed on the four intensity ratios from the technical replicates with a significance level of 0.01. This was less conservative than Bonferroni multiple testing correction while still lowering the number of false positives. Genes with a significant p-value had ratios that were statistically different from one (zero in the log domain). The one-sample t-test was repeated for the other biological replicates in the experimental comparison. Using DecisionSite, the three lists of p-values were compared and genes with significant p-values in at least two of the three biological replicates were noted. This process was repeated for the remaining comparisons of experimental groups.
Quantitative real-time RT–PCR
Total RNA was isolated from primary microglia using the TriZol reagent and treated with DNAse1 (both from Invitrogen) prior to use in real-time RT–PCR studies. The experimental procedure was performed as previously described with minor modifications (Esen et al. 2004). Briefly, TLR2, CD14, and GAPDH primers and TAMRA TaqMan probes were synthesized by Applied Biosystems (Foster City, CA, USA; Table 1). The reverse transcription (RT) reaction was carried out using the iScript cDNA™ synthesis kit (Bio-Rad) according to the manufacturer’s instructions. The real-time PCR reactions were performed in a total reaction volume of 25 μL using the iCycler™ kit (Bio-Rad) containing a final concentration of 400 nM of forward and reverse primers, 200 nM of Taqman probe, and 1 μL of cDNA from the RT step and analyzed using the iCycler IQ™ multicolor real-time PCR detection system (Bio-Rad). The levels of TLR2 and CD14 gene expression following S. aureus and/or 15d-PGJ2 treatment were calculated after normalizing cycle thresholds against the ‘housekeeping’ gene GADPH and are presented as the fold-induction (2–Δ ΔCt) value relative to unstimulated microglia (mean ± SD).
Table 1.
Primers and probes used for quantitative real-time RT–PCR
| Genes | Forward primers | Reverse primers | Probes | Amplicon length |
|---|---|---|---|---|
| GAPDH | GGGAAGCTCACTGGCATGG | CTTCTTGATGTCATCATACTTGGCAG | TCCTACCCCCAATGTGTCCGTCGTG | 105 |
| TLR2 | GGAACTGTCGGAGGTAGAGTTCG | TTTCTACTTTACCCAGCTCGCTCA | ACCCTCAATGGGCTCGGCGATTTC | 102 |
| CD14 | CCAGTCAGCTAAACTCGCTCAATC | TCCAGCCTGTTGTAACTGAGATCC | AAGGGCTGCCAGCCAAGCTCAGC | 106 |
Statistics
Significant differences between experimental groups were determined by using the Student’s t-test at the 95% confidence interval.
Results
Mouse microglia express PPAR-γ
PPAR-γ serves as a receptor for several agonists capable of inhibiting inflammatory gene expression in macrophages/microglia including 15d-PGJ2 (Jiang et al. 1998; Ricote et al. 1998a; Petrova et al. 1999; Bernardo et al. 2000; Drew and Chavis 2001). PPAR-γ protein expression has been demonstrated in monocytes/macrophages (Ricote et al. 1998b; Shu et al. 2000; Thieringer et al. 2000); however, to date, only one group has reported PPAR-γ protein expression in primary rat microglia (Bernardo et al. 2000, 2003). To determine whether primary mouse microglia expresses PPAR-γ and if receptor levels were influenced by cell activation, western blots were performed. Primary microglia constitutively expressed PPAR-γ (Fig. 1) and, interestingly, treatment with either 15d-PGJ2 or S. aureus had no effect on PPAR-γ levels. Similar results were obtained with N9 microglia (data not shown). The finding that microglia express detectable PPAR-γ suggested that these cells may be sensitive to the inhibitory effects of PPAR-γ agonists.
Fig. 1.
