

Abstract
The increasing prevalence of Staphylococcus aureus strains isolated from hospital- and community-acquired respiratory tract infections is an important public health concern worldwide. The majority of S. aureus strains produce staphylokinase, a plasminogen activator capable of inactivating neutrophil α-defensins and of impairing phagocytosis via opsonin degradation. Cathelicidin antimicrobial peptides are present at sites of infection before the release of neutrophil α-defensins. Therefore, we hypothesized that staphylokinase interacts with cathelicidin during the early pathogenesis of S. aureus airway infection. In a mouse intranasal infection model, cathelicidin was strongly up-regulated in the airways during the development of staphylococcal pneumonia. In vitro, cathelicidin bound directly to staphylokinase and augmented staphylokinase-dependent plasminogen activation and fibrinolysis at concentrations consistent with those detected in the airways during infection. These data suggest that staphylokinase production may be a novel virulence mechanism by which S. aureus exploits cathelicidin to promote fibrinolysis, leading to enhanced bacterial dissemination and invasive infection.
Staphylococcus aureus causes a wide range of infections, including arthritis, toxic shock syndrome, sepsis, bacteremia, and pneumonia [1]. Worldwide, the prevalence of S. aureus strains isolated from hospital- and community-acquired respiratory tract infections has steadily increased over the past few decades [2, 3]. Approximately 60% of the healthy human population is permanently or intermittently colonized with S. aureus, which constitutes a major risk factor for the development of invasive S. aureus infections [4, 5].
Host innate immunity is essential for recognizing invading pathogens and preventing the infection of mucosal surfaces [6]. S. aureus produces various virulence factors that promote survival in the host environment [7]. The majority of S. aureus clinical isolates synthesize the plasminogen activator staphylokinase (SAK), which has recently been shown to contribute to S. aureus evasion of host innate immune defenses [8, 9]. SAK is a potent fibrinolytic agent that forms complexes with plasminogen to generate plasmin activity that preferentially degrades fibrin [10, 11]. SAK interactions with plasminogen have been proposed to facilitate bacterial colonization and dissemination by mediating fibrin clot lysis and host tissue degradation [11, 12]. In addition, SAK has been linked to impaired phagocytosis of S. aureus by neutrophils through the plasmin-mediated degradation of IgG and complement protein C3b on the bacterial surface [9]. Furthermore, SAK has been shown to interact with human neutrophil α-defensins, resulting in both the inhibition of SAK fibrinolytic activity and the inactivation of α-defensin antimicrobial activity [8, 13].
In the respiratory tract, antimicrobial proteins and peptides (AMPs)—including lysozyme, lactoferrin, cathelicidins, and defensins—act as essential innate immune effectors against bacterial infection [14–18]. Although lysozyme and neutrophil α-defensins are the most prominent AMPs detected in human airways [14, 19], S. aureus is highly resistant to their bactericidal activity [20–22]. By contrast, cathelicidin demonstrates potent antistaphylococcal activity [23–25], as well as additive or synergistic activity with lysozyme, lactoferrin, and defensins [17, 23, 26, 27]. Cathelicidin expression is up-regulated in the airways during bacterial infection [28] and has been detected in airway surface fluid, bronchoalveolar lavage fluid (BALF), alveolar macrophages, neutrophils, and airway epithelial cells [16, 23, 29]. Furthermore, a mouse pulmonary infection model has been used to demonstrate that local overexpression of the human cathelicidin LL-37 decreases bacterial loads in airways, whereas systemic LL-37 overexpression decreases mortality after bacterial challenge [30]. In addition, adenovirus-mediated gene transfer of LL-37 in a cystic fibrosis xenograft model restored bactericidal activity against S. aureus [31]. Thus, cathelicidin appears to play an important role in innate immune defense in the airway.
Cathelicidin demonstrates bactericidal activity against S. aureus and is present at sites of infection before the release of α-defensins by recruited neutrophils. Because SAK has been implicated in S. aureus evasion of innate immune defenses, we further hypothesized that SAK might interact with cathelicidin during the early pathogenesis of S. aureus airway infection. Here, we show that cathelicidin binds directly to SAK and enhances plasminogen activation and fibrinolysis. Concentrations of cathelicidin required for interaction with SAK in vitro were consistent with those detected in the airways during the development of staphylococcal pneumonia. These data suggest that SAK production might serve as a virulence mechanism by which S. aureus exploits cathelicidin to promote fibrinolysis, resulting in bacterial dissemination and invasive infection.
MATERIALS AND METHODS
Reagents
Recombinant SAK was purchased from ProSpec-Tany TechnoGene. Human Glu-plasminogen and chromogenic plasmin-specific substrate H-D-Val-Leu-Lys-para-nitroanilide (S-2251) were purchased from Chromogenix. H-D-Pro-Phe-Arg chloromethylketone (PPACK) was purchased from Bachem. Recombinant mouse plasminogen (mPLG) and rabbit anti-mPLG antibody were purchased from Molecular Innovations. LL-37 and mouse cathelicidin (mCRAMP) peptides and rabbit anti-mCRAMP antibody were gifts from R. Gallo (University of California, San Diego). Human α-defensin–1 was purchased from Peptides International. Affinity-purified goat anti–rabbit IgG horseradish peroxidase (HRP)–conjugated antibody was purchased from BioRad. 3,3′,5,5′-Tetramethylbenzidine (TMB) peroxidase substrate was purchased from KPL. SuperSignal West Pico chemiluminescent substrate was purchased from Pierce.
Bacterial strains
S. aureus strains 8325-4 (SAK negative) and RN6390 (SAK-positive derivative of 8325-4) were used for the present studies [32, 33]. S. aureus strains were grown overnight to stationary phase in sterile tryptic soy broth at 37°C with aeration. SAK production was assessed in bacterial supernatants by measuring plasminogen activation capacity using standard enzymatic assays [8].
Mouse pneumonia model and BALF collection
C57BL/6 mice were infected intranasally under anesthesia with 3–5 ×108 cfu of S. aureus JP1, a human blood isolate obtained from the microbiology laboratory of Veterans Affairs Puget Sound Health Care System [34]. Mock-infected control mice were inoculated intranasally with PBS. Mice were killed at 0.5 or 6 h after infection, and lungs were lavaged with PBS to recover BALF, which was stored at −70°C before analysis.
