

Abstract
Polycyclic aromatic hydrocarbons (PAH) are one of the major carcinogens in tobacco smoke. They are metabolically activated through different routes to form either diol-epoxides, PAH o-quinones, or radical cations and each of which has been proposed to be an ultimate carcinogen. To study how PAH metabolites mutate p53, we used a yeast reporter gene assay based on the p53 transcriptional activity. Colonies expressing wt p53 turn white (ADE +) and those expressing mutant p53 turn red (ADE −). We examined the mutagenicity of three o-quinones, benzo[a]pyrene-7,8-dione, benz[a]anthracene-3,4-dione and dimethylbenz[a]anthracene-3,4-dione, and compared them with (±) anti-benzo[a]pyrene diol epoxide ((±)-anti-BPDE) within the same system. The PAH o-quinones tested gave a dose-dependent increase in mutation frequency in the range of 0.160 – 0.375 μM quinone, provided redox-cycling conditions were used. The dominant mutations were G to T transversions (>42%), and the incidence of hotspot mutations in the DNA-binding domain was more than twice than would be expected by a random distribution. The dependence of G to T transversions on redox-cycling implicates 8-oxo-dGuo as the lesion responsible, which is produced under identical conditions (Chem. Res. Toxicol. (2005) 18: 1027). A dose-dependent mutation frequency was also observed with (±)-anti-BPDE but at micromolar concentrations (0–20 μM). The mutation pattern observed was G to C (63%) > G to A (18%) > G to T (15%) in umethylated p53 and was G to A (39%) > G to C (34%) > G to T (16%) in methylated p53. The preponderance of G mutations is consistent with the formation of anti-BPDE-N2-dGuo as the major adduct. The frequency of hotspots mutated by (±)-anti-BPDE was essentially random in umethylated and methylated p53, suggesting that 5′-CpG-3′ islands did not direct mutations in the assay. These data suggest that smoking may cause mutations in p53 by formation of PAH o-quinones which produce reactive oxygen species. The resultant 8-oxo-dGuo yields a pattern of mutations but not a spectrum consistent with that seen in lung cancer; we suggest that the emergence of the spectrum requires biological selection.
Keywords: Aldo-keto reductase, polycyclic aromatic hydrocarbon, ortho-quinones, reactive oxygen species
Introduction
Smoking is the leading cause of lung cancer, and is responsible for about 90% of the estimated 170,000 lung cases in the United States each year (1). One of the major classes of carcinogens in tobacco smoke is polycyclic aromatic hydrocarbons (PAH). These compounds must undergo metabolic activation to be mutagenic. Three pathways for PAH activation have been shown to lead to reactive metabolites. The reactive metabolites include the anti-diol-epoxides (P450A1/P4501B1 derived), reactive and redox-active o-quinones (Aldo-Keto Reductase (AKR) derived), and radical cations (P450 peroxidase derived) (2–7). Anti-diol-epoxides (8–11), PAH o-quinones (12–14) and radical cations can all covalently modify DNA (15,16).
Ultimately, the activated metabolites must cause change-of-function mutations to activate oncogenes or inactivate the tumor suppressor genes responsible for lung cancer. These reactive intermediates most likely target p53 since it is the most commonly mutated tumor suppressor gene in human lung cancer (17). p53 is also mutated in many other tumors. However, three properties distinguish p53 mutations in lung cancer from other tumors (18). The first feature is the predominance of G to T transversions in p53. Other types of cancers show different mutational patterns, generally dominated by G to A transitions, suggesting that they have been exposed to different carcinogens. This is the most unambiguous signature of lung cancer. The second property, called a strand bias, is that guanine residues are preferentially mutated in the non-transcribed strand (19). The third is mutation of a number of hotspot codons which account for about 50% of all the reported mutations. The most common hotspots are codons 157, 158, 179, 248 and 273 and the majority are G to T transversions, but since most of these are mutated in other cancers, this feature is not as unique to lung cancer (17,19). These observations have led to the search for mutagenic mechanisms initiated by cigarette smoke that can cause this profile in p53.