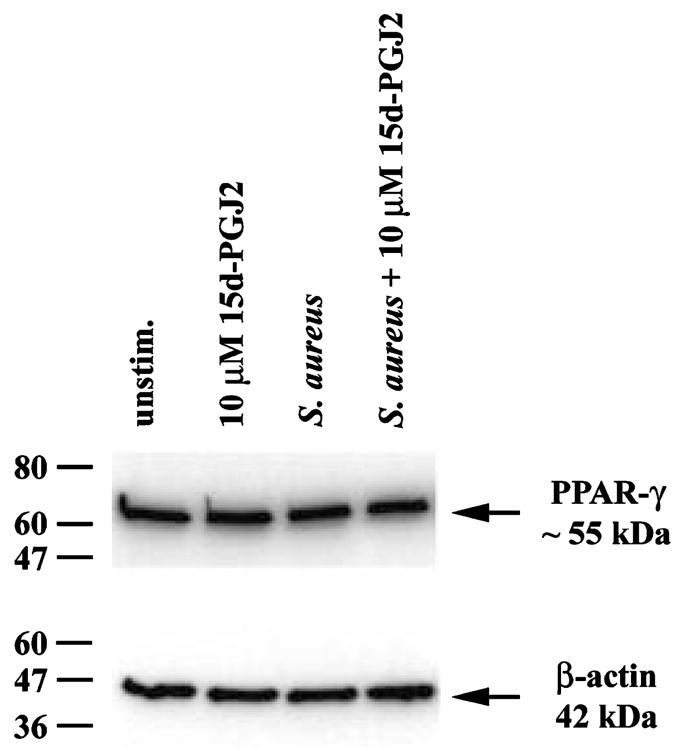
Microglia express PPAR-γ. Primary microglia were stimulated with 107 heat-inactivated Staphylococcus aureus in the presence or absence of 10 μM 15d-PGJ2. Protein extracts from whole cell lysates were prepared 24 h following stimulation and 10 μg of total protein from each sample was run on a 4–10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) gel as described in Materials and methods. Following transfer, membranes were probed with antibodies specific for PPAR-γ2 (55 kDa) or β-actin (42 kDa) to verify uniformity in gel loading. Results are representative of two independent experiments.
The cyclopentenone prostaglandin 15d-PGJ2 attenuates cytokine and CC chemokine production from S. aureus-stimulated primary microglia
To date, the effects of the natural PPAR-γ agonist 15d-PGJ2 on S. aureus-dependent microglial activation have not been investigated. Therefore, we evaluated the ability of 15d-PGJ2 to modulate the expression of several proinflammatory mediators known to be produced by microglia following S. aureus exposure (Kielian et al. 2002b). Stimulation of primary microglia with heat-inactivated S. aureus led to cellular activation typified by the production of IL-1β, TNF-α, and IL-12 p40 (Fig. 2). Pre-treatment of microglia with 15d-PGJ2 for 1 h prior to bacterial stimulation led to a dose-dependent inhibition of all three mediators, with maximal inhibition observed at a concentration of 20 μM (Figs 2a–c, respectively). Cell viability experiments revealed that 15d-PGJ2 was not toxic to primary microglia at any of the doses examined, indicating that the reduction in proinflammatory cytokine production following 15d-PGJ2 treatment was not the result of cell death (Fig. 2d). Similar findings were obtained with the N9 microglial cell line (data not shown); however, the effective dose of 15d-PGJ2 required for maximal inhibition of proinflammatory cytokine production without significant toxicity in these cells was much lower (2.5 μM) compared to primary microglia (20 μM).
Fig. 2.
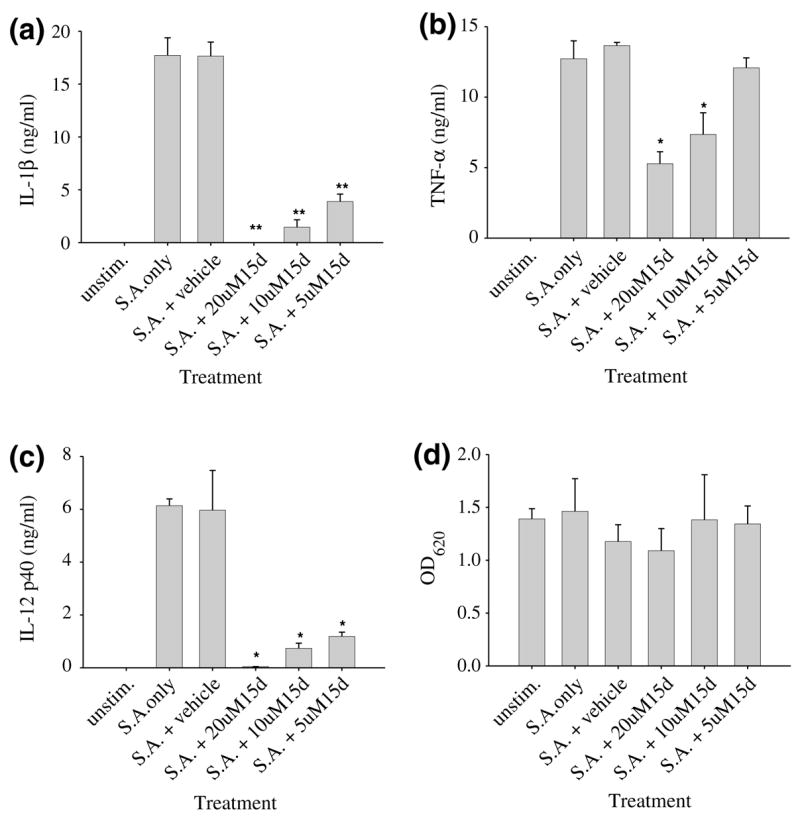
15d-PGJ2 is a potent inhibitor of proinflammatory cytokine expression in Staphylococcus aureus-stimulated primary microglia. Primary microglia were seeded in 96-well plates at 2 × 105 cells per well and incubated overnight. The following day, microglia were pre-treated with vehicle alone or various concentrations of 15d-PGJ2 for 1 h prior to stimulation with 107 heat-inactivated S. aureus. Supernatants were collected 24 h following S. aureus exposure whereupon IL-1β (a), TNF-α (b), and IL-12 p40 (c) release was quantified by ELISA. Results are presented as the amount of cytokine (ng) per ml of culture supernatant (mean ± SEM). Microglial viability was assessed using a standard MTT assay and the raw OD570 absorbance values are reported (mean ± SEM, d). Significant differences between microglia pretreated with vehicle + S. aureus versus cells exposed to the various concentrations of 15d-PGJ2 + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of eight independent experiments.