Analysis of host protein levels in BALF
Triplicate wells of 96-well microtiter plates were coated overnight at 4°C with BALF samples. Wells were washed between incubations with 0.05% Tween 20/PBS. Plates were incubated with anti-mPLG or anti-mCRAMP antibody diluted in 1% bovine serum albumin (BSA)/Tris for 1 h, followed by incubation with HRP-conjugated secondary antibody for 1 h. TMB peroxidase substrate was added, and reactions were stopped with 1 mol/L H2SO4. Absorbance was measured at 450 nm using a Spectro-Max 190 spectrophotometer with SOFTmaxPRO software (version 3.1.1; Molecular Devices). Levels of mPLG and mCRAMP in BALF samples were quantitated by comparison with standard curves.
Effects of SAK on plasminogen activation in BALF
The ability of SAK to mediate plasminogen activation in the airway environment was assessed by incubating BALF samples in the presence or absence of SAK (10 μg/mL) for 10 min at 37°C before the addition of S-2251 (1 mmol/L). Plasminogen activation was assessed over the course of 24 h at 37°C by measuring absorbance at 405 nm. Cleavage of S-2251 provides a direct indicator of plasminogen activation to plasmin.
Analysis of cathelicidin processing
To further evaluate the state of cathelicidin activation in the airways during infection, BALF samples were analyzed by Western blotting. Samples were normalized for protein concentrations (BCA Protein Assay; Pierce), mixed with SDS loading buffer, and incubated for 10 min at 95°C. Synthetic mCRAMP was used as a positive control. Samples were loaded onto 10%–20% Tris-Tricine-SDS gels and run for 1 h at 120 V, followed by transfer to nitrocellulose membranes for 1 h at 100 V on ice. Membranes were blocked and incubated with anti-mCRAMP antibody, followed by HRP-conjugated secondary antibody for 1 h each, before they were developed with chemiluminescent reagents. Membranes were washed between incubations with 0.05% Tween 20/PBS.
Analysis of SAK-cathelicidin complex formation
Complex formation between mCRAMP and SAK was assessed essentially as described elsewhere [13]. Briefly, triplicate wells of 96-well microtiter plates were coated overnight at 4°C with 500 ng SAK or 1% BSA in 0.1 mol/L carbonate buffer (pH 9.6). Wells were washed between incubations with 0.05% Tween 20/PBS. Plates were blocked for 1 h with 1% BSA/Tris (pH 8.0), followed by incubation with mCRAMP (0.01–10 μg/mL) for 1 h. SAK-mCRAMP complexes were detected by incubation with anti-mCRAMP antibody, followed by HRP-conjugated secondary antibody for 1 h each. TMB peroxidase substrate was added, and reactions were stopped with 1 mol/L H2SO4. Absorbance was measured at 450 nm, and specific binding of mCRAMP to SAK was calculated as the difference in absorbance between SAK-coated and BSA-coated wells. To evaluate the effect of α-defensins on SAK-mCRAMP complex formation, wells were incubated with 5 μg/mL α-defensin for 1 h at 37°C before or during incubation with mCRAMP.
Effects of cathelicidin on SAK-dependent plasminogen activation
In vitro assays for plasminogen activation were performed essentially as described elsewhere [8, 35]. Briefly, human plasminogen (40 nmol/L) was incubated with SAK (for 10 min) or with supernatants from stationary-phase S. aureus (overnight). S-2251 (1 mmol/L) was added after plasminogen activation, and absorbance was measured at 405 nm over time. To investigate the effects of host AMPs on SAK-dependent plasminogen activation, SAK or S. aureus supernatants were incubated alone or in combination with cathelicidin (LL-37 or mCRAMP), α-defensin, or PPACK inhibitor (100 μmol/L) for 30 min at 37°C before the addition of plasminogen. Initial studies used 10 μg/mL AMPs; further cathelicidin dose-response experiments assessed concentrations ranging from 1 to 100 μg/mL. The percentage of plasminogen activation was calculated relative to SAK or SAK-producing S. aureus supernatant alone.
Effects of cathelicidin on SAK-dependent fibrinolysis
In vitro assays for measuring fibrinolysis were performed essentially as described elsewhere [13, 36]. Briefly, human fibrinogen (10 μmol/L), human plasminogen (500 nmol/L), and either SAK (4 nmol/L) or S. aureus supernatants were added to trip—thrombin licate wells of 96-well plates. CaCl2 (2 mmol/L) and α (1 nmol/L) were added to initiate fibrin clotting. Changes in turbidity were assessed over time as absorbance at 400 nm. The initial increase in turbidity indicates clot formation; the subsequent decrease in turbidity reflects fibrinolysis. To investigate the effects of cathelicidin on SAK-dependent fibrinolysis, SAK or S. aureus supernatants were incubated with mCRAMP for 30 min at 37°C before fibrinolysis was measured.
Statistical analysis
Results are expressed as means ± SDs of triplicate results of at least 3 independent experiments. Comparisons were made using either the paired Student’s t test or, when >2 groups were compared, analysis of variance followed by Tukey’s post hoc test (Prism, version 4.02; GraphPad). P < .05 was considered significant for all experiments.
RESULTS
Increased host plasminogen levels in the airways during staphylococcal pneumonia
To assess changes in host airway protein content during the development of S. aureus pneumonia, mice were inoculated intranasally with either PBS or S. aureus, and BALF was collected 0.5 and 6 h after infection. When a modified ELISA system was used, mPLG expression was found to increase over time in the airways of both mock- and S. aureus–infected mice but was significantly higher at both time points in response to S. aureus infection (P < .001; figure 1A). To further evaluate whether plasminogen present in the airways could be activated by SAK, BALF samples collected from mock-and S. aureus–infected mice were incubated in the presence or absence of SAK before plasminogen activation was assessed. Consistent with the up-regulation of plasminogen observed during the development of staphylococcal pneumonia, the presence of exogenous SAK strongly increased plasminogen activation in BALF samples (P < .01 for control mice at 0.5 h; P < .001 for all others; figure 1B). Thi effect was particularly apparent in BALF collected 6 h after S. aureus infection, in which the addition of SAK generated a 9-fold increase in plasminogen activation.
Figure 1.