Of the different PAH metabolites, the most extensively studied has been the anti-diol-epoxide of benzo[a]pyrene ((±)-anti-BPDE). This is formed by the combined action of P4501A1/P4501B1 and epoxide hydrolase. (±)-anti-BPDE is mutagenic in a number of paradigms. Interestingly, it will form adducts preferentially at many of the hotspots in p53 including 157, 248 and 249 (20) in one study, and 157, 248, and 273 in another study (21). Evidence exists that this sequence specificity is governed by 5′-methylation of 5′-CpG-3′ islands. (22,23). (±)-anti-BPDE predominantly causes G to T transversions in most mutagenesis studies (20,24,25). Taken together this suggests that (±)-anti-BPDE is an ultimate carcinogen by forming adducts at specific sites in p53 to cause G to T transversions. However, formal proof of this hypothesis is lacking since, to date, a direct assay for measuring p53 mutagenesis in vivo has not been developed that has reproduced both the pattern of mutations (type of base changes) and spectrum of mutations (location of mutations by codon) observed in lung cancer.
Reactive oxygen species (ROS) may play a role in lung carcinogenesis. ROS exposure causes the formation of 8-oxo-2′-deoxyguanosine (8-oxo-dGuo), which if not repaired can cause G to T transversions. Loss of heterozygosity of hOGG1, an enzyme that excises 8-oxo-dGuo, is observed in nearly 100% of small cell lung cancers (26). This suggests that during lung cancer development cells may have an increased mutational load due to the formation of 8-oxo-dGuo. ROS damaged bases may also occur via the metabolic activation of PAH.
Dihydrodiol dehydrogenases (AKR1A1, AKR1C1–AKR1C4) from the AKR superfamily can oxidize PAH trans-dihydrodiols to catechols (6). The catechols can then undergo two 1 electron oxidation events leading to the generation of ROS (27). This autooxidation generates a PAH o-quinone. Since PAH o-quinones are Michael acceptors they can form stable covalent adducts such as BP-7,8-dione-N2-dGuo and BP-7,8-dione-N6-dAdo which can hydrate and cyclize (12,14). They can also form unstable covalent depurinating adducts such as BP-7,8-dione-N7-Gua (13). When reducing equivalents are present, PAH o-quinones can be reduced back to the catechol. In this way trace amounts of PAH o-quinones can establish futile redox-cycles that will amplify ROS. Thus PAH o-quinones can form covalent adducts with deoxyguanosine and deoxyadenosine residues but they can also generate ROS which can form 8-oxo-dGuo (28). Using an aldehyde reactive probe assay to detect aldehydic sites that were revealed after base-excision repair, it was found that the rank order of lesions produced by PAH o-quinones was 8-oxo dGuo ≫ oxidized pyrimidines = abasic sites (29). These reactions are likely to occur in smokers because AKRs are consistently overexpressed in NSCLC and SCLC, bronchial epithelial cells of NSCLC or oral cancer cells which are tobacco related cancers (30–33).
Studies of the p53 mutations from over 20,000 tumor samples compiled into two databases provides a resource that has led to a number of proposed mechanisms for carcinogenesis. Yet testing the mechanisms has been difficult in mammalian cells because the p53 mutant cells are not easily identified. We have employed a p53 mutagenicity assay which scores for functional p53 based on a yeast reporter system (34,35). Yeast offer some advantages to studying mutagenesis over bacterial systems. The primary advantage is that the assay system utilizes the major activity of p53, its transcriptional function to isolate biologically relevant mutations. The yeast are especially well suited for studies of reactive oxygen mediated damage as they express homologs of OGG1, the DNA glycosylase that excises 8-oxo-dGuo. Deletion of yeast OGG1causes a high incidence of spontaneous G to T transversions (36). In the yeast mutagenesis assay, wild-type p53 cDNA is exposed to a mutagen in vitro. The treated cDNA is co-transfected with the gapped vector where in vivo, the two DNA fragments undergo homologous recombination to form a functional plasmid that expresses p53. When wild type p53 is expressed, it binds to a stably integrated reporter gene in which a p53 dependent promoter drives the expression of the ADE2 reporter, and the host yeast strain turns white. When mutant p53 is expressed it fails to bind to the promoter, the ADE2 reporter is not expressed, and the yeast colonies turn red. This assay thus reports the transcriptional competency of p53. Using this in vitro assay we showed that BP-7,8-dione was 80 times more mutagenic than (±)-anti-BPDE provided redox-cycling conditions were present. The mutational pattern observed with BP-7,8-dione showed a predominance of G to T transversions that were abolished with ROS scavengers. These data suggested that the primary lesion responsible for these mutations could be 8-oxo-2′-deoxyguanosine and that PAH o-quinone mediated ROS production may contribute to p53 mutation. Under identical redox-cycling conditions, PAH o-quinones were shown to produce significant amounts of 8-oxo-deoxyguanosine (28).
