

Abstract
Abnormal wound healing encompasses a wide spectrum, from chronic wounds to hypertrophic scars. Both conditions are associated with an abnormal cytokine profile in the wound bed. In this study, we sought to understand the dynamic relationships between myofibroblast differentiation and mechanical performance of the collagen matrix under tissue growth factor–β (TGF-β) and tumor necrosis factor–α (TNF-α) stimulation. We found TGF-β increased α-smooth muscle actin (α-SMA) and TNF-α alone decreased the basal α-SMA expression. When TGF-β1 and TNF-α were both added, the α-SMA expression was suppressed below the baseline. Real-time PCR showed that TNF-α suppresses TGF-β1-induced myofibroblast (fibroproliferative) phenotypic genes, for example, α-SMA, collagen type 1A, and fibronectin at the mRNA level. TNF-α suppresses TGF-β1-induced gene expression by affecting its mRNA stability. Our results further showed that TNF-α inhibits TGF-β1-induced Smad-3 phosphorylation via Jun N-terminal kinase signaling. Mechanical testing showed that TNF-α decreases the stiffness and contraction of the lattices after 5 days in culture. We proposed that changes in α-SMA, collagen, and fibronectin expression result in decreased contraction and stiffness of collagen matrices. Therefore, the balance of cytokines in a wound defines the mechanical properties of the extracellular matrix and optimal wound healing.
INTRODUCTION
Myofibroblasts are a specialized subgroup of cells with features of both fibroblasts and smooth muscle cells. The most significant marker for myofibroblasts is the expression of α-smooth muscle actin (α-SMA) within cytoplasmic stress fibers. However, myofibroblasts also show increased production of collagens I, III, V, fibronectin, and proteoglycan (Serini and Gabbiani, 1999), proteins that characterize fibrotic disease. It has been widely believed that the myofibroblast’s ability to generate contractile forces is due to the accumulation of cytoplasmic actins and cellular fibronectin, resulting in reorganization of the extracellular matrix (ECM) (Tomasek et al., 2002).
Tissue growth factor–β1 (TGF-β1) is a key cytokine in normal wound healing and fibrosis (Roberts et al., 2003). It is known to induce α-SMA expression in fibroblasts both in vitro and in vivo (Desmouliere et al., 1993; Roy et al., 2001). TGF-β1 also stimulates ECM synthesis, especially collagen I, and inhibits the activity of matrix metalloproteinases by decreasing their expression and upregulating tissue inhibitor of metalloproteinase expression (Gabbiani, 2003). Therefore, it controls two important activities of granulation tissue: tissue contraction and ECM remodeling.
The TGF-β family exerts its effects mainly through Smad signal transduction pathways (Zhang and Laiho, 2003). The signal transduction starts with ligand-binding activation of the receptor complex, followed by phosphorylation of the receptor-activated Smad2 and -3 by the type I receptor. The phosphorylated R-Smads then associate with Smad4 and migrate into the nucleus, where they modulate the transcription of a large number of genes. The Smad signaling pathway is implicated in TGF-β1 induction of α-SMA expression and formation of actin-stress fibers (Edlund et al., 2002, 2004; Evans et al., 2003).
Tumor necrosis factor–α (TNF-α) is a potent inflammatory cytokine expressed during the inflammatory phase of wound healing. TNF-α, secreted by inflammatory cells, both inhibits ECM synthesis and activates matrix metalloproteinases (Singer and Clark, 1999). Recent studies have demonstrated that inflammatory cytokines have antagonistic activities against TGF-β1 (Mauviel et al., 1993; Abraham et al., 2000). For example, TNF-α antagonizes TGF-β1-induced upregulation of type I and III collagens in fibroblasts through a Jun N-terminal kinase (JNK)-dependent regulatory mechanism in mouse fibroblasts (Verrecchia et al., 2003). Mariani et al. (1999) showed that TNF-α suppresses both tropoelastin and α-SMA expression in rat lung fibroblasts. The antagonist relationship between TGF-β1 and TNF-α may play an important role in maintaining tissue homeostasis and ECM deposition.
The increased level of α-SMA found in myofibroblast is potentially important for the contractile features of these cells under normal tissue repair and fibrotic conditions. Fibroblasts embedded in collagen matrix under mechanical stress have been shown to exert mechanical force on the collagen lattice, leading to reorganization of the matrix and alignment of the cells along the principal strain of the lattice (Delvoye et al., 1991; Kolodney and Wysolmerski, 1992; Eastwood et al., 1994). However, few studies have attempted to measure the mechanical properties of collagen lattice after matrix reorganization. Therefore, an increased knowledge of the induction and suppression of α-SMA expression correlating with the mechanical properties of the ECM would be of interest. On the basis of several findings of antagonistic activity of TNF-α on TGF-β1 activity, we sought to ascertain (i) the antagonistic activity of TNF-α on TGF-β1 induction of α-SMA expression, (ii) the role of the mitogenic-activated protein kinase (MAPK) signaling in TNF-α suppression of TGF-β1-induced α-SMA expression, (iii) whether TNF-α antagonistic activity on TGF-β1 is through inhibition of Smad3 phosphorylation, and (iv) the correlation between mechanical and molecular response of fibroblasts in collagen matrix to TGF-β1 and TNF-α actions by measuring the rate of lattice contraction and the stiffness of the collagen lattice. We examined the above objectives in human fibroblasts embedded in collagen 1 matrix, which remained attached to the substratum of the culture plate, also known as stressed fibroblast-populated collagen matrix.
RESULTS
TGF-β1 induces α-SMA expression and TNF-α suppresses its induction in human dermal fibroblasts
Desmouliere et al. (1992) have shown that TGF-β1 induces α-SMA expression in both quiescent and growing human dermal fibroblasts in monolayer. By contrast, TNF-α is known to have no effect on α-SMA expression in granulation tissue. However, TNF-α mediates antagonistic activity against TGF-β1 in synthesis of collagen types I and III. Therefore, we examined whether TNF-α exhibits the same antagonistic activity against TGF-β1-induced upregulation of α-SMA. To simulate the microenvironment of dermis, isolated human dermal fibroblasts at early passages were embedded in three-dimensional type-I collagen matrices attached to the culture plate designated for stressed fibroblast-populated collagen lattices (FPCLs). To recapitulate the wound environment or scar formation, stressed FPCLs were treated with TGF-β1. In the absence of TGF-β1 treatment, the stressed FPCLs showed minimal expression of α-SMA. As expected, TGF-β1 significantly increased α-SMA expression above that of the non-stimulated fibroblasts, consistent with previous published works (Figure 1). In contrast to previous reports of TNF-α having no effect on α-SMA expression in fibroblasts in a mouse wound model (Desmouliere et al., 1992), here we showed that TNF-α alone suppressed α-SMA below the baseline in human dermal fibroblasts. Interestingly, during treatment with both TGF-β1 (1 ng/ml) and TNF-α (up to 2 ng/ ml), TNF-α reversed the ability of TGF-β1 to upregulate α-SMA. We also examined the expression of vimentin protein, a member of the intermediate filament family, under the same experimental conditions and did not see any effects attributable to TGF-β1 or TNF-α. The vimentin protein acted as an internal control along with glyceraldehydes-3-phosphate dehydrogenase (GAPDH) protein.
Figure 1. TNF-α suppresses TGF-β1 induction in human dermal fibroblasts.
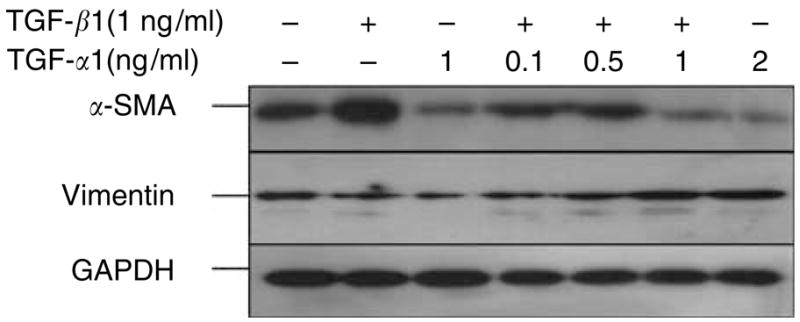
Normal human dermal fibroblasts (3 × 105 cells/ml) were seeded in collagen I matrix (1 mg/ml). The fibroblast-populated collagen lattices (FPCLs) were treated with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), or combinations of TGF-β1 (1 ng/ml) and increasing concentrations of TNF-α for 96 hours under 0.1% serum conditions. Representative Western blots for α-SMA, vimentin, and GAPDH are shown.