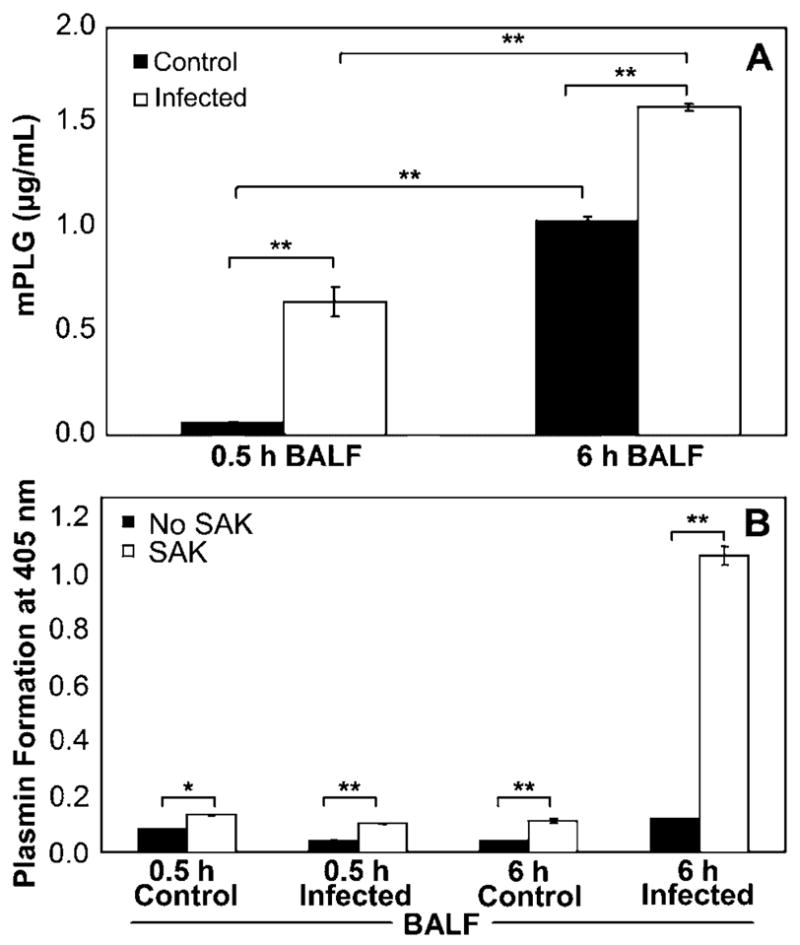
Increased host plasminogen levels in the airways during staphylococcal pneumonia. A, Detection of plasminogen (mPLG) in bronchoalveolar lavage fluid (BALF) samples collected from C57BL/6 mice at 0.5 or 6 h after intranasal inoculation with either Staphylococcus aureus or PBS by modified ELISA using rabbit anti-mPLG antibody and goat anti-rabbit horseradish peroxidase–conjugated antibody. Absorbance was measured at 450 nm. Concentrations of mPLG were quantitated by comparison with a standard curve. B, BALF samples incubated in the absence or presence of recombinant staphylokinase (SAK; 10 μg/mL) for 15 min at 37°C. S-2251 chromogenic substrate (1 mmol/L) was added, and mPLG activation measured as absorbance at 405 nm for up to 24 h at 37°C. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. **P < .001, *P < .01, analysis of variance with Tukey’s post hoc test.
Up-regulation and processing of host cathelicidin in the airways during staphylococcal pneumonia
Because neutrophil α-defensins are not synthesized by mice, the expression of mCRAMP was quantitated in BALF collected at 0.5 and 6 h from mice infected intranasally with either PBS or S. aureus. Using a modified ELISA system, cathelicidin expression was shown to be significantly and specifically up-regulated in the airways at 6 h after S. aureus infection, whereas no response was observed in mock-infected mice (P < .001; figure 2A). Furthermore, the cathelicidin present during S. aureus infection was detected as both an unprocessed precursor (~18 kDa) and mature AMP (~5 kDa) by Western blot analysis (figure 2B).
Figure 2.
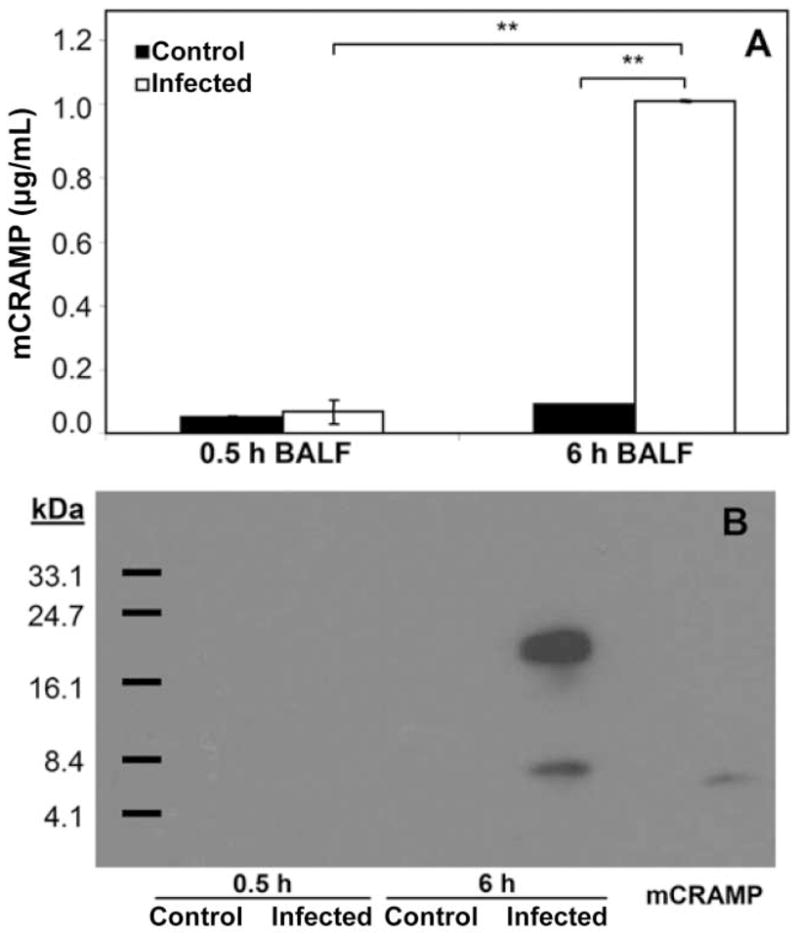
Up-regulation and processing of host cathelicidin in the airways during staphylococcal pneumonia. A, Detection of cathelicidin (mCRAMP) in bronchoalveolar lavage fluid (BALF) samples collected from C57BL/6 mice at 0.5 or 6 h after intranasal inoculation with either Staphylococcus aureus or PBS by modified ELISA using rabbit anti-mCRAMP antibody and goat anti-rabbit horseradish peroxidase–conjugated antibody. Absorbance was measured at 450 nm. Concentrations of mCRAMP were quantitated by comparison with a standard curve. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. **P < .001, analysis of variance with Tukey’s post hoc test. B, Western blot analysis of mCRAMP expression in BALF samples normalized for total protein content using the antibodies described in panel A. Synthetic mCRAMP peptide was used as a positive control.