Using the same yeast reporter system another study showed that (±)-anti-BPDE mutated umethylated and methylated p53 to yield a pattern of mutations similar to that observed in lung cancer (24). However, neither study gave a mutational spectrum similar to that seen in the disease.
In this study, we compare the potency of mutagens, pattern of mutations and spectrum of mutations of three PAH o-quinones, benzo[a]pyrene-7,8-dione, benz[a]anthracene-3,4-dione and dimethylbenz[a]anthracene-3,4-dione, and compare them with a representative diol epoxide ((±)anti-BPDE). We also examined the effect of methylation on (±)-anti-BPDE mutagenesis. By all three measurements the PAH o-quinones yielded results closer to those observed in lung cancer than results with (±)-anti-BPDE, even when methylated DNA was tested. We note however, that while the o-quinones were able to mutate hotspots about 25% of time, this frequency was somewhat less than the ~50% found in the p53 databases, suggesting that biological selection in the tumor cells may play a major role in determining the spectrum of fixed mutations.
Materials and methods
Chemicals and Reagents
Adenine, L-leucine, L-tryptophan, were purchased from Sigma (St. Louis, MO). YEASTMAKER Yeast Transformation and Plasmid Isolation Kit and all yeast culture media were obtained from CLONTECH (Palo Alto, CA). BP-7,8-dione and benz[a]anthracene-3,4-dione and dimethylbenz[a]anthracene-3,4-dione were synthesized according to the published routes (37). N-methyl-N′-nitro-N-nitroso-guanidine (MNNG) and (±)-anti-BPDE were obtained from the National Cancer Institute, Chemical Carcinogen Standard Reference Repository (Midwest-Research Institute, Kansas City, Missouri). The purity of all PAH-metabolites was assessed by LC/MS. Other reagents were of the highest grade available. Caution: All PAHs are potentially hazardous and should be handled in accordance with “NIH Guidelines for the Laboratory Use of Chemical Carcinogens”.
Yeast Strains, Media and Plasmids
The ade reporter yeast strain, yIG397, and gap-repair expression vector pss16 were kindly provided by Dr. Richard Iggo (Swiss Institute for Experimental Cancer Research, 1066 Epalinges, Basel, Switzerland) (38). Basic methods for yeast manipulations were carried out as described (39). Liquid media contained 0.67% yeast nitrogen base, 2% dextrose, 1% casamino acids and 20 μg/mL of adenine. Solid media for prototrophic selection of appropriate plasmids contained 0.67% yeast nitrogen base, 2% dextrose, 2% agar with complete additions minus the relevant amino acids and nucleosides to select for auxotrophic markers. p53 cDNA was obtained by RT-PCR from RNA isolated from HepG2 cells and its sequence confirmed by dideoxysequencing. For assays, a 1.2 kb EcoR1 fragment was gel isolated and then purified using QIA quick gel extraction kit (Qiagen Sciences, MD). The purified p53 cDNA fragment was methylated in vitro using the CpG specific SssI DNA methylase (New England Biolabs) using S-adenosyl-L-methionine as methyl donor. A control reaction omitted S-adenosyl-L-methionine. The extent of methylation was validated by demonstrating complete digestion of an aliquot of the methlyation reaction product with HpaII. After methylation, the fragment was isolated using a QIAquick PCR purification kit and used in mutagenesis assays.