We next undertook to determine whether TNF-α could suppress TGF-β1-promoted formation of the stress fibers that characterize myofibroblasts. Stressed FPCLs were cultured in the presence or absence of 1 ng/ml TGF-β1 or TGF-β1 combined with TNF-α (1 ng/ml each) for 5 days, followed by fixation. The lattices were immunostained for α-SMA (Figure 2a and b). The immunofluorescence, using monoclonal antibody–recognizing human α-SMA, showed that TGF-β1 increased stress fibers containing α-SMA compared with the control group. However, when we combined TGF-β1 with TNF-α, the expression of actin stress fibers was suppressed back to the basal levels. Furthermore, TGF-β1-treated lattices had significantly larger stress fibers compared with lattices treated with TGF-β1 and TNF-α. We also examined the actin cytoskeleton and morphology of fibroblasts in stress FPCL under the stimulation of the same cytokines by staining with rhodamine-conjugated phalloidin, a florescence probe that binds F-actin (Figure 2e–h). Under resting condition of 0.1% fetal bovine serum (FBS), stressed FPCL demonstrated F-actin stress fibers as expected. The mechanical stress within the FPCL-induced stress fibers has been shown in granulation tissue (Tomasek et al., 2002). Following stimulation with TGF-β1 (1 ng/ml), fibroblasts within stressed FPCL became larger and polygonal compared with unstimulated control cells. Furthermore, the cells demonstrated larger actin fibers with increasing thickness and the number of stress fibers. For stressed FPCLs treated with TNF-α (10 ng/ml) and TNF-α (10 ng/ml) plus TGF-β1 (1 ng/ml), cells had a small spindle-shaped appearance and no significant stress F-actin fibers. These findings provide further support for the hypothesis that TNF-α can suppress TGF-β1 promotion of stress fibers and cytoskeletal changes essential for myofibroblasts differentiation.
Figure 2. TNF-α suppresses TGF-β1 promotion of myofibroblast structural elements.
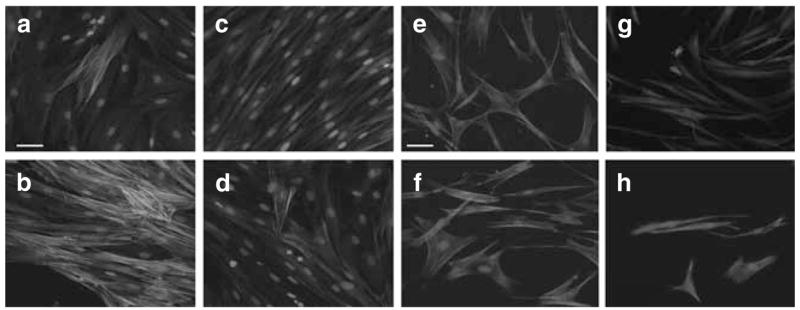
(a–d) Representative images of the immunohistochemistry staining for α-SMA. Normal human dermal fibroblasts (1 × 105 cells) were seeded in collagen I matrix (1 mg/ml), which remained attach to the culture plate throughout the cytokine stimulation period. α-SMA was visualized using mouse monoclonal α-SMA FITC conjugated. (a) FPCL under control conditions at 96 hours. (b) FPCL treated with TGF-β1 (1 ng/ml) for 96 hours. (c) FPCL treated with TNF-α (1 ng/ml), and (d) FPCL treated with combination of TGF-β1 (1 ng/ml) and TNF-α (1 ng/ml). Bars = 50 μm (panels a–d). (e–h) Rhodamine-conjugated phalloidin staining for F-actin to examine the assembly of stress fibers. (e) FPCL under control conditions at 96 hours. (f) FPCL treated with TGF-β1 (1 ng/ml) for 96 hours. (g) FPCL treated with TNF-α (1 ng/ml), and (h) FPCL treated with a combination of TGF-β1 (1 ng/ml) plus TNF-α (1 ng/ml). Bars = 50 μm (panels e–h).
During skin wound repair, fibroblasts go through a transition from fibroblast to myofibroblast under the influences of mechanical stress, cytokines, and an array of ECM molecules. The transition between fibroblasts and myofibroblasts results in fibroblast–ECM matrix remodeling and thus leads to a change in the matrix mechanical tension. Few studies have attempted to measure directly this mechanical tension within the ECM of a healing wound. For this, we used an indentation assay to measure the stiffness of the collagen matrix under different cytokine stimulations. In Figure 3, stressed fibroblast collagen lattices were constructed and induced with TGF-β1, TNF-α, or a combination of TGF-β1 and TNF-α for 4 days. The lattices were then tested and the Young’s modulus (representative of the mechanical stiffness of the collagen matrix) was determined. The results showed that TGF-β1 stimulation caused an insignificant increase in the stiffness (Young’s modulus of 2,010 ± 280 Pa) compared with the control with P = 0.16 (Young’s modulus of 1,740 ± 185 Pa). However, TNF-α treatment alone or combined with TGF-β1 resulted in a significantly decreased stiffness of the matrices with P = 0.01 (Young’s modulus of 1,260 and 1,300 ± 80 Pa, respectively).
Figure 3. TNF-α affects the mechanical response of the fibroblast collagen constructs.
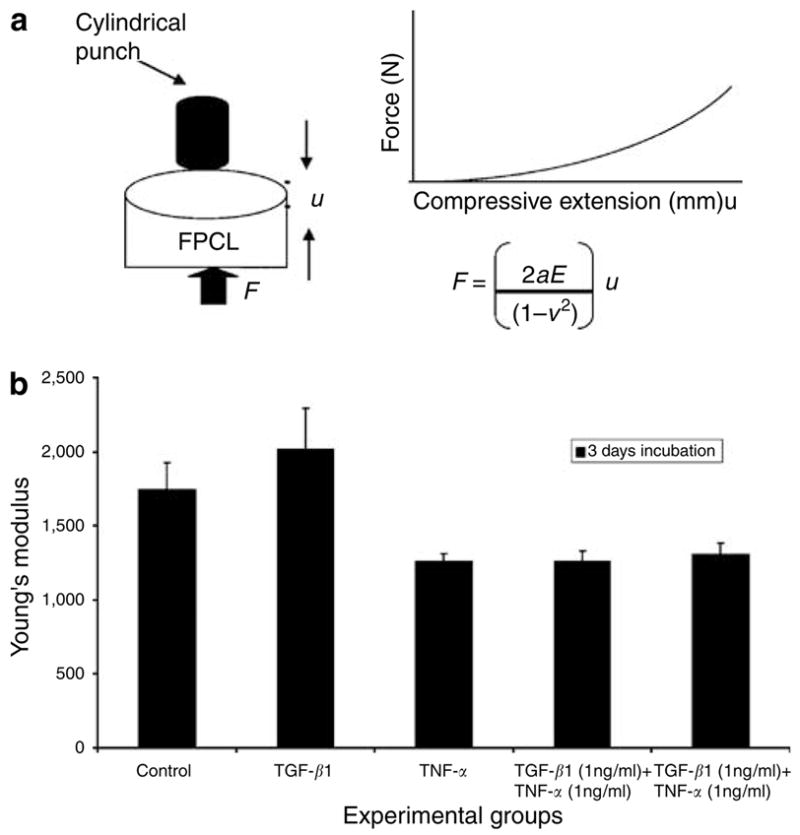
The mechanical response of FPCL treated with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), and a combination of TGF-β1 and TNF-α were measured using compressive indentation assay. In the indentation assay, normal fibroblasts (3 × 105 cells/cm3) were seeded in collagen I matrix (1.5 mg/ml) and induced with cytokines for 72 hours. The constructs underwent indentation measurements. (a) Schematic of the mechanical test and equation used for calculating Young’s modulus. (b) The data are expressed as Young’s modulus for each experimental group.