Direct dose-dependent binding of cathelicidin to SAK
Because cathelicidin was up-regulated in the mouse airways during the development of staphylococcal pneumonia, we evaluated the ability of cathelicidin to directly interact with SAK. Binding of mCRAMP to immobilized SAK was assessed using a polyclonal antibody recognizing mCRAMP. Specific binding to SAK occurred in a dose-dependent manner within the range of 0.1–10 μg/mL mCRAMP (P < .01; figure 3A). These concentrations overlap the levels of cathelicidin detected in BALF samples from S. aureus–infected mice. Interestingly, incubation of SAK with neutrophil α-defensins, either before or during incubation with cathelicidin, did not diminish the ability of cathelicidin to form complexes with SAK, which suggests that these AMPs occupy distinct binding sites on SAK (figure 3B).
Figure 3.
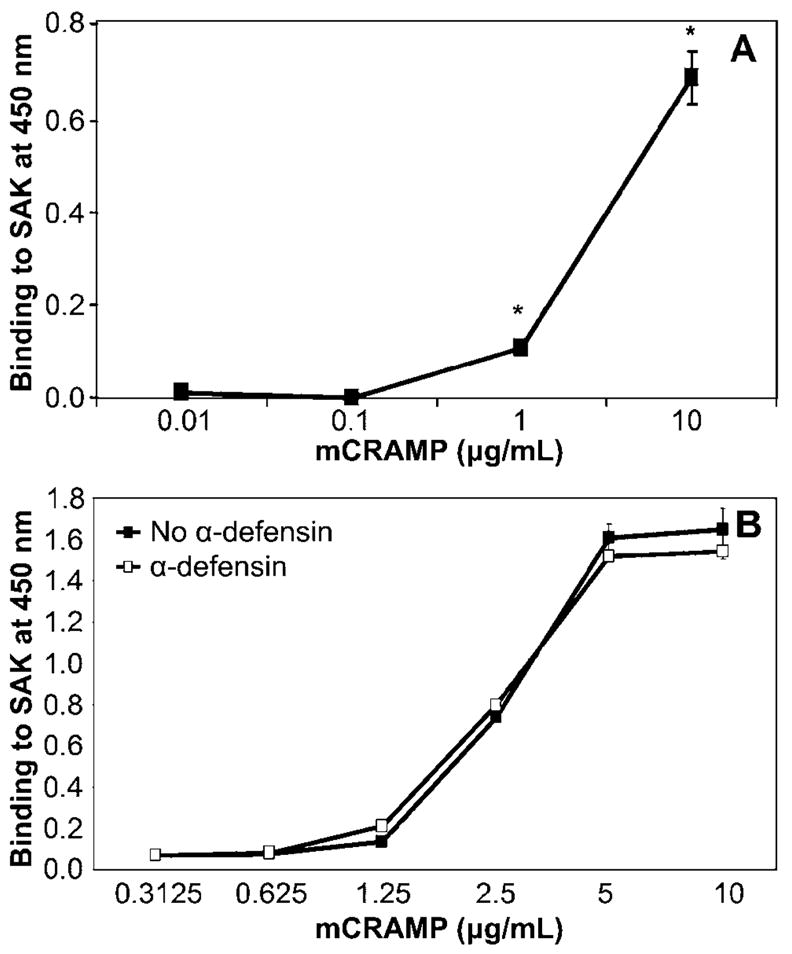
Direct dose-dependent, binding of cathelicidin to staphylokinase (SAK). SAK (500 ng) and 1% bovine serum albumin (BSA) in carbonate buffer were immobilized in 96-well plates. Concentrations of cathelicidin (mCRAMP) ranging from 0.01 to 10 μg/mL were added to wells in the absence (A) or presence (B) of 5 μg/mL α-defensin. Bound mCRAMP was detected using anti-mCRAMP antibody and horseradish peroxidase–conjugated secondary antibody. Absorbance was measured at 450 nm. Specific binding was calculated as the difference in absorbance between SAK- and BSA-coated wells. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. *P < .01, Student’s t test.
Augmentation of SAK-dependent plasminogen activation by cathelicidin
Because interactions between SAK and neutrophil α-defensins have been shown to inhibit SAK-dependent plasminogen activation, we initially hypothesized a similar inhibitory role for cathelicidin. However, the presence of high levels of cathelicidin in mouse airways during the development of staphylococcal pneumonia, combined with the significant increase in plasminogen activation after the addition of exogenous SAK to BALF collected from these mice, argued against the inhibition of SAK-dependent plasminogen activation by cathelicidin. In contrast to neutrophil α-defensins, cathelicidin augmented the activation of human plasminogen both by recombinant SAK (P < .01 for each time point; figure 4A) and by supernatants obtained from stationary-phase cultures of SAK-producing S. aureus (P < .001 for each time point; figure 4B). Supernatants from SAK-negative S. aureus did not activate plasminogen, and, as expected, no effect was observed for pre-treatment with cathelicidin (data not shown). In addition, neither cathelicidin nor α-defensin demonstrated any inherent ability to activate plasminogen in the absence of SAK (data not shown). Human and mouse cathelicidins, LL-37 and mCRAMP, respectively, demonstrated nearly identical stimulatory effects on SAK-dependent plasminogen activation. Incubation with cathelicidin also enhanced SAK-dependent activation of mouse plasminogen, albeit to levels lower than that observed for human plasminogen (data not shown). Interestingly, incubation of SAK with both cathelicidin and α-defensin, which would occur in the presence of neutrophils at sites of S. aureus infection in humans, resulted in the complete abrogation of SAK-dependent plasminogen activation. PPACK, a synthetic proteinase inhibitor that interferes with plasminogen activation, served as a positive control for the inhibition of SAK activity.
Figure 4.