p53 Reporter Gene Assay
Unmethylated or methylated p53 cDNA (500 ng) was treated for 1 h at 37°C with the indicated concentrations of (±)-anti-BPDE in 100 mM potassium phosphate (pH 7.0) in a reaction volume of 50 μl. Then 10 μl of the reaction mixture plus 40 μg of carrier DNA (herring testis DNA) was precipitated with 2.5 volumes of ethanol and 0.3 M sodium acetate at −80°C for 1–2 h. The DNA was isolated by microcentrifugation and washed twice with 70% ethanol. The pellets were then dried and suspended in TE buffer and used in yeast transformations. Where indicated, incubations containing PAH o-quinones were supplemented with 100 μM CuCl2 plus 1 mM NADPH. Incubations were performed for 2 h at 37°C. After reactions, a mixture of 50–100 ng of the pSS16 gapped-vector (digested with HindIII/StuI, and purified by gel electrophoresis), 50–100 ng of treated p53 cDNA, and 100 μg carrier DNA (herring testis) were co-transformed into the yeast host strain yIG397 (grown to a OD600 0.6–0.9) using the lithium acetate procedure according to the YEASTMAKER™ Yeast transformation System Kit (CLONTECH). The cells were collected, resuspended in 150 μL of TE buffer and plated on synthetic minimal medium minus leucine plus minimal adenine (5 μg/mL) and incubated for 3 days at 35 °C. The yIG397 strain has an ADE2 reporter gene under the control of the p21 promoter stably integrated into its genome. Wild-type p53 stimulates the reporter gene expression whereas change-in-function mutations of p53 do not. Yeast colonies expressing wild-type p53 are white and yeast colonies expressing mutant p53 are red. Red colonies were clearly identifiable after three days at 30 °C but the color is more intense after an additional 2 days at 4 °C. The mutation incidence was expressed as: [(number of red colonies − number of spontaneous red colonies)/total number of colonies] × 100.
Recovery of p53 Plasmids from Yeast and DNA Sequencing
p53 expression plasmids were rescued from transformed yeast following lysis with glass beads (Sigma, St. Louis, MO), as described (39). Briefly, yeast colonies were cultured in 5 mL SC-leu for 2 days. An aliquot of the culture medium (1.5 mL) was transferred to a microfuge tube, the yeast was collected by centrifugation at 10,000 rpm for 10 s and resuspended in 0.1 mL 50 mM Tris-HCl (pH 8.0), 62.5 mM Na2EDTA, 2.5 M LiCl, and 4% Triton X-100. After addition of 0.1 mL of phenol:chloroform (1:1, v/v) and 0.2 g of acid-washed glass beads (Sigma, St. Louis, MO) the cells were disrupted by vigorous vortexing for 2 min. The upper aqueous phase was removed following centrifugation at 12,000 rpm for 2 min. The DNA was precipitated, by adding 0.2 ml ethanol. After washing with 70% ethanol and drying, the DNA was dissolved in 20 μl Tris-EDTA pH 8.0. Plasmid DNA was checked for the presence of a p53 insert by restriction digestion with Sac I and Kpn I. Plasmid DNA (1 μl) containing insert was used for transformation into XL-1 blue electro-competent E.coli cells (Stratagene, Cedar Creek, TX). The transformation was carried out using a Bio-Rad (Hercules, CA) Gene Pulser Electroporator according to the instruction manual.
The DNA binding domain of p53 (codons 102 to 292) was sequenced on both strands with S6 (5′-dCTGGGACAGCCAAGTCTGT-3′) and R6 (5′-dCCTCATTCAGCTCTCGGAA-3′) primers. These primers allow double stranded sequencing from amino acids 126 to 339 encompassing almost all of the DNA-binding domain. Sequencing was performed on an Applied Biosystems 373A automated sequencer in the DNA sequencing facility at the Cell Center at the University of Pennsylvania School of Medicine.