TNF-α suppresses TGF-β1-induced gene expression at the mRNA level
The next question is whether TNF-α suppression of TGF-β1-induced upregulation of α-SMA, collagen type I, and fibronectin occurred at the mRNA level (Figure 4). In these experiments, we used quantitative real-time reverse transcriptase-PCR to determine the levels of expression of α-SMA, fibronectin, and collagen 1A. We normalized the quantified mRNA of each gene to its GAPDH mRNA and the results show an increase in folds. We showed that TGF-β1 induced increases in collagen type I, fibronectin, and α-SMA mRNA levels after 24 hours of cytokine treatments and declined by 48 hours (Figure 4a). However, when fibroblasts were treated with TNF-α plus TGF-β1, the mRNA levels of all three genes were suppressed. In Figure 4b, TGF-β1-treated matrices had an eightfold increase in mRNA expression compared to only a twofold increase in the matrices treated with TGF β1 combined with TNF-α (P<0.001). Fibronectin mRNA showed an 18-fold increase at 24 hours in TGF-β1 treatment compared with only a threefold increase when TNF-α was combined with TGF-β1 (P < 0.01; Figure 4c). The same trend was observed with collagen type 1A mRNA. At 24 hours, collagen 1A mRNA increased sixfold with the TGF-β1 treatment groups, and decreased to 1.5-fold in the groups treated with TGF-β1 plus TNF-α (P = 0.01; Figure 4d). These results demonstrate consistent suppression by TNF-α of TGF-β1-induced myofibroblast phenotypic genes, at the mRNA level.
Figure 4. TNF-α suppresses TGF-β1-induced gene expression at the mRNA level.
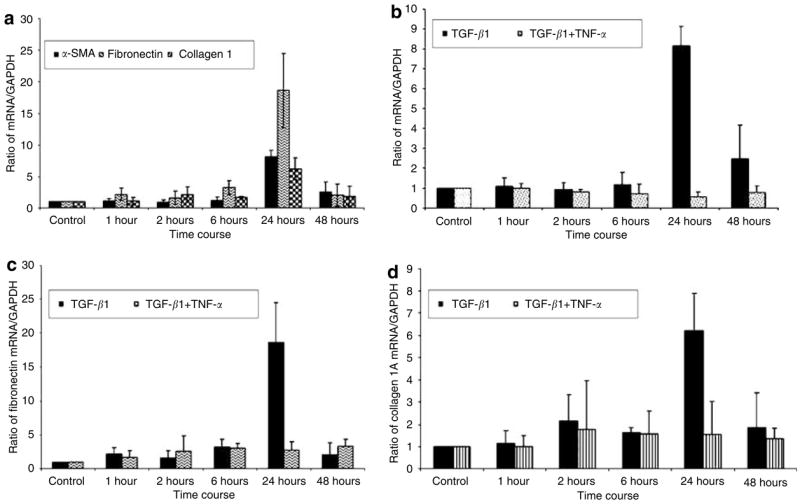
Fibroblast-populated collagen lattice (FPCL) was prepared at a final cell density of 1.5 × 106 cells/ml in collagen (1 mg/ml) and a 4 ml gel cell mixture was plated in 6 cm2 plate. After polymerization, the FPCL was induced with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), and TGF-β1 (1 ng/ml) plus TNF-α (1 ng/ml) in DMEM containing 0.1% FBS. Total RNA was extracted and reverse-transcribed into cDNA. cDNA was analyzed for the mRNA expression of α SMA, fibronectin, collagen 1A, and GAPDH by real-time PCR. Results were expressed as a ratio of target gene to GAPDH. Results from three independent experiments are shown with means ± SE (a) Results for TGF-β1 induction of α-SMA, fibronectin, and collagen 1A. (b–d) TNF-α suppression of α SMA, fibronectin, and collagen 1A, respectively.
TNF-α suppresses TGF-β1-induced gene expression by affecting mRNA stability
To elucidate possible post-transcriptional mechanisms that could explain TNF-α negative effects on α-SMA mRNA levels seen in our quantitative reverse transcriptase-PCR experiments, we measured α-SMA mRNA stability (Figure 5). Earlier studies showed that TGF-β1 stabilizes collagen I, fibronectin, and thrombospondin mRNA in mouse 3T3 cells, with maximal increase at 16–24 hours after induction (Penttinen et al., 1988). However, the effect of TGF-β1 and TNF-α on α-SMA mRNA stability has not previously been examined. In these experiments, human dermal fibroblasts were stimulated with TGF-β1 or TGF-β1 combined with TNF-α for 24 hours. At the end of 24-hour cytokine induction, actinomycin D (2.5 μg/ml) was added to the media and total RNA was harvested at 0, 2, 4, 6, 8, and 24 hours. Real-time quantitative reverse transcriptase-PCR was used to measure α-SMA mRNA levels. The results showed that TGF-β1 stabilized α-SMA mRNA levels with t1/2 of 53 hours. Interestingly, TNF-α destabilized α-SMA transcripts 10-fold, with t1/2 of only 5 hours. The result suggests that TNF-α can abrogate TGF-β-induced α-SMA production through destabilization of its mRNA.
Figure 5. TNF-α affects α-SMA mRNA stability.
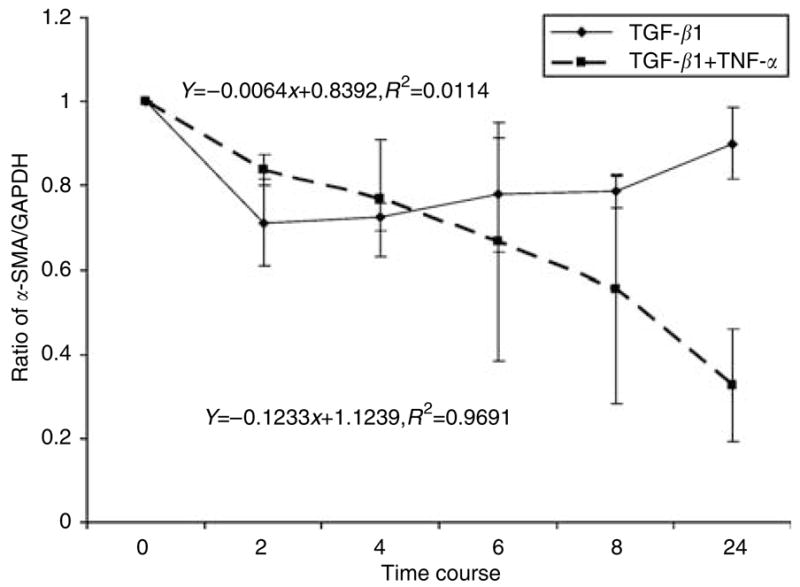
Adult human dermal fibroblasts were grown in monolayer to confluence and serum-starved for 16 hours before induction with TGF-β1 (1 ng/ml) or TGF-β1 (1 ng/ml) plus TNF-α (1 ng/ml) for 24 hours. At the end of 24 hours, actinomycin D (2.5 μg/ ml) was added and total RNA was harvested at different time points for real-time PCR analysis. The graph represents a ratio of α-SMA and GAPDH mRNA versus time from three independent experiments with SD.