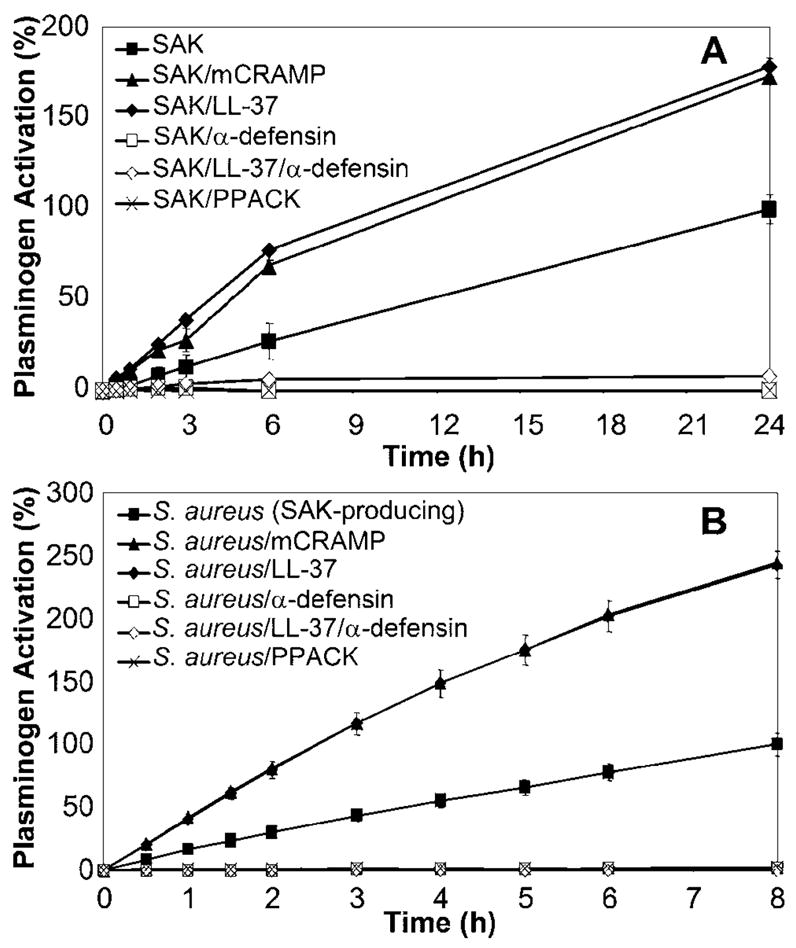
Augmentation of staphylokinase (SAK)–dependent plasminogen activation by cathelicidin. Recombinant SAK (A) or supernatants from stationary-phase cultures of SAK-producing Staphylococcus aureus (B) were incubated alone or in the presence of 10 μg/mL cathelicidins mCRAMP or LL-37 or of α-defensin for 30 min at 37°C. H-D-Pro-Phe-Arg chloromethylketone (PPACK; 100 μmol/L) served as a positive control for the inhibition of SAK-dependent plasminogen activation. Human plasminogen (40 nmol/L) was added and incubated with SAK samples at 37°C for either 10 min (SAK) or overnight (S. aureus supernatants). After the addition of S-2251 substrate, plasminogen activation was monitored over time at 37°C as absorbance at 405 nm. Data are shown as the percentage of plasminogen activation relative to that of SAK or SAK-producing S. aureus supernatant alone. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. Incubation with mCRAMP or LL-37: P < .01 (A) or P < .001 (B) for each time point, Student’s t test.
Dose-dependent stimulation of SAK-dependent plasminogen activation by cathelicidin
To confirm that the stimulation of SAK-dependent plasminogen activation by cathelicidin was not due to the use of subinhibitory peptide concentrations, further experiments were conducted to evaluate the dose and species dependence of the observed effects. Cathelicidin levels ranging from 1 to 100 μg/mL were tested to encompass concentrations above and below that shown initially to enhance SAK-dependent plasminogen activation (10 μg/mL). In the presence of 10 and 100 μg/mL mCRAMP, significant increases in both the rate and threshold of SAK-dependent plasminogen activation were observed (P < .01 for each time point; figure 5). In a manner nearly identical to mCRAMP, LL-37 also augmented SAK-dependent plasminogen activation in a dose-dependent manner at equivalent concentrations (data not shown). The augmentation of SAK-dependent plasminogen activation by cathelicidin was saturable; the highest concentration tested (100 μg/mL) produced a maximum stimulatory effect.
Figure 5.
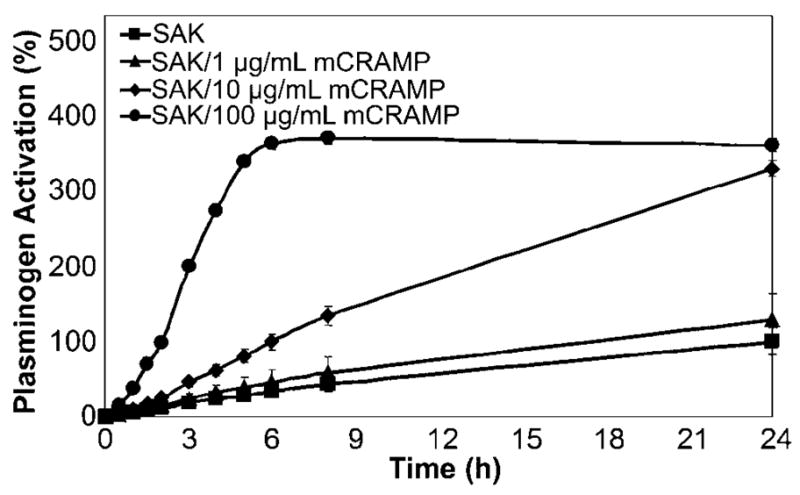
Dose-dependent stimulation of staphylokinase (SAK)–dependent plasminogen activation by cathelicidin (mCRAMP). Recombinant SAK was incubated with various concentrations of mCRAMP ranging from 1 to 100 μg/mL for 30 min at 37°C before assessment of plasminogen activation at 405 nm as described in the text. Data are the percentage plasminogen activation relative to that of SAK alone. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. Incubation with 10 or 100 μg/mL mCRAMP: P < .01 for each time point, Student’s t test.
Augmentation of SAK-dependent fibrinolysis by cathelicidin
Plasminogen activation by SAK generates the active enzyme plasmin, which specifically degrades fibrin clots. Because cathelicidin was found to potently augment SAK-dependent plasminogen activation, its ability to stimulate fibrinolysis was further studied in vitro. Using supernatants from SAK-producing S. aureus, cathelicidin significantly enhanced the rate of SAK-dependent fibrinolysis (P < .001 at 65 min; P = .05 at 45 and 80 min; figure 6). Supernatants from SAK-negative S. aureus did not initiate fibrin clot degradation, and no effect was observed that was due to cathelicidin alone in the absence of SAK (data not shown).