Analysis of chemokine expression revealed that 15d-PGJ2 significantly inhibited the release of the CC chemokines MIP-1β /CCL4 and MCP-1/CCL2 from S. aureus-stimulated primary microglia (Figs 3a and b), whereas it had no effect on the production of the CXC chemoattractants MIP-2/CXCL2 or KC/CXCL2 (Figs 3c and d). Again, no toxic effects were observed with any of the concentrations of 15d-PGJ2 tested (data not shown). Similar results were obtained with the N9 microglial cell line in response to S. aureus (data not shown). Collectively, these findings indicate that 15d-PGJ2 inhibits a specific subset of proinflammatory genes expressed by S. aureus-stimulated microglia and is not a global inhibitor of microglial activation.
Fig. 3.
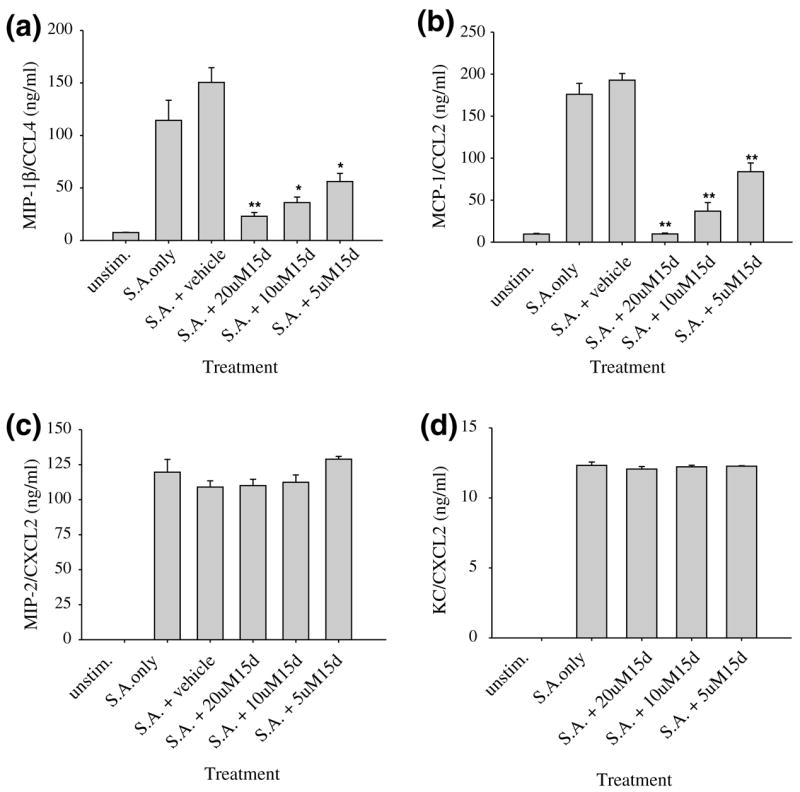
15d-PGJ2 is a selective inhibitor of CC but not CXC chemokine release from Staphylococcus aureus-stimulated primary microglia. Primary microglia were seeded in 96-well plates at 2 × 105 cells per well and incubated overnight. The following day, microglia were pre-treated with vehicle alone or various concentrations of 15d-PGJ2 for 1 h prior to stimulation with 107 heat-inactivated S. aureus. Supernatants were collected 24 h following S. aureus exposure whereupon MIP-1β /CCL4 (a), MCP-1/CCL2 (b), MIP-2/CXCL2 (c), and KC/CXCL2 (d) release was quantified by ELISA. Results are presented as the amount of chemokine (ng) per ml of culture supernatant (mean ± SEM). Significant differences between microglia pre-treated with vehicle + S. aureus versus cells exposed to the various concentrations of 15d-PGJ2 + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of eight independent experiments.