TNF-α decreases the contraction of FPCLs
We also examined the correlation between TNF-α suppression of α-SMA expression and the contractile force within the collagen lattice (Figure 6). We used the attached and delayed released contraction model, which has been shown to represent the actual contraction force exerted by fibroblasts (Tomasek et al., 1992). The FPCL was cast in high-density −500,000 fibroblasts per milliliter – lattice, which remained attached throughout the cytokine stimulation period. After 4 days of cytokine stimulation, the stressed FPCL was mechanically released from the culture well. It contracted rapidly within 60 minutes, and the percent gel contraction was calculated as area of gel at the end of contraction divided by area of gel before release from the culture plate. Serum-starved treatments (0.1% FBS) showed considerably less contraction than fully stimulated treatments (5% FBS); 55 vs 25% gel contraction, respectively. TGF-β1 (1 ng/ml + 0.1% FBS)-treated FPCL showed significant contraction compared with the serum-starved controls; 25 vs 55%. FPCL treated with TNF-α (10 ng/ml in 0.1% FBS) alone showed minimal contraction equivalent to the untreated controls; 64 vs 55%. However, treatment of FPCL with TGF-β1 combined with TNF-α showed minimal contraction compared with treatments with TGF-β1 alone; 55 vs 25%. We observed similar results using a free-floating gel contraction assay model (data not shown). Thus, TNF-α abolished TGF-β1-induced myofibroblast contractility.
Figure 6. TNF-α abolishes TGF-β1-induced myofibroblast contractility.
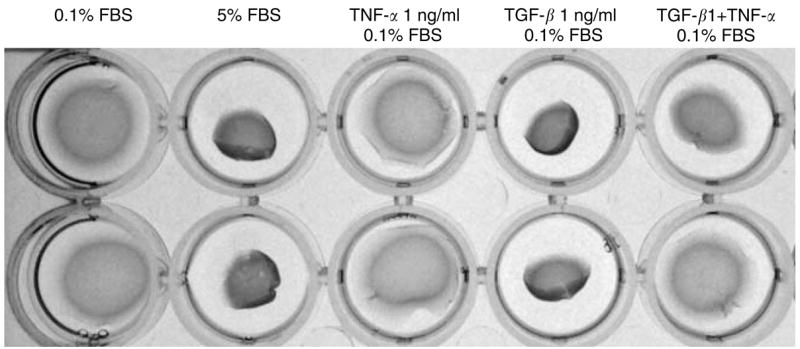
The stressed fibroblast-populated collagen lattice (FPCL) was prepared in a final cell density of 500,000 cells/ml plus collagen concentration of (1.5 mg/ml) and 0.6 ml volume per gel. The stressed FPCLs were treated under different conditions for 4 days. At the end of treatment, the stressed FPCLs were mechanically released from culture plate and allowed to contract rapidly within 1 hour. The pictures were taken at the end of contraction.
Differential participation of protein kinase (MAPK) cascades in TNF-α antagonistic effect on TGF-β1 induction of α-SMA expression
Activation of TNF receptors leads to recruitment of tumor necrosis factor receptor–associated factor family members, and leads to activation of multiple signal transduction pathways such as NF-κB, JNK, p38 MAP, and phosphoinosi-tide 3-kinase. To gain insight into the cellular mechanisms by which TNF-α suppresses TGF-β1-induced α-SMA expression, we focused on the functions of two major downstream MAPK cascades, p38 MAPK and JNK.
First, we tested whether TNF-α treatment would activate the JNK pathway. As shown in Figure 7a, TNF-α treatment leads to phosphorylation of JNK at Thr 183 and Tyr 185, using mouse monoclonal antibody in human dermal fibroblasts in stressed FPCL. This indicates that intracellular JNK activation is present in primary adult human fibroblast cells in response to TNF-α.
Figure 7. TNF-α decreases TGF-β1 induction of α-SMA expression in a JNK-dependent pathway.
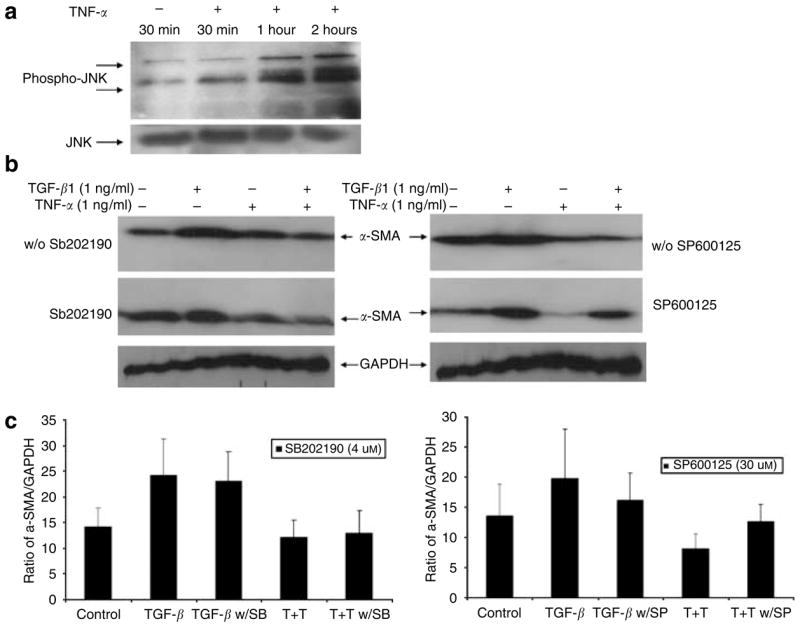
(a) Human dermal fibroblasts were grown in monolayer to confluence and serum starved overnight followed by induction with TNF-α. Total protein was harvested at indicated time and Western blot analysis was performed for phosphorylated JNK and JNK proteins. (b) FPCL was prepared in a final cell density of 300,000 cells/ml plus collagen concentration of 1 mg/ml and induced with different cytokine combinations with or without SB202190 and SP600125 inhibitors for 96 hours. Cell lysates were analyzed for α-SMA and GAPDH expression using Western blot. (c) The graph represents mean α-SMA expression as a ratio of GAPDH obtained by densitometric analysis from three independent experiments.
Utilizing chemical inhibitors for p38 MAPK and JNK, we examined the effect of TNF-α on these signaling pathways and their possible association with suppression of α-SMA expression. The cells were grown in stressed collagen I matrix and induced with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), and TGF-β1 (1 ng/ml) together with TNF-α (1 ng/ml). The chemical inhibitors were added at the same time as the cytokines. After 5 days of treatment, the cells were harvested and Western blots were used to examine α-SMA. As shown in Figure 7b and c, blockade of the p38 MAPK pathway by highly specific chemical inhibitor SB202190 (4 μM) did not affect TNF-α suppression of TGF-β1-induced α-SMA expression compared with the untreated group. SB202190 also had no effect on TGF-β1 upregulation of α-SMA expression. However, blockade of JNK pathway by SP600125 (30 μM) resulted in reversal of TNF-α suppression. The cells treated with SP600125 showed no suppression effect by TNF-α. These results suggest that the JNK pathway plays a central role in mediating TNF-α suppression of α-SMA expression.
TNF-α inhibits Smad3 phosphorylation in normal human dermal fibroblasts
Smad signaling is considered the principal signaling pathway for TGF-β. Smads affect the transcription of specific genes through direct or indirect binding to their promoters. As previous studies have demonstrated specific interaction between the MAPK cascade and TGF-β-driven Smad signaling (Verrecchia et al., 2003), we wanted to determine if TNF-α inhibits phosphorylation of Smad3. Adult human dermal fibroblasts were grown in monolayer to confluence and serum starved for 16 hours before cytokine induction. Cells were then induced with TGF-β1 (1 ng/ml) alone or with a combination of TGF-β1 (1 ng/ml) and TNF-α (1 ng/ml). As shown in Figure 8, TGF-β1 activated Smad-3 phosphorylation after 30 minutes of induction. In contrast, TNF-α inhibited phosphorylation of Smad3 by TGF-β1. Therefore, it is likely that TNF-α suppresses TGF-β1-induced α-SMA expression by inhibiting Smad-3 phosphorylation via a JNK-dependent pathway (Figure 9).
Figure 8. TNF-α inhibits Smad3 phosphorylation in normal human dermal fibroblasts.

To test whether TNF-α affects the TGF-β1 Smad signaling pathway, human dermal fibroblasts were grown in monolayer and induced with TGF-β1 (1 ng/ml) or TGF-β1(1 ng/ml) combined with TNF-α (1 ng/ml). Cell lysates were analyzed for phosphorylated Smad3 and GAPDH using Western blot analysis.