Data analysis
To assess differences in the incidence of mutation across groups, a Wilcoxon rank sum test was performed. The following analysis was carried out to determine the significance of the base substitutions observed: There are 12 possible base substitutions that can occur. Each of the four bases could be mutated to any one of the remaining three. Since the mutagenicity assay does not distinguish whether the initial hit occurred on the coding or non-coding strand the number of possible base substitutions is effectively reduced to six: G →A (transition), G→C (transversion), G→T (transversion), A→G (transition), A→C (transversion) and A→T (transversion). The incidence of other mutations were also considered in the analysis, insertions and deletions, as well as none found. The none-found group represents false-positives. These most likely represent mutations that exist in p53 that lie outside the boundaries of the sequencing primers. Thus, there are 9 possible mutation outcomes that can occur. If these outcomes occur randomly, then all 9 should occur with equal frequency (1/9 = 11.1%). To test whether the observed distribution deviates from the assumed random distribution, a goodness of fit χ2 test, with 8 degrees of freedom, was performed.
Comparisons performed also asked the question of whether the frequency of mutations for the solvent control were similar or different with respect to those seen with a mutagen (e.g., (±)-anti-BPDE or BP-7,8-dione), and were conducted using a chi-square test or a Fisher’s exact test when required because of small sample sizes. Significance of coding strand bias was assessed by comparing the observed percentage of mutation in the coding strand with an incidence of 50%. All tests were two sided and conducted at a significance level of 0.05.
Results
PAH o-Quinones as Redox-Active Mutagens for p53
The p53 mutagenicity assay uses a yeast reporter system to score for a functional protein based on p53 transcriptional activation of the yeast ADE2 gene (34,35,38). Wild-type p53 cDNA is first exposed to a mutagen in vitro and then the treated fragment is co-transfected with a gapped vector in yeast. In vivo, the two DNA fragments undergo homologous recombination to form a functional plasmid that expresses p53. Wild type p53 binds to a stably integrated ADE2 reporter gene driven by a p53 dependent promoter. Expression of ADE, in an otherwise ade2- background generates a white yeast colony. Transcriptionally incompetent p53 mutants fail to transcribe ADE2 leading to red colonies. Plasmid DNA is then extracted and sequenced. The assay generates three types of data, potency, patterns and spectrum. From the frequency of red colonies, the potency of mutagens can be compared. The DNA sequence is then analyzed to obtain the two additional parameters. Analysis of the specific base pair change yields the pattern of mutations while plots of mutations against codon number yields the spectrum of mutations.
The compounds tested are shown in Figure 1. Three are o-quinones benz[a]anthracene-3,4-dione (BAQ or BA-3,4-dione) and dimethylbenz[a]anthracene-3,4-dione (DMBAQ or DMBA-3,4-dione). We previously analyzed benzo[a]pyrene-7,8-dione (BPQ or BP-7,8-dione) and N-methyl-N′-nitro-N-nitroso-guanidine (MNNG) in this assay system, so they were used as control compounds for the experiments in this study (34). At concentrations as high as 20 μM only modest mutation frequencies (1–2% red colonies) were observed from all three o-quinones. By contrast when the system was supplemented with NADPH and CuCl2 a concentration dependent increase in mutation frequency was observed using nanomolar concentrations of o-quinone (31–375 nM), in good agreement with previous data using BPQ (Figure 2). These data suggest that redox-cycling is essential to reveal the mutagenic potential of the PAH o-quinones.
Figure 1.

Metabolic formation of diol epoxides and PAH o-quinones.
Figure 2.