Figure 6.
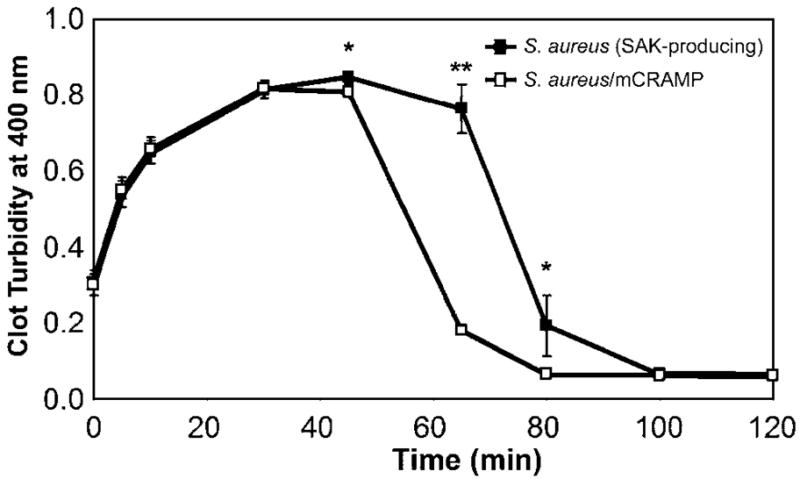
Augmentation of staphylokinase (SAK)–dependent fibrinolysis by cathelicidin (mCRAMP). Supernatants from stationary-phase cultures of SAK-producing Staphylococcus aureus were incubated alone or in the presence of mCRAMP (10 μg/mL) for 30 min at 37°C. To initiate fibrin clot formation, CaCl2 (2 mmol/L) and α-thrombin (1 nmol/L) were added to human fibrinogen (10 μmol/L), human plasminogen (500 nmol/L), and S. aureus supernatants or supernatant/mCRAMP mixtures. Fibrin clot formation and lysis were assessed by measuring changes in turbidity over time at 25°C as absorbance at 400 nm. Data are the mean ± SD of triplicate values and are representative of 3 independent experiments. **P < .001, *P = .05, Student’s t test.
DISCUSSION
Dysregulation of fibrinolysis is an important characteristic of airway infections such as pneumonia [37]. Although the pathogenesis of acute inflammatory lung disease is typically characterized by fibrin deposition due to local and systemic inhibition of fibrinolysis, the initial host response to bacteremia actually involves increased fibrinolytic activity due to the release of plasminogen activators [38]. S. aureus produces its own plasminogen activator, SAK, which has been hypothesized to promote host immune evasion and bacterial virulence by degrading fibrin clots to allow systemic spread [11]. However, SAK production by clinical isolates has not been shown to correlate with S. aureus invasiveness [39]. Therefore, we set out to explore the involvement of SAK in the dysregulation of fibrinolysis observed during staphylococcal pneumonia.
Plasminogen levels increased in the airways in response to S. aureus infection, and dramatic plasminogen activation was observed after the addition of SAK to infected mouse BALF. These data stand in contrast to the absolute species specificity among bacterial plasminogen activators that has been reported elsewhere [40–42]. Therefore, we predicted that additional host proteins present in the airway environment during infection may alter or stabilize the conformation of mouse plasminogen in such a way that it more closely resembles human plasminogen, thereby enabling SAK to circumvent this species specificity. Previous studies have demonstrated that the ability of Streptococcus pyogenes to produce streptokinase capable of activating mouse plasminogen contributed to bacterial invasiveness in a mouse skin infection model [43]. Furthermore, mouse skin passage of S. pyogenes has been shown to generate increased streptokinase expression and activity [44]. Therefore, bacterial pathogens might respond to specific host signals in a manner that alters plasminogen binding and activation in vivo.
AMPs are an important component of innate immunity at mucosal surfaces. During early stages of infection, cathelicidin is synthesized by resident alveolar macrophages and airway epithelial cells; neutrophils are recruited later and produce cathelicidin and α-defensins [16, 45]. We detected increased levels of both unprocessed precursor and mature cathelicidin in the airways during the development of staphylococcal pneumonia, when mice exhibit clinical symptoms such as hunched posture, labored breathing, and diminished activity. The presence of high levels of cathelicidin and the ability of SAK to enhance plasminogen activation in the airway environment led us to propose a novel relationship between SAK and cathelicidin that could potentially promote SAK-dependent fibrinolysis during the early pathogenesis of S. aureus airway infection. The results of in vitro binding studies and enzymatic assays further demonstrated that cathelicidin directly binds to SAK and augments SAK-dependent plasminogen activation and fibrinolysis at concentrations consistent with those detected in the airways during staphylococcal pneumonia. Previous studies have shown that the presence of human plasminogen in a mouse model of S. pyogenes skin infection led to enhanced plasminogen activation and fibrinolysis, resulting in an increased bacterial dissemination and mortality that was dependent on streptokinase production [46, 47]. Therefore, SAK might serve as a virulence factor by which S. aureus exploits cathelicidin to enhance fibrinolysis and promote dissemination and invasive infection.
Human and mouse cathelicidin demonstrated nearly identical stimulatory effects on SAK-dependent plasminogen activation, whereas human neutrophil α-defensins displayed an opposite, inhibitory role, consistent with previous reports [13]. The unique effects of these AMPs might be attributable to structural differences resulting in the utilization of distinct binding sites on SAK. LL-37 and mCRAMP form α-helices in solution, whereas defensins form β-sheet structures [48]. SAK contains both features—a central α-helix positioned above a 5-stranded β-sheet [49]. The α-helix contains the major regions of SAK interaction with plasminogen, and several α-defensin binding sites have been predicted to overlap plasminogen binding sites on SAK, which might account for the disruption of plasminogen activation by α-defensins [13]. Our data showing that α-defensins do not disrupt SAK-cathelicidin interactions support our hypothesis that cathelicidin binds to distinct sites on SAK that do not interfere with plasminogen-binding regions and perhaps even stabilize such interactions. The complete abrogation of SAK-dependent plasminogen activation after exposure to both cathelicidin and α-defensins, which would occur when neutrophils are present at sites of S. aureus infection in humans, offers further insight into the regulation of SAK-dependent fibrinolysis by different families of host AMPs. Additional insight into the mechanism by which cathelicidin augments SAK-dependent plasminogen activation and fibrinolysis can be gained from a recent study that demonstrated that relaxation of plasminogen conformation can result in enhanced fibrinolysis [50]. Ongoing studies will focus on further characterizing the structural requirements for SAK-cathelicidin interactions and the mechanism by which cathelicidin enhances SAK activity.