Figure 9.
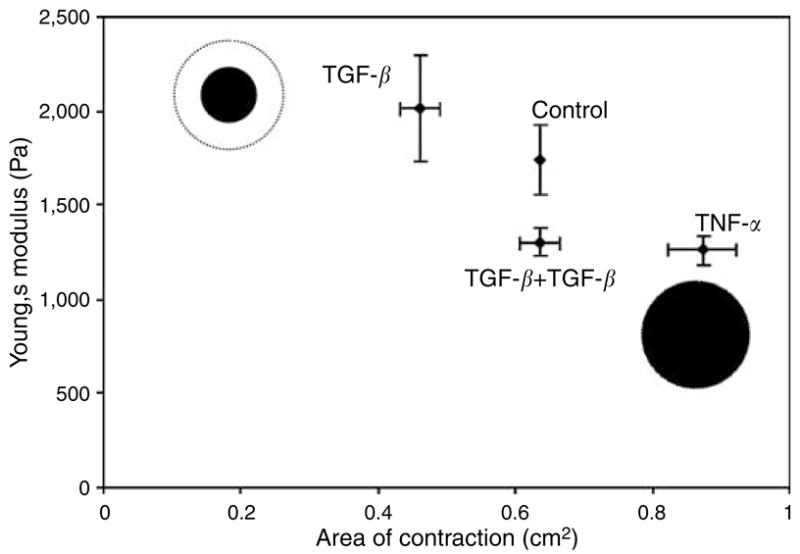
Correlation of collagen gel matrix stiffness, seen in indentation assay, with the rate of gel contraction.
DISCUSSION
Cellular phenotypic changes within the wound environment play an essential role in response to injury. The best studied example of this is the transformation of fibroblasts into myofibroblasts, which are present in both normal and fibrotic wound healing. The key feature of these activated fibroblasts or myofibroblasts is the expression of α-SMA (Darby et al., 1990). The transition from fibroblast to myofibroblast also involves the acquisition of stress fibers composed of cytoplasmic actins and fibronectin under the influences of both cytokines and mechanical stress within the wound (Gabbiani, 2003).
Previous investigators have described TGF-β1 induction of myofibroblast differentiation in both in vitro and in vivo wound healing models (Ronnov-Jessen and Petersen, 1993; Hinz et al., 2001). As we confirmed in our study, TGF-β1 is an inducer of α-SMA expression and myofibroblast phenotype. Further, we showed that TGF-β1 stimulates α-SMA expression at the mRNA level through stabilization of α-SMA mRNA stability with t1/2 of 56 hours. Previous investigators have only examined the effects of TNF-α on α-SMA expression using an in vivo murine model (Rubbia-Brandt et al., 1991; Desmouliere et al., 1992). In those studies, TNF-α was locally infused into mouse wounds and the local granulation tissues showed limited α-SMA expression compared to other cytokines, that is, TGF-β, GM-CSF, and heparin. From those experiments, the authors concluded that TNF-α had no effect on α-SMA expression. In this study, we used a more defined model and found that the situation is far more complex and interesting. Treatment of human dermal fibroblasts in a stressed three-dimensional collagen matrix with TNF-α suppresses α-SMA expression below the unstimulated controls. Of much greater interest, we documented that TNF-α exposure abrogates TGF-β1 induction of α-SMA expression at the post-transcriptional level by destabilizing α-SMA mRNA. This response is not limited to this particular protein. We are the first to report that TNF-α suppresses TGF-β1-promoted increases of two other structural proteins, collagen 1A and fibronectin. It is of interest that mRNA levels for α-SMA, collagen I, and fibronectin declined after 48 hours, despite the continued presence of TGF-β1. Whether this resulted from downregulation of TGF-β receptors or other intracellular mechanisms served to blunt TGF-β1-induced increases in mRNA levels is not yet known.
There is increasing evidence that suggests that members of the stress-activated kinase pathways, JNK and P38 MAPK, are involved in the interaction between these two cytokines. As described above, they appear to mediate TNF-α antagonistic activity against TGF-β1-induced α-SMA expression. The JNK pathway has been shown to be involved in TNF-α antagonistic activity against TGF-β1-driven Smad3 and -4 collagen1A1 and collagen 3A1 gene expression (Verrecchia et al., 2003). Another study showed that treatment of human prostate carcinoma cells with a MAPK p38 inhibitor resulted in inhibition of TGF-β1-induced actin reorganization (Edlund et al., 2002). These reports helped to trigger our interest in the interaction of the signaling pathways during cytokine-induced myofibroblast differentiation. To investigate these phenomena, we utilized two pharmacological inhibitors. Our results showed that an inhibitor of p38 MAPK (SB203580) did not affect TNF-α suppression of TGF-β1-induced α-SMA expression. By contrast, an inhibitor of JNK (SP600125) reversed the suppression effect of TNF-α on TGF-β1-induced α-SMA expression. The results suggest that the JNK pathway plays a central role in mediating TNF-α suppression of α-SMA expression.
Because Smad signaling plays a key role in transmitting TGF-β1 signals, we investigated the effect of TNF-α on Smad3 phosphorylation. The results clearly showed that TNF-α inhibits phosphorylation of Smad3. There is no consensus in the literature on which members of the Smad family are responsible for TGF-β1-induced α-SMA expression. One study found that overexpression of Smad2, but not Smad3, was responsible for upregulation of α-SMA mRNA and reorganization of actin cytoskeleton in human lung fibroblast (Evans et al., 2003). However, in vivo studies of mice showed that the recruitment of inflammatory cells, including neutrophiles, mast cells, and myofibroblasts, in skin lesions after irradiation is TGF-β1/activin/Smad3-dependent (Ashcroft et al., 1999). Smad3 also has an important function in the upregulation of TGF-β1 at the site of injury. Loss of Smad3 in wild-type skin and Smad3-null skin shows reduction in scarring (Flanders et al., 2003). Furthermore, overexpression of Smad3 markedly increases α-SMA protein expression (Hu et al., 2003). It appears that there is enough evidence to suggest that inhibition of Smad3 and not Smad2 is the key mediator of pathogenic effects of TGF-β1 in fibrosis. Our study also suggests that TNF-α decreases α-SMA expression via inhibition of Smad3 phosphorylation.
Several studies have demonstrated that TGF-β1 increases tensile strength during wound healing (Mustoe et al., 1987; Connors et al., 2000; Korenkov et al., 2005). For example, Mustoe et al. (1987) found that TGF-β1 improved both the breaking strength and the rate of healing in rats’ incisional wounds. In contrast, exogenous administration of TNF-α has been shown to both enhance and attenuate local collagen synthesis and wound-breaking strength of cutaneous wounds in mice and rats (Salomon et al., 1991; Maish et al., 1998; Lee et al., 2000). Unfortunately, these studies have all been performed in animals or using non-human tissues. Our study used the stressed FPCL model to examine the interaction of these mediators, as it probably occurs in the local tissue microenvironment. Further, it is among the few attempts to correlate the molecular response with mechanical behavior induced by fibroblasts. In our project, we measured two mechanical parameters of collagen lattice: contraction rate and stiffness of the matrix. Our mechanical data correlate with the molecular data in that increasing α-SMA, collagen 1A, and fibronectin mRNA levels under TGF-β1 stimulation resulted in increasing stiffness and contraction of the matrix. Conversely, stress FPCL treated with TNF-α together with TGF-β1 had lower mRNA levels of α-SMA, collagen 1A, and fibronectin. As expected, these FPCL matrices demonstrated negligible contraction as well as lower tensile strength compared with TGF-β1 treatment groups. It is of interest that we found the same contraction response using the Bell free-floating lattice contraction assay (Ehrlich and Rittenberg, 2000), in which TNF-α significantly reduced contraction forces induced by TGF-β1 (data not shown). TNF-α can reverse the stimulation of TGF-β1-induced collagen lattice contraction in stressed as well as free-floating collagen lattice.