15d-PGJ2 selectively attenuates surface marker expression in S. aureus-stimulated microglia
We have recently demonstrated that S. aureus enhances the expression of numerous microglial cell surface markers involved in microbial recognition and antigen presentation, including TLR2, TLR6, CD40, CD80, CD86, and MHC class II (Kielian et al. 2002). The ability of 15d-PGJ2 to modulate the expression of these receptors could serve as a means to regulate microglial and T cell activation during gram-positive bacterial infections in the CNS. For these studies, we assessed the effects of 15d-PGJ2 on surface marker expression using N9 microglia as the numbers of cells required for flow cytometric analysis prohibited the use of primary cells, the latter of which can only be obtained in limited quantities. Similar to its selective inhibitory effects on chemokine expression, 15d-PGJ2 was only capable of inhibiting microglial CD40, TLR2, CD14, and MHC class II expression following S. aureus exposure, whereas CD80, CD86, and TLR6 were not affected (Fig. 4 and data not shown). Importantly, the observed changes in TLR2 and CD14 receptor expression were also confirmed at the RNA level by quantitative real-time RT–PCR analysis in primary microglia (Fig. 5), suggesting that the results obtained with N9 cells accurately reflect changes occurring in primary microglia. These findings suggest that 15d-PGJ2 may be capable of influencing microbial recognition and antigen presentation in the CNS.
Fig. 4.
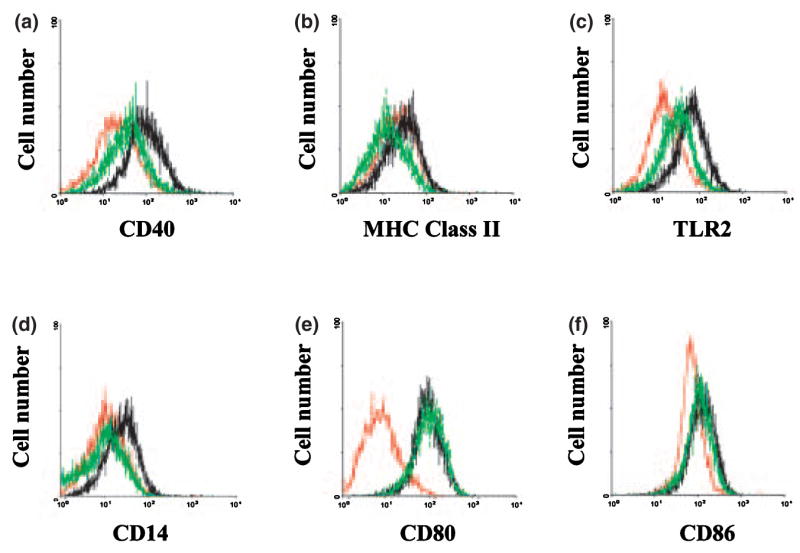
15d-PGJ2 selectively attenuates surface marker expression in Staphylococcus aureus-stimulated microglia. N9 microglia were cultured at 2 × 106 cells in 100-mm dishes and incubated overnight. On the following day, microglia were either pre-treated with 2.5 μM 15d-PGJ2 alone (red lines), 2.5 μM of 15d-PGJ2 for 1 h prior to stimulation with 108 heat-inactivated S. aureus (green lines), or exposed to S. aureus only (black lines). Cells were collected 24 h following stimulation and CD40 (a), MHC Class II (b), TLR2 (c), CD14 (d), CD80 (e), and CD86 (f) receptor expression was evaluated at the cell surface using flow cy-tometric analysis as described in Materials and methods. Results are representative of three independent experiments.
Fig. 5.