In summary, the present study provides the first evidence that cathelicidin AMPs bind directly to a bacterial plasminogen activator, SAK, resulting in the augmentation of plasminogen activation and fibrinolysis by S. aureus. These data sharply contrast the inhibition of SAK activity by human neutrophil α-defensins, which suggests that host AMPs, which are produced by various cell types present at different stages of infection, exert distinct effects on SAK-dependent fibrinolysis (figure 7). Furthermore, the results demonstrate that SAK is capable of activating mouse plasminogen in the airway environment, which suggests that factors present in vivo might relax the species specificity of SAK-dependent plasminogen activation. Combined, these data provide evidence that S. aureus is able to exploit cathelicidin produced by resident airway cells to augment SAK-dependent fibrinolysis. Enhanced fibrinolysis might be a novel virulence mechanism by which S. aureus avoids confinement in the airways by fibrin deposition, leading to dissemination and systemic infection.
Figure 7.
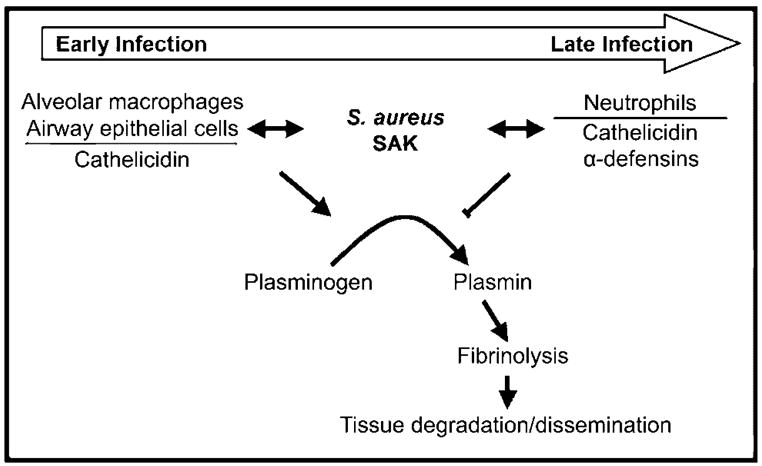
Opposite effects of host antimicrobial peptides on staphylokinase (SAK)–dependent fibrinolysis. Cathelicidin, which is produced by resident alveolar macrophages and airway epithelial cells during early stages of infection, enhances SAK-dependent plasminogen activation and fibrinolysis. Increased fibrinolysis may lead to host tissue degradation within the airways, as well as to bacterial dissemination to the bloodstream. As infection progresses, recruited neutrophils release both cathelicidin and α-defensins. This results in the inhibition of SAK-dependent plasminogen activation and fibrinolysis, which may serve to confine the pathogen and control infection.
Acknowledgments
National Institutes of Health (grants AI007509 to M.H.B. and HL073996 to C.E.R.).
Footnotes
Potential conflicts of interest: none reported.
References
- 1.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis. 2001;7:178–82. doi: 10.3201/eid0702.010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diekema DJ, Pfaller MA, Schmitz FJ, et al. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin Infect Dis. 2001;32(Suppl 2):S114–32. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- 4.Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10:505–20. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group N Engl J Med. 2001;344:11–6. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- 6.Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- 7.Fedtke I, Gotz F, Peschel A. Bacterial evasion of innate host defenses—the Staphylococcus aureus lesson. Int J Med Microbiol. 2004;294:189–94. doi: 10.1016/j.ijmm.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 8.Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol. 2004;172:1169–76. doi: 10.4049/jimmunol.172.2.1169. [DOI] [PubMed] [Google Scholar]
- 9.Rooijakkers SH, van Wamel WJ, Ruyken M, van Kessel KP, van Strijp JA. Antiopsonic properties of staphylokinase. Microbes Infect. 2005;7:476–84. doi: 10.1016/j.micinf.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Collen D, Lijnen HR. Staphylokinase, a fibrin-specific plasminogen activator with therapeutic potential? Blood. 1994;84:680–6. [PubMed] [Google Scholar]
- 11.Bokarewa MI, Jin T, Tarkowski A. Staphylococcus aureus: staphylokinase. Int J Biochem Cell Biol. 2005;38:504–9. doi: 10.1016/j.biocel.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Parry MA, Zhang XC, Bode I. Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem Sci. 2000;25:53–9. doi: 10.1016/s0968-0004(99)01521-2. [DOI] [PubMed] [Google Scholar]
- 13.Bokarewa M, Tarkowski A. Human alpha-defensins neutralize fibrinolytic activity exerted by staphylokinase. Thromb Haemost. 2004;91:991–9. doi: 10.1160/TH03-11-0696. [DOI] [PubMed] [Google Scholar]
- 14.Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. 2002;109:693–7. doi: 10.1172/JCI15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Travis SM, Conway BA, Zabner J, et al. Activity of abundant antimicrobials of the human airway. Am J Respir Cell Mol Biol. 1999;20:872–9. doi: 10.1165/ajrcmb.20.5.3572. [DOI] [PubMed] [Google Scholar]
- 16.Agerberth B, Grunewald J, Castanos-Velez E, et al. Antibacterial components in bronchoalveolar lavage fluid from healthy individuals and sarcoidosis patients. Am J Respir Crit Care Med. 1999;160:283–90. doi: 10.1164/ajrccm.160.1.9807041. [DOI] [PubMed] [Google Scholar]
- 17.Singh PK, Tack BF, McCray PB, Jr, Welsh MJ. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am J Physiol Lung Cell Mol Physiol. 2000;279:L799–805. doi: 10.1152/ajplung.2000.279.5.L799. [DOI] [PubMed] [Google Scholar]
- 18.Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T. Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol. 2002;169:6985–91. doi: 10.4049/jimmunol.169.12.6985. [DOI] [PubMed] [Google Scholar]
- 19.Schnapp D, Harris A. Antibacterial peptides in bronchoalveolar lavage fluid. Am J Respir Cell Mol Biol. 1998;19:352–6. doi: 10.1165/ajrcmb.19.3.3384. [DOI] [PubMed] [Google Scholar]
- 20.Bera A, Herbert S, Jakob A, Vollmer W, Gotz F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol. 2005;55:778–87. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- 21.Schroder JM. Epithelial antimicrobial peptides: innate local host response elements. Cell Mol Life Sci. 1999;56:32–46. doi: 10.1007/s000180050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;388:416. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 23.Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA. 1998;95:9541–6. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Midorikawa K, Ouhara K, Komatsuzawa H, et al. Staphylococcus aureus susceptibility to innate antimicrobial peptides, beta-defensins and CAP18, expressed by human keratinocytes. Infect Immun. 2003;71:3730–9. doi: 10.1128/IAI.71.7.3730-3739.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorschner RA, Lopez-Garcia B, Peschel A, et al. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 2006;20:35–42. doi: 10.1096/fj.05-4406com. [DOI] [PubMed] [Google Scholar]