We further correlated the rate of contraction with the stiffness of the gels by plotting the Young’s modulus obtained from the indentation assay against the final area of contraction (Figure 9). Two main results are evident: (i) with the TGF-β treatments, the stiffness increased along with the degree of contraction (i.e., a lower final area) as compared with the untreated control and (ii) in contrast, with the TNF-α treatment, the stiffness decreased along the degree of contraction as compared with the control. These results suggest that the contraction and stiffness are coupled. Namely, as contraction proceeds, there is a consequent increase in stiffness, roughly corresponding to an increasing density of the collagen matrix. In other words, the observed increase/decrease in stiffness is the result of a greater/lesser degree of contraction. Furthermore, we found that TGF-β1 induced marked alternation in the cell phenotype and reorganization of the actin cytoskeleton, as demonstrated in our rhodamine phalloidin staining. Larger organized stress fibers in fibroblasts within stressed FPCL treated with TGF-β1 directly correlates with a greater degree of contraction as well as an increase in matrix stiffness. Conversely, TNF-α combined with TGF-β1 treatments resulted in a decreased assembly of stress fibers, which correlates with reduction in stiffness and contraction of collagen matrix. Recent work by Goffin et al. (2006) demonstrated α-SMA as a mechano-sensitive protein that is recruited to stress fibers under high tension. They illustrated this phenomenon by growing primary rat lung myofibroblasts on micro-pattern silicone substrates of a given thickness (80 μm) and varying stiffnesses, ranging from normal soft tissue to plastic. They demonstrated that α-SMA recruitment to stress fibers of cultured myofibroblasts requires a substrate stiffness of approximately 16 kPa. This is significantly different from the findings in our study, in which α-SMA differentiation occurred in matrices with stiffnesses of 1,700–2,200 Pa. There are many factors that would contribute to the differences in our results. First, we use primary human dermal fibroblasts embedded in three-dimensional collagen 1 matrices, as opposed to rat lung myofibroblasts grown on top of micro-pattern silicone substrates. Second, we stimulated our human dermal fibroblasts with TGF-β1 and showed myofibroblast differentiation. We then measured the matrix stiffness. In our experiments, we did not examine the redistribution of α-SMA to stress fibers. Lastly, the substrates’ elastic moduli in the work by Goffin et al. (2006) are the result of microscopic measurements, whereas ours are macroscopic measurements of normal soft tissue. The stiffness of matrices in our experiments exhibited elastic moduli of 1,700–2,200 Pa, which is consistent with recently published results from Clark et al. (Ghosh et al., 2007). Clark et al. showed that adult human dermal fibroblasts grown on hydrogels with a stiffness of 4,270 Pa exhibited linear, stretched arrays of F-actin microfilaments with uniform diameter similar to that of our fibroblasts treated with TGF-β1. Our substrate stiffness is also similar to the elastic modulus of normal soft tissue, 1–20 kPa (Bao and Suresh, 2003).
In view of our results, we propose a functional relationship between mechanics and molecular influences of TGF-β1 and TNF-α on ECM reorganization in human dermal fibroblasts as follows: TNF-α mediates its antagonistic effects on TGF-β1 through the JNK pathway. Inhibition of Smad3 phosphorylation leads to a decrease in transcription of TGF-β1 response genes, that is, collagen 1A, fibronectin, and α-SMA. As a consequence of these molecular changes, TNF-α suppresses TGF-β1-induced myofibroblast differentiation and cytoskeletal changes necessary for efficient matrix contraction coupled with matrix tensile strength. The model is summarized in Figure 10.
Figure 10. A proposed mechanism of TNF-α suppression of TGF-β1-induced gene expression.
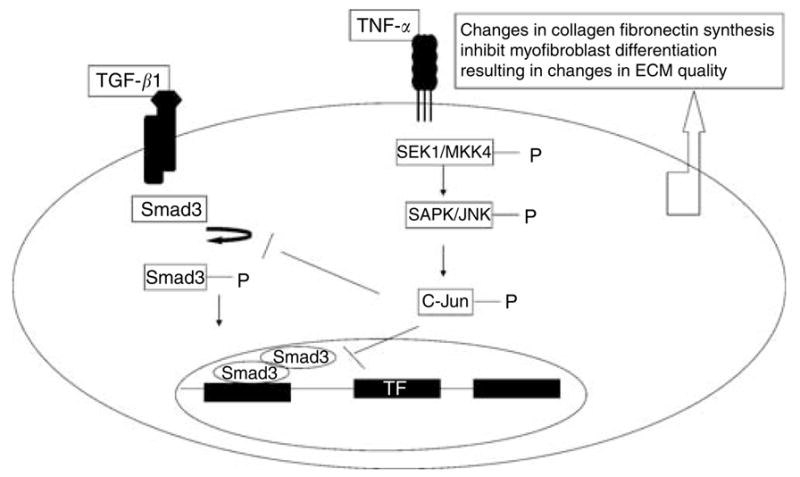
TNF-α inhibits TGF-β1-induced collagen 1a, fibronectin, and α-SMA expression by inhibiting Smad3 phosphorylation via JNK pathway. Inhibition of Smad3 phosphorylation precluded formation of Smad3 and -4 complex and thus prevented it from regulating the target gene. The overall effect of TNF-α is inhibition of TGF-β1-induced myofibroblast differentiation, leading to softer and less contracted matrix.
The data in this paper could support a molecular and mechanical mechanism for inflammation-induced abnormal wound healing. Normally, TNF-α is not present during the re-epithelialization and ECM reorganization phases of wound healing (Singer and Clark, 1999). Normal cutaneous wound healing is a coordinated sequence of events involving clotting, inflammation, remodeling, and re-epithelization. In the inflammation phase, infiltrating inflammatory cells secrete growth factors (i.e., platelet-derived growth factor and TGF-β) and cytokines (TNF-α) essential for cleansing the wounded area of foreign particles and bacteria and initiating myofibroblast-dependent wound contraction (Singer and Clark, 1999). Healing proceeds only after the inflammation has subsided. Under pathologic conditions, such as in chronic wounds, the cascade of wound healing is disrupted and the wounds are locked into a state of inflammation characterized by abundant neutrophil infiltration with associated reactive oxygen species and inflammatory cytokines (TNF-α). Stacey et al. found very high level of TNF-α and low levels of TGF-β1 in chronic wounds fluid compared to healing wounds (Trengove et al., 2000). The results from our study showed high levels of TNF-α in suppression of TGF-β1-stimulated α-SMA, collagen, and fibrin synthesis. TNF-α-suppressed myofibroblast differentiation and cytoskeletal changes required for normal matrix contraction. Therefore, chronic wounds with a high level of TNF-α would fail to contract in a normal fashion. The granulation tissues in chronic wounds are often characterized as fragile. We have demonstrated here that matrices treated with TNF-α (10 ng/ml) have lower tensile strength than normal untreated matrices. Continued inflammation, mediated by TNF-α, might prevent normal matrix deposition and myofibroblast-dependent wound contraction mediated by TGF-β1 in normal wound healing.
MATERIALS AND METHODS
Reagents
Monoclonal Anti-α-SMA was purchased from Sigma (St Louis, MO). Mouse monoclonal anti-α-SMA conjugated with FITC was purchased from Sigma. Mouse anti-vimentin monoclonal antibody was purchased from Chemicon International (Temecula, CA). Mouse monoclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) (G9), rabbit polyclonal anti-phospho Smad3, and-rabbit polyclonal anti-JNK were purchased from Cell Signaling Technology (Beverly, MA). Recombinant human TNF-α and TGF-β1 were purchased from R&D Systems (Minneapolis, MN). Purified collagen (Vitrogen 100) was purchased from Cohesion Technology (Palo Alto, CA). Both selective inhibitor SB202190 and SP600125 were purchased from Sigma.