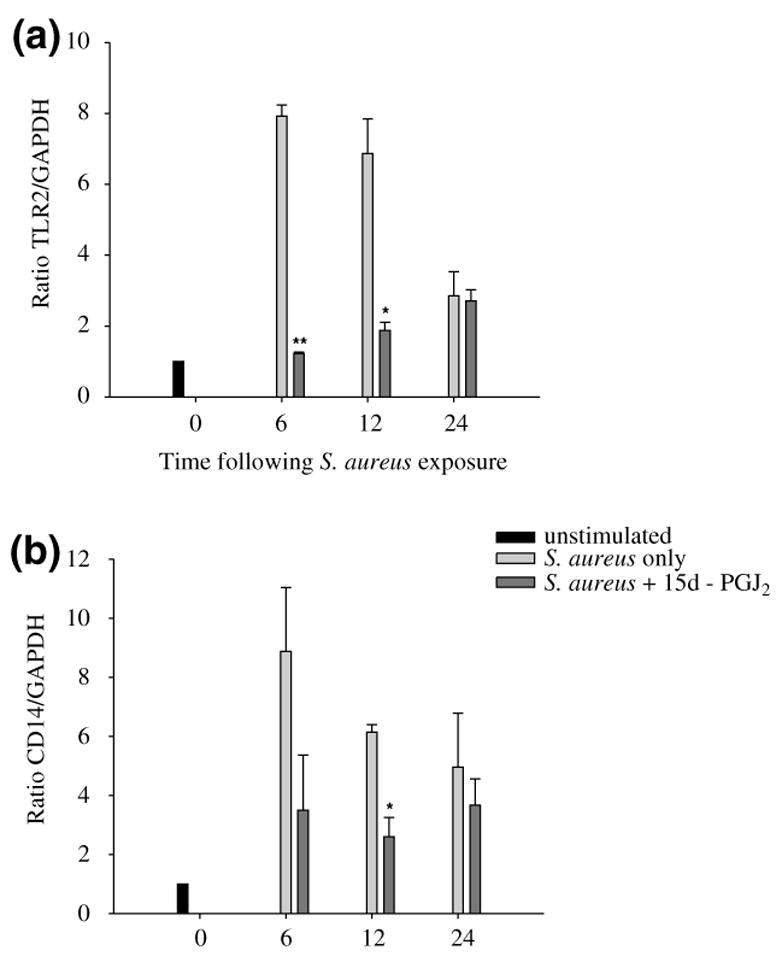
15d-PGJ2 is capable of attenuating TLR2 and CD14 mRNA expression in primary microglia following Staphylococcus aureus exposure. Primary microglia were pre-treated with 20 μM 15d-PGJ2 or vehicle alone for 1 h prior to stimulation with 107 heat-inactivated S. aureus. Total RNA was isolated from microglia at the indicated time points and analyzed for TLR2 (a) and CD14 (b) mRNA expression by quantitative real-time RT–PCR as described in Materials and methods. The levels of TLR2 and CD14 gene expression following S. aureus and/or 15d-PGJ2 treatment were calculated after normalizing cycle thresholds against the ‘housekeeping’ gene GADPH and are presented as the fold-induction (2–ΔΔCt) value relative to unstimulated microglia (mean ± SD). Significant differences between microglia stimulated with S. aureus alone versus cells treated with 20 μM 15d-PGJ2 + S. aureus are denoted with asterisks (*p < 0.05, **p < 0.001). Results are representative of three independent experiments.
Expanded analysis of changes in gene expression influenced by the PPAR-γ agonist 15d-PGJ2 in S. aureus-stimulated primary microglia using microarray analysis
To broaden our analysis of inflammatory mediators modulated by 15d-PGJ2 in S. aureus-stimulated primary microglia, a targeted microarray approach was utilized. The time- and cost-restrictive nature of microarray analysis prevented the evaluation of a wide range of time points; therefore, we chose to examine the effects of 15d-PGJ2 at 6 h following S. aureus stimulation to identify immediate changes in inflammatory gene expression. As shown in Table 2, S. aureus led to the induction of a wide array of genes in primary microglia whose expression was significantly augmented compared to vehicle treated cells. The predominant mediators expressed by S. aureus-stimulated microglia were cytokines and chemokines. A direct comparison of primary microglia stimulated with S. aureus in the presence or absence of 15d-PGJ2 on the same microarray only revealed a total of six genes that were differentially expressed and statistically significant (Table 2), the majority of which were chemokines. Importantly, 15d-PGJ2 treatment had no effect on the expression of several genes included in the array, suggesting that this compound selectively altered responses by activated microglia. These findings confirm our results demonstrating that 15d-PGJ2 is capable of modulating the expression a subset of proinflammatory mediators produced by primary microglia following S. aureus exposure.
Table 2.