- 26.Nagaoka I, Hirota S, Yomogida S, Ohwada A, Hirata M. Synergistic actions of antibacterial neutrophil defensins and cathelicidins. Inflamm Res. 2000;49:73–9. doi: 10.1007/s000110050561. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Niyonsaba F, Ushio H, et al. Synergistic effect of antibacterial agents human beta-defensins, cathelicidin LL-37 and lysozyme against Staphylococcus aureus and Escherichia coli. J Dermatol Sci. 2005;40:123–32. doi: 10.1016/j.jdermsci.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Schaller-Bals S, Schulze A, Bals R. Increased levels of antimicrobial peptides in tracheal aspirates of newborn infants during infection. Am J Respir Crit Care Med. 2002;165:992–5. doi: 10.1164/ajrccm.165.7.200110-020. [DOI] [PubMed] [Google Scholar]
- 29.Beisswenger C, Bals R. Antimicrobial peptides in lung inflammation. Chem Immunol Allergy. 2005;86:55–71. doi: 10.1159/000086651. [DOI] [PubMed] [Google Scholar]
- 30.Bals R, Weiner DJ, Moscioni AD, Meegalla RL, Wilson JM. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun. 1999;67:6084–9. doi: 10.1128/iai.67.11.6084-6089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bals R, Weiner DJ, Meegalla RL, Wilson JM. Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. J Clin Invest. 1999;103:1113–7. doi: 10.1172/JCI6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novick R. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology. 1967;33:155–66. doi: 10.1016/0042-6822(67)90105-5. [DOI] [PubMed] [Google Scholar]
- 33.Peng HL, Novick RP, Kreiswirth B, Kornblum J, Schlievert P. Cloning, characterization, and sequencing of an accessory gene regulator (agr) in Staphylococcus aureus. J Bacteriol. 1988;170:4365–72. doi: 10.1128/jb.170.9.4365-4372.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–81. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 35.Christner RB, Boyle MD. Role of staphylokinase in the acquisition of plasmin(ogen)-dependent enzymatic activity by staphylococci. J Infect Dis. 1996;173:104–12. doi: 10.1093/infdis/173.1.104. [DOI] [PubMed] [Google Scholar]
- 36.Wolberg AS, Gabriel DA, Hoffman M. Analyzing fibrin clot structure using a microplate reader. Blood Coagul Fibrinolysis. 2002;13:533–9. doi: 10.1097/00001721-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Choi G, Schultz MJ, van Till JW, et al. Disturbed alveolar fibrin turnover during pneumonia is restricted to the site of infection. Eur Respir J. 2004;24:786–9. doi: 10.1183/09031936.04.00140703. [DOI] [PubMed] [Google Scholar]
- 38.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia—a review. Crit Care Med. 2006;34:871–7. [PubMed] [Google Scholar]
- 39.Jin T, Bokarewa M, McIntyre L, et al. Fatal outcome of bacteraemic patients caused by infection with staphylokinase-deficient Staphylococcus aureus strains. J Med Microbiol. 2003;52:919–23. doi: 10.1099/jmm.0.05145-0. [DOI] [PubMed] [Google Scholar]
- 40.Gladysheva IP, Turner RB, Sazonova IY, Liu L, Reed GL. Coevolutionary patterns in plasminogen activation. Proc Natl Acad Sci USA. 2003;100:9168–72. doi: 10.1073/pnas.1631716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada K, Ueshima S, Tanaka M, Fukao H, Matsuo O. Analysis of plasminogen activation by the plasmin-staphylokinase complex in plasma of [alpha]2-antiplasmin-deficient mice. Blood Coagul Fibrinolysis. 2000;11:645–55. doi: 10.1097/00001721-200010000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Marcum JA, Kline DL. Species specificity of streptokinase. Comp Biochem Physiol B. 1983;75:389–94. doi: 10.1016/0305-0491(83)90345-0. [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Ploplis VA, French EL, Boyle MD. Interaction between group A streptococci and the plasmin(ogen) system promotes virulence in a mouse skin infection model. J Infect Dis. 1999;179:907–14. doi: 10.1086/314654. [DOI] [PubMed] [Google Scholar]
- 44.Rezcallah MS, Boyle MD, Sledjeski DD. Mouse skin passage of Streptococcus pyogenes results in increased streptokinase expression and activity. Microbiology. 2004;150:365–71. doi: 10.1099/mic.0.26826-0. [DOI] [PubMed] [Google Scholar]
- 45.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23:327–33. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 46.Khil J, Im M, Heath A, et al. Plasminogen enhances virulence of group A streptococci by streptokinase-dependent and streptokinase-independent mechanisms. J Infect Dis. 2003;188:497–505. doi: 10.1086/377100. [DOI] [PubMed] [Google Scholar]
- 47.Sun H, Ringdahl U, Homeister JW, et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–6. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 48.Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. 2002;110:823–31. doi: 10.1067/mai.2002.129801. [DOI] [PubMed] [Google Scholar]
- 49.Ohlenschlager O, Ramachandran R, Guhrs KH, Schlott B, Brown LR. Nuclear magnetic resonance solution structure of the plasminogen-activator protein staphylokinase. Biochemistry. 1998;37:10635–42. doi: 10.1021/bi980673i. [DOI] [PubMed] [Google Scholar]
- 50.Kikuchi T, Hasumi K. Enhancement of plasminogen activation by surfactin C: augmentation of fibrinolysis in vitro and in vivo. Biochim Biophys Acta. 2002;1596:234–45. doi: 10.1016/s0167-4838(02)00221-2. [DOI] [PubMed] [Google Scholar]