Cell Cultures
Normal human dermal fibroblasts were obtained from adult human skin discarded after reconstructive surgery. All experiments involving human tissue were approved by the Institutional Review Board and adhered to the Declaration of Helsinki Principles. Patient consents were obtained as outlined in the USC IRB no.999061. Early passage cells (passages 2–4) were used in all experiments. The isolated fibroblasts were cultured in DMEM containing 10% FBS with antibiotics (Gibco, Carlsbad, CA). Cells were cultured until they were confluent. FPCLs were prepared at a final cell density of 300,000 cells/cm3 with a collagen concentration of 1 mg/ml. A drop of 250 μl of cell/gel suspension was placed onto a 24-well tissue culture plate, which remained attached throughout the culture period. After polymerization for 1 hour at 37°C, the stressed FPCL was treated with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), or combinations of TGF-β1 (1 ng/ml) and TNF-α (1 and 2 ng/ml) in 1.5 ml of DMEM containing 0.1% FBS. This cell culture condition was used in our Western blot and immunohistochemistry staining analysis.
Contraction assay
Human dermal fibroblasts were embedded in type I collagen at the final concentration of 5 × 105 cells and collagen concentration of 1.5 mg/ml. A volume of 600 μl per gel was fabricated in a 24-well plastic culture dish, which ensured that the gel would remain attached throughout the culture period. The cells were cultured in 10% FBS for 20 hours after polymerization. After 20 hours of recovery in 10% FBS, the stressed FPCLs were incubated in 0.5% FBS with TGF-β1 (1 ng/ml), TNF-α (1 ng/ml), and a combination of TGF-β1 (1 ng/ml) and TNF-α (1 ng/ml). After 4 days in culture, the stressed FPCLs were mechanically released from the plate and rapid contraction was recorded in 1 hour.
Western blot
The stressed FPCLs were induced with different cytokines for 96 hours. Cell extracts were prepared with SDS sample buffer and boiled for 3 minutes at 95°C. Equal amounts of cell lysates were separated on 12% SDS-PAGE gel. After electrophoresis, the separated proteins were transferred to a nitrocellulose membrane. The membrane was blocked with Tris-buffered saline (50 mM Tris, pH 7.5, and 100 mM NaCl) containing 5% non-fat milk for 1 hour and then incubated with the primary antibody at 4°C overnight. The blots were subsequently washed with Tris-buffered saline–Tween-20 (0.1%) and then incubated with an appropriate horseradish peroxidase-conjugated secondary antibody from Pierce Biotechnology (Rockford, IL) in saline-Tween. Proteins were visualized using Femto-enhanced chemiluminescence according to the manufacturer’s instructions.
Immunohistochemistry staining
To analyze the effects of TGF-β1 and TNF-α on α-SMA protein expression, human dermal fibroblasts (100,000 cells) were grown in collagen I lattice (1 mg/ml). The stressed lattices were stimulated with designated cytokines for 96 hours and then fixed with methanol and treated with Triton-100 (0.15%). After blocking with 5% BSA, the lattice was incubated with mouse monoclonal anti-α-SMA FITC conjugate (1:200 dilution). The nucleus was counter stained with propidium iodine.
The procedure for staining cells with rhodamine-conjugated phalloidin is as follows: after 96 hours of stimulation with different treatment conditions, the stressed FPCLs were fixed with 35% methanol for 10 minutes followed by TritonX100 (0.12%) for 10 minutes. Phalloidincy3 (Sigma) at 20 μg/ml was used for staining in phosphate-buffered saline overnight. The FPCLs were then washed with phosphate-buffered saline and the nuclei were counterstained with 4′,6-diamidino-2-phenylindole at 1 μg/ml for 10 minutes. The stained FPCLs were visualized under the microscope with an attached digital camera (Nikon, AG Heinze Precision Microoptics, Chandler, AZ).
Measurement of α-SMA mRNA message stability
Normal human fibroblasts were grown to confluence in 10 cm2 plates in DMEM with 10% FBS. Confluent fibroblasts were serum-starved in DMEM containing 0.1% FBS for 16 hours, followed by induction with TGF-β1 (1 ng/ml) or TGF-β1 (1 ng/ml) combined with TNF-α (1 ng/ml) for 24 hours. At the end of 24 hours of cytokine stimulation, actinomycin D (2.5 μg/ml) was added and cells were scraped at the indicated time points. Total RNA was extracted, and real-time PCR was performed for α-SMA and GAPDH. t1/2 was calculated as the amount of time required for the α-SMA message to decrease 50% from initiation.
Total RNA extraction and cDNA synthesis
RNA was extracted from cells grown in stressed collagen type I (1 mg/ml) lattices using Trizol (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. RNA was quantified spectrometrically. Starting from 2 μg RNA, 20 μl cDNA was synthesized using random primers and SuperScript III Platinum as well as a CellsDirect two-step quantitative reverse transcriptase PCR kit from Invitrogen (Carlsbad, CA).
Real-time PCR
The cDNA samples were diluted 20 times. Five microliters of each sample was used to measure mRNA levels of α-SMA, collagen 1, fibronectin, and glyceraldehyde-3-phosphate dehydrogenase-GAPDH using the qPCR Mastermix Plus for SYBR Green from VWR. Reactions were performed and monitored using the ABI Prism 7700 Sequence Detection System (Perkin Elmer, Waltham, MA). The following primers were used: α-SMA: 5′-CCGACCGAATGCAGAAG GA-3′, sense and 5′-ACAGAGTATTTGCGCTCCGAA-3′, antisense (Knerr et al., 2001); collagen I: 5′-CAGCCGCTTCACCTACAGC-3′, sense and 5′-TTTTGTATTCAATCACTGTCTTGCC-3′, antisense (Frank et al., 2002); fibronectin: 5′-GGAGAATTCAAGTGTGACCCT CA-3′, sense and 5′-TGCCACTGTTCTCCTACGTGG-3′, antisense (Eikmans et al., 2003); GAPDH: 5′-GAAGGTGAAGGTCGGAGT-3′, sense and 5′-GAAGATGGTGATGGGATTTC-3′, antisense. Expression levels for each gene of interest were calculated by normalizing the quantified mRNA amount to the GAPDH mRNA. Each sample was assessed in triplicate.
Compressive indentation mechanical assay
FPCLs, 1.0 ml (by vol), were prepared to the final cell concentration of 300,000 plus collagen concentration of 1.5 mg/ml within standard 24-well culture plates. The gels remain attached to the dish throughout the testing period. We characterized the mechanical performance of the attached FPCL using an established compressive indentation experimental protocol (Mooney et al., 2006). Specifically, after removal of the excess phosphate-buffered saline solution from each well, a 3.0 mm circular, flat-ended glass punch was pressed into the surfaces of the FPCLs at a controlled displacement rate of 10 mm/minute to a final displacement, u, of 3 mm using an Instron 3365 Universal Testing machine equipped with a 2.5 N load cell. The force, F, and displacement, u, data were recorded and analyzed to determine the Young’s modulus, a key elastic coefficient of the FPCL governing its stiffness. This was done by fitting a linear equation to the data between 0.0 and 0.5 mm and analyzing the slope of the line in accordance with the Boussinesq elastic solution for the flat punch indentation geometry:
where a is the punch radius (1.5 mm), v is the Poisson’s ratio of the FPCL, and F and u are the applied force and displacement, respectively (Figure 2a–d). A Poisson’s ratio of 0.25 was assumed for the FPCL. The experimental force–displacement data were analyzed to yield the Young’s modulus (SI unit is Pa) of the FPCL as a function of experimental condition. All experiments were carried out in triplicate and data points and error bars in the figure represent averages and standard deviations.
Statistical analysis
All values are presented as mean ± SD Statistical analysis of differences between triplicate sets of experiments were assessed using a Student’s t-test, assuming double-sided independent variance with P < 0.05 considered significant.