Microarray analysis demonstrating changes in inflammatory mediator expression in primary microglia following exposure to S. aureus ± 15d-PGJ2
|
Staphylococcus aureus only versus
|
||||
|---|---|---|---|---|
| Methyl acetate control | S. aureus + 15d-PGJ2 | |||
| Category | Gene name* | Fold change† | Gene name | Fold change‡ |
| Chemokine | CCL2/MCP-1 | 8.1 | ||
| CCL3/MIP-1α | 6.3 | |||
| CCL4/MIP-1β | 11.7 | CCL4/MIP-1β | −3.3 | |
| CCL5/RANTES | 15.1 | |||
| CCL7/MCP-3 | 22.9 | |||
| CCL12/MCP-5 | 8.5 | CCL12/MCP-5 | −2.0 | |
| CCL24/eotaxin-2 | 4.1 | CCL24/eotaxin-2 | −1.6 | |
| CXCL2/MIP-2 | 25.3 | |||
| CX3CL1/fractalkine | 3.4 | |||
| Cytokine | IL-1β | 28.4 | IL-1β | −2.0 |
| IL-15 | 3.3 | |||
| IL-18 | 3.6 | |||
| TNF-α | 6.1 | |||
| TNFSF14 | −1.4 | |||
| VEGF B | −1.5 | |||
| FGF4 | 4.0 | |||
| Receptor | CCR5 | 4.5 | ||
| CXCR2 | 5.5 | CXCR2 | −2.2 | |
| IL-1 Ra | 8.2 | |||
| CD14 | 5.6 | |||
| TLR3 | 17.7 | |||
| LAM RI | −1.7 | |||
| MRC type I | −1.0 | |||
| MMP/TIMP | MMP-13 | 5.9 | MMP-13 | −2.0 |
| TIMP-1 | 1.3 | |||
| TIMP-2 | 1.1 | |||
| Apoptosis | Casp-1 | 3.0 | ||
| Casp-4 | 6.2 | |||
| Casp-12 | 13.0 | |||
| PG pathway | PTGS1 | −2.5 | ||
| PTGS2 | 10.2 | |||
| Signal transduction | NF-kBib | 2.7 | ||
| Stat-6 | 4.4 | |||
| SOCS-1 | 7.2 | |||
| c-fos IGF | 3.2 | |||
| Miscellaneous | iNOS | 4.6 | ||
MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; RANTES, regulated upon activation T cell expressed and secreted; IL, interleukin; TNFRSF, tumor necrosis factor receptor superfamily; TLR, Toll-like receptor; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinases; Casp, caspase; PTGS, prostaglandin-endoperoxide synthase; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; SOCS, suppressor of cytokine signaling; NOS, nitric oxide synthase; LAM, laminin receptor; MRC, macrophage man-nose receptor;
(+) = increased in S. aureus-treated primary microglia; (–)= increased in methyl acetate-treated primary microglia;
(+)= increased in S. aureus + 15d-PGJ2-treated primary microglia; (–)= increased in S. aureus-treated primary microglia.
Discussion
The present work has established that 15d-PGJ2 is a selective inhibitor of S. aureus-dependent microglial activation and is not a global suppressor, suggesting that it may have therapeutic benefits in the treatment of CNS bacterial infections if administered subsequent to bacterial neutralization. Microarray analysis revealed that the majority of genes that were affected by 15d-PGJ2 in S. aureus-treated primary microglia were chemokines. Importantly, 15d-PGJ2 had no effect on CXCL2/MIP-2 but significantly inhibited IL-1β expression, confirming our findings at the protein level. In general, our microarray results revealed that 15d-PGJ2 did not exert dramatic effects on proinflammatory mediator mRNA expression. This finding is intriguing because we have demonstrated that 15d-PGJ2 is a potent inhibitor of numerous cytokines, chemokines, and surface markers on microglia at the protein level. These results may be explained by the limited sensitivity of microarray analysis compared to other approaches used to evaluate changes in mRNA expression, as our microarray procedure does not involve an initial RNA amplification step. This possibility is supported by the fact that using quantitative real-time RT–PCR analysis, a technique that incorporates an initial amplification step, 15d-PGJ2 was found to significantly attenuate the S. aureus-dependent increase in TLR2 and CD14 mRNA expression in primary microglia confirming the changes observed at the protein level by flow cytometric analysis. In addition, the inhibitory effects of 15d-PGJ2 on TLR2 and CD14 mRNA expression detected with real-time RT–PCR were observed at 6 h following S. aureus stimulation, the same time point used for our microarray analysis. Because both our microarray and real-time RT–PCR studies utilized primary microglia, the failure to detect changes in these surface markers following 15d-PGJ2 treatment in the former suggests that these discrepancies can best be explained by the differential sensitivities of each technique. Therefore, our microarray results may have actually underestimated the number of genes influenced by 15d-PGJ2 in S. aureus-treated primary microglia.