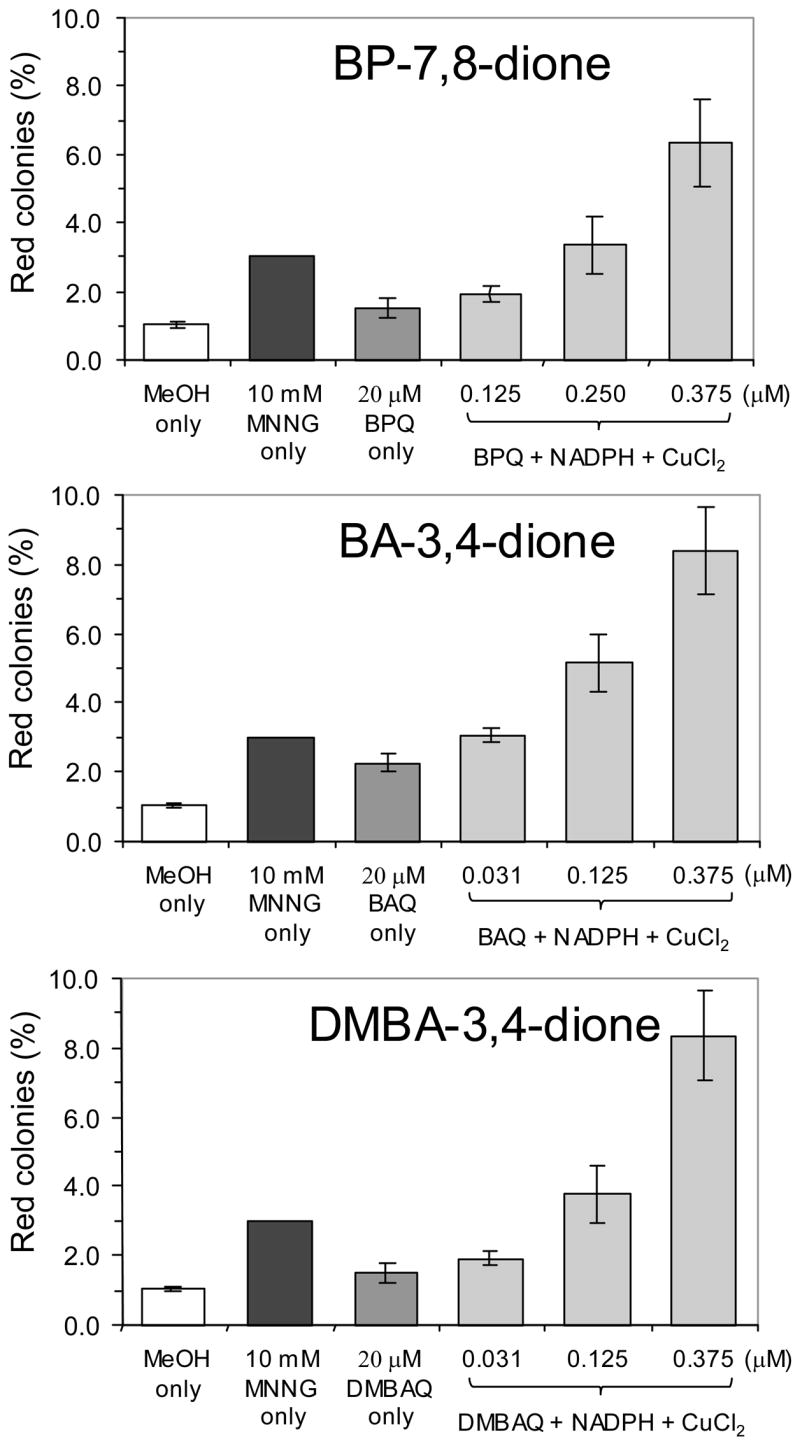
Comparison of the mutation frequencies of PAH o-quinones. Data is presented as percentage of red colonies vs. the treatment indicated. Incubation conditions are described in Materials and Methods.