Abbreviations
- α-SMA
α-smooth muscle actin
- ECM
extracellular matrix
- FBS
fetal bovine serum
- FPCL
fibroblast-populated collagen lattices
- JNK
Jun N-terminal kinase
- MAPK
mitogenic-activated protein kinase
- TGF-β1
tissue growth factor-β1
- TNF-α
tumor necrosis factor-α
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A. Tumor necrosis factor alpha suppresses the induction of connective tissue growth factor by transforming growth factor-beta in normal and scleroderma fibro-blasts. J Biol Chem. 2000;275:15220–5. doi: 10.1074/jbc.275.20.15220. [DOI] [PubMed] [Google Scholar]
- Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–6. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- Bao G, Suresh S. Cell and molecular mechanics of biological materials. Nat Mater. 2003;2:715–25. doi: 10.1038/nmat1001. [DOI] [PubMed] [Google Scholar]
- Connors D, Gies D, Lin H, Gruskin E, Mustoe TA, Tawil NJ. Increase in wound breaking strength in rats in the presence of positively charged dextran beads correlates with an increase in endogenous transforming growth factor-beta1 and its receptor TGF-betaRI in close proximity to the wound. Wound Repair Regen. 2000;8:292–303. doi: 10.1046/j.1524-475x.2000.00292.x. [DOI] [PubMed] [Google Scholar]
- Darby I, Skalli O, Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63:21–9. [PubMed] [Google Scholar]
- Delvoye P, Wiliquet P, Leveque JL, Nusgens BV, Lapiere CM. Measurement of mechanical forces generated by skin fibroblasts embedded in a three-dimensional collagen gel. J Invest Dermatol. 1991;97:898–902. doi: 10.1111/1523-1747.ep12491651. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Rubbia-Brandt L, Grau G, Gabbiani G. Heparin induces alpha-smooth muscle actin expression in cultured fibroblasts and in granulation tissue myofibroblasts. Lab Invest. 1992;67:716–26. [PubMed] [Google Scholar]
- Eastwood M, McGrouther DA, Brown RA. A culture force monitor for measurement of contraction forces generated in human dermal fibroblast cultures: evidence for cell-matrix mechanical signaling. Biochim Biophys Acta. 1994;1201:186–92. doi: 10.1016/0304-4165(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13:902–14. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Smad7 is required for TGF-beta-induced activation of the small GTPase Cdc42. J Cell Sci. 2004;117:1835–47. doi: 10.1242/jcs.01036. [DOI] [PubMed] [Google Scholar]
- Ehrlich HP, Rittenberg T. Differences in the mechanism for high- versus moderate-density fibroblast-populated collagen lattice contraction. J Cell Physiol. 2000;185:432–9. doi: 10.1002/1097-4652(200012)185:3<432::AID-JCP14>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Eikmans M, Baelde HJ, Hagen EC, Paul LC, Eilers PH, De Heer E, et al. Renal mRNA levels as prognostic tools in kidney diseases. J Am Soc Nephrol. 2003;14:899–907. doi: 10.1097/01.asn.0000056611.92730.7b. [DOI] [PubMed] [Google Scholar]
- Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Major CD, Arabshahi A, Aburime EE, Okada MH, Fujii M, et al. Interference with transforming growth factor-beta/Smad3 signaling results in accelerated healing of wounds in previously irradiated skin. Am J Pathol. 2003;163:2247–57. doi: 10.1016/s0002-9440(10)63582-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, Heim M, Jakob M, Barbero A, Schafer D, Bendik I, et al. Real-time quantitative RT-PCR analysis of human bone marrow stromal cells during osteogenic differentiation in vitro. J Cell Biochem. 2002;85:737–46. doi: 10.1002/jcb.10174. [DOI] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Ghosh K, Pan Z, Guan E, Ge S, Liu Y, Nakamura T, et al. Cell adaptation to a physiologically relevant ECM mimic with different viscoelastic properties. Biomaterials. 2007;28:671–9. doi: 10.1016/j.biomaterials.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259–68. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–41. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- Knerr I, Dittrich K, Miller J, Kummer W, Rosch W, Weidner W, et al. Alteration of neuronal and endothelial nitric oxide synthase and neuropeptide Y in congenital ureteropelvic junction obstruction. Urol Res. 2001;29:134–40. doi: 10.1007/s002400000165. [DOI] [PubMed] [Google Scholar]
- Kolodney MS, Wysolmerski RB. Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J Cell Biol. 1992;117:73–82. doi: 10.1083/jcb.117.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenkov M, Yuecel N, Koebke J, Schierholz J, Morsczeck C, Tasci I, et al. Local administration of TGF-beta1 to reinforce the anterior abdominal wall in a rat model of incisional hernia. Hernia. 2005;9:252–8. doi: 10.1007/s10029-005-0341-y. [DOI] [PubMed] [Google Scholar]
- Lee RH, Efron DT, Tantry U, Stuelten C, Moldawer LL, Abouhamze A, et al. Inhibition of tumor necrosis factor-alpha attenuates wound breaking strength in rats. Wound Repair Regen. 2000;8:547–53. doi: 10.1046/j.1524-475x.2000.00547.x. [DOI] [PubMed] [Google Scholar]
- Maish GO, III, Shumate ML, Ehrlich HP, Cooney RN. Tumor necrosis factor binding protein improves incisional wound healing in sepsis. J Surg Res. 1998;78:108–17. doi: 10.1006/jsre.1998.5315. [DOI] [PubMed] [Google Scholar]
- Mariani TJ, Arikan MC, Pierce RA. Fibroblast tropoelastin and alpha-smooth-muscle actin expression are repressed by particulate-activated macrophage-derived tumor necrosis factor-alpha in experimental silicosis. Am J Respir Cell Mol Biol. 1999;21:185–92. doi: 10.1165/ajrcmb.21.2.3641. [DOI] [PubMed] [Google Scholar]
- Mauviel A, Qiu Chen Y, Dong W, Evans CH, Uitto J. Transcriptional interactions of transforming growth-factor-beta with pro-inflammatory cytokines. Curr Biol. 1993;3:822–31. doi: 10.1016/0960-9822(93)90216-b. [DOI] [PubMed] [Google Scholar]
- Mooney RG, Costales CA, Freeman EG, Curtin JM, Corrin AA, Lee JT, et al. Indentation micromechanics of three-dimensional fibrin/collagen biomaterial scaffolds. J Mater Res. 2006;21:2023–34. [Google Scholar]
- Mustoe TA, Pierce GF, Thomason A, Gramates P, Sporn MB, Deuel TF. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science. 1987;237:1333–6. doi: 10.1126/science.2442813. [DOI] [PubMed] [Google Scholar]
- Penttinen RP, Kobayashi S, Bornstein P. Transforming growth factor beta increases mRNA for matrix proteins both in the presence and in the absence of changes in mRNA stability. Proc Natl Acad Sci USA. 1988;85:1105–8. doi: 10.1073/pnas.85.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann NY Acad Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. [PubMed] [Google Scholar]
- Roy SG, Nozaki Y, Phan SH. Regulation of alpha-smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. Int J Biochem Cell Biol. 2001;33:723–34. doi: 10.1016/s1357-2725(01)00041-3. [DOI] [PubMed] [Google Scholar]
- Rubbia-Brandt L, Sappino AP, Gabbiani G. Locally applied GM-CSF induces the accumulation of alpha-smooth muscle actin containing myofibroblasts. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;60:73–82. doi: 10.1007/BF02899530. [DOI] [PubMed] [Google Scholar]
- Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–83. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- Salomon GD, Kasid A, Cromack DT, Director E, Talbot TL, Sank A, et al. The local effects of cachectin/tumor necrosis factor on wound healing. Ann Surg. 1991;214:175–80. doi: 10.1097/00000658-199108000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J, Burslem F, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen. 1999;7:442–52. doi: 10.1046/j.1524-475x.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Wagner EF, Mauviel A. Distinct involvement of the Jun-N-terminal kinase and NF-kappaB pathways in the repression of the human COL1A2 gene by TNF-alpha. EMBO Rep. 2002;3:1069–74. doi: 10.1093/embo-reports/kvf219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Laiho M. On and off: proteasome and TGF-beta signaling. Exp Cell Res. 2003;291:275–81. doi: 10.1016/j.yexcr.2003.07.007. [DOI] [PubMed] [Google Scholar]