Recently, 15d-PGJ2 has been shown to attenuate the expression of a variety of immune response genes in monocytes/macrophages through PPAR-γ-independent mechanisms including inhibition of I-κB kinase activation (Castrillo et al. 2000; Rossi et al. 2000), alkylation of NF-κB-rel proteins (Straus et al. 2000) and covalent modification and oligomerization of c-Jun (Perez-Sala et al. 2003) which interferes with their DNA binding activities, and inhibition of AP-1 and Stat-1 activity (Kerppola et al. 1993; Ricote et al. 1998a). Although these findings have not yet been demonstrated in microglia, the fact that macrophages and microglia share many phenotypical and functional characteristics suggests that the majority of 15d-PGJ2 effects on microglia may also occur through PPAR-γ-independent pathways. We are currently initiating studies to delineate the mechanism(s) by which 15d-PGJ2 modulates S. aureus-dependent proinflammatory mediator expression in microglia.
Our findings demonstrating the inhibitory effects of 15d-PGJ2 on S. aureus-dependent microglial activation are similar to those observed following LPS stimulation. Specifically, 15d-PGJ2 has been shown to inhibit LPS-induced TNF-α (Bernardo et al. 2000; Drew and Chavis 2001), MHC class II (Bernardo et al. 2000), and CD40 (Diab et al. 2002) expression in microglia. Importantly, our study has provided a more comprehensive analysis on the effects of 15d-PGJ2 on microglial activation with the inclusion of microarray analysis. However, our results demonstrating that PPAR-γ levels remained constant following microglial activation with S. aureus differ from Bernardo et al. (2000) who reported that stimulation of primary rat microglia with LPS and 15d-PGJ2 alone were found to decrease and enhance PPAR-γ expression, respectively. The reasons for these discrepancies are not known but may be explained by the nature of the stimuli used, the origin of microglia, the kinetics at which inflammatory mediator expression was evaluated, and/or the concentrations of 15d-PGJ2 tested.
Elevated levels of endogenous 15d-PGJ2 have been associated with the resolution of inflammation in vivo, suggesting that it functions as a negative feedback regulator of the inflammatory response (Gilroy et al. 1999; Lawrence et al. 2002). Whether the concentrations of 15d-PGJ2 utilized in our studies accurately reflect levels achieved during the chronic stages of inflammation in vivo is not known. However, 15d-PGJ2 may approach these concentrations within distinct microdomains in the CNS, although this remains to be demonstrated. Evidence to support a modulatory role for 15d-PGJ2 in the context of brain abscess is provided by our findings that 15d-PGJ2 effectively inhibited MHC class II and CD40 expression on S. aureus-stimulated microglia, two receptors with important functions in antigen presentation to CD4+ T cells. It is possible that the downregulation of these receptors on microglia following 15d-PGJ2 treatment results in the failure to stimulate S. aureus-specific T cells in the CNS parenchyma due to the absence of an effective co-stimulatory signal. In addition, the attenuation of TLR2 expression by 15d-PGJ2, a receptor that has been shown to play an important role in the recognition of gram-positive bacteria by mononuclear phagocytes (Takeuchi et al. 1999; Underhill et al. 1999a, 1999b; Hajjar et al. 2001), could serve to limit microglial responsiveness to S. aureus and its cell wall products during the later stages of brain abscess development when bacterial burdens are negligible (Baldwin 2004). Therefore, preventing chronic microglial activation by 15d-PGJ2 or synthetic PPAR-γ agonists may help to resolve inflammation earlier, resulting in smaller abscesses and less damage to surrounding normal brain parenchyma.
Acknowledgments
The authors would like to thank Patrick Mayes and Anessa Haney for excellent technical assistance and Dr Marjorie Beggs in the UAMS Microarray Core Facility for her expertise with microarray design and execution. This work was supported by grants from the National Institutes of Health (NS40730 and MH65297 to TK and NS42860 to PDD), the National Multiple Sclerosis Society (RG 3198A1 to PDD), and the BRIN program of the National Center for Research Resources (P20 RR-16460). MM was supported by an Arkansas BRIN Undergraduate Summer Student Fellowship (P20 RR-16460).