Mutational Patterns Observed with PAH o-Quinones
To analyze the pattern and spectrum of mutations, we rescued the plasmid DNA from red colonies, propagated the plasmids in E. coli and then sequenced the DNA binding domain of p53. DNA was not isolated from BPQ treated cells, but the earlier data are plotted for comparison. In most instances (~80%), only single point mutations were detected in the DNA binding domain. The changes in the coding sequence are plotted with complementary base changes below the base line. For each of the PAH o-quinones, the pattern was dominated by G>T transversions (or its complement C>A) (44% for BAQ and 42% for DMBAQ) (p < 0.0001) and by G>A transitions (or its complement C>T) (28% for BAQ and 36% for DMBAQ) (Figure 3). These values compare favorably with the incidence of G to T transversions seen previously with BP-7,8-dione (46%). There was a small strand bias with 13 G>T transversions vs 12 C>A transversions for BAQ (p=.8415) and 18 G>T transversions vs 11 C>A transversions for DMBAQ (p=.0278), suggesting that transcription coupled repair was uncommon. The mutation pattern obtained from BAQ and DMBAQ were thus essentially indistinguishable from BPQ and all were remarkably similar to the patterns observed in lung cancer.
Figure 3.
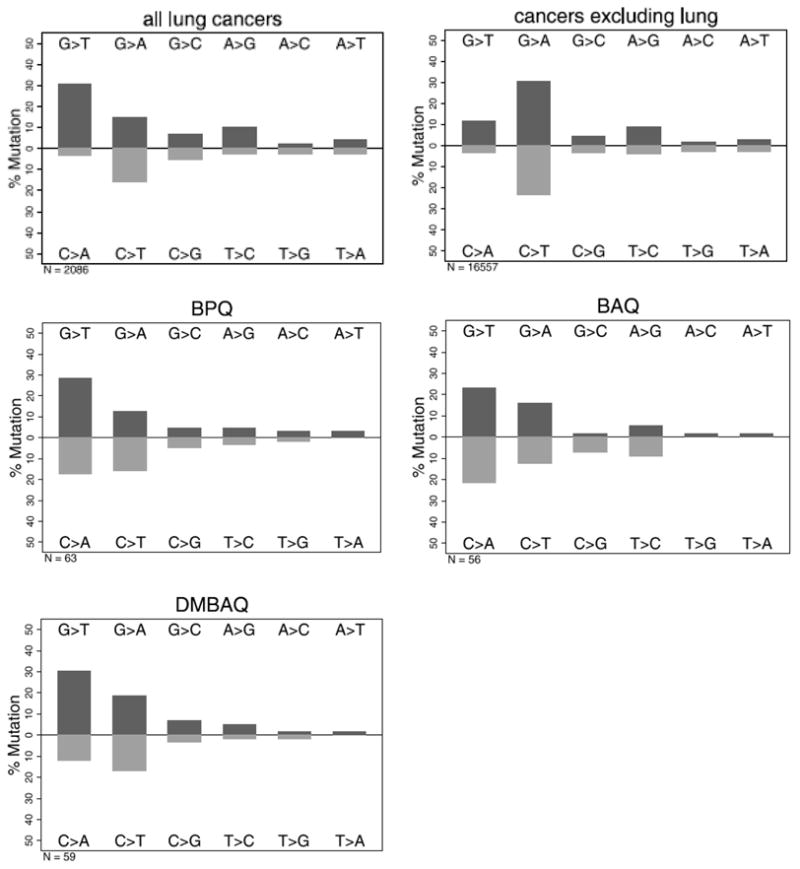
Patterns of mutations induced in p53 by PAH o-quinones. Data are presented as % mutation for the given base change. The twelve possible changes are reduced to six when complimentary changes are plotted together since the original base lesion cannot be determined from the mutant sequence. For comparison, the base substitution data for lung cancer (2,086 total substitutions) and all other cancers excluding lung cancer (16,557 total substitutions) are shown.
Mutational Spectra with PAH o-Quinones
For each of the PAH o-quinones tested as mutagens, the occurrence of the single point mutations were plotted against codon number to obtain a mutational spectrum (Figure 4). The region sequenced was from codons 126 to 339 and encompasses most of the DNA binding domain (amino acids 102 to 292). An analysis of the IARC database release R10 identified 23 codons that account for ~50% of the reported mutations in lung cancers. If the 23 hotspots were assumed to be mutated randomly, then they would be predicted to be hit 23/213 or 10.8% of the time. We observed 10/56 (18%) hotspot mutations with BAQ (p=0.09) and 15/59 (25%) hotspot mutations with DMBAQ (p=.0003), which were similar to the frequency we