Abbreviations used
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- DMEM
Dulbecco’s modified Eagle medium
- EAE
experimental auto-immune encephalomyelitis
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- GFAP
glial fibrillary acidic protein
- GM–CSF
granulocyte–macrophage colony-stimulating factor
- IMDM
Isovec’s minimal defined medium
- LAL
Limulus amebocyte lysate assay
- LPS
lipopolysaccharide
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NO
nitric oxide
- PBS
phosphate-buffered saline
- PPAR
peroxisome proliferator-activated receptors
- PVDF
polyvinylidene difluoride
- SDS–PAGE
sodium dodecyl sulfate poly-acrylamide gel electrophoresis
References
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Baldwin A, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-γ) and its natural ligand 15-deoxy-Δ12,14-prostaglandin J2 in the regulation of microglial functions. Eur J Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Ajmone-Cat MA, Levi G, Minghetti L. 15-deoxy-Δ12,14-prostaglandin J2 regulates the functional state and the survival of microglial cells through multiple molecular mechanisms. J Neurochem. 2003;87:742–751. doi: 10.1046/j.1471-4159.2003.02045.x. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Inhibition of IκB kinase and IκB phosphorylation by 15-deoxy-Δ (12,14)-prostaglandin J(2) in activated murine macrophages. Mol Cell Biol. 2000;20:1692–1698. doi: 10.1128/mcb.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Δ (12,14)-prostaglandin J(2) ameliorates experimental autoimmune encep-halomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Drew PD, Chavis JA. The cyclopentone prostaglandin 15-deoxy-Δ (12,14) prostaglandin J2 represses nitric oxide, TNF-α, and IL-12 production by microglial cells. J Neuroimmunol. 2001;115:28–35. doi: 10.1016/s0165-5728(01)00267-3. [DOI] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. Am J Pathol. 1992;141:1299–1307. [PMC free article] [PubMed] [Google Scholar]
- Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol. 2002;72:233–238. [PMC free article] [PubMed] [Google Scholar]
- Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Kerppola TK, Luk D, Curran T. Fos is a preferential target of glucocorticoid receptor inhibition of AP-1 activity in vitro. Mol Cell Biol. 1993;13:3782–3791. doi: 10.1128/mcb.13.6.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-γ agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315–325. doi: 10.1002/jnr.10501. [DOI] [PubMed] [Google Scholar]
- Kielian T, Hickey WF. Chemokines and Neural Inflammation in Experimental Brain Abscesses. Elsevier Science BV; Amsterdam: 2002. pp. 217–224. [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001b;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Cheung A, Hickey WF. Diminished virulence of an α-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect Immunol. 2001a;69:6902–6911. doi: 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, co-stimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–956. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. quiz. 780–761. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Perez-Sala D, Cernuda-Morollon E, Canada FJ. Molecular basis for the direct inhibition of AP-1 DNA binding by 15-deoxy-Δ12,14-prostaglandin J2. J Biol Chem. 2003;278:51251–51260. doi: 10.1074/jbc.M309409200. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Δ12,14-prosta-glandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-α expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Ricote M, Huang J, Fajas L, et al. Expression of the peroxisome proliferator-activated receptor gamma (PPAR-γ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low-density lipoprotein. Proc Natl Acad Sci USA. 1998b;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998a;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Microglial interleukin-1α expression in brain regions in Alzheimer’s disease: correlation with neuritic plaque distribution. Neuropathol Appl Neurobiol. 1995;21:290–301. doi: 10.1111/j.1365-2990.1995.tb01063.x. [DOI] [PubMed] [Google Scholar]
- Shu H, Wong B, Zhou G, Li Y, Berger J, Woods JW, Wright SD, Cai TQ. Activation of PPAR-α or -γ reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267:345–349. doi: 10.1006/bbrc.1999.1968. [DOI] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Thieringer R, Fenyk-Melody JE, Le Grand CB, et al. Activation of peroxisome proliferator-activated receptor γ does not inhibit IL-6 or TNF-α responses of macrophages to lipopolysaccharide in vitro or in vivo. J Immunol. 2000;164:1046–1054. doi: 10.4049/jimmunol.164.2.1046. [DOI] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Tran EH, Hardin-Pouzet H, Verge G, Owens T. Astrocytes and microglia express inducible nitric oxide synthase in mice with experimental allergic encephalomyelitis. J Neuroimmunol. 1997;74:121–129. doi: 10.1016/s0165-5728(96)00215-9. [DOI] [PubMed] [Google Scholar]
- Underhill D, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999a;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci USA. 1999b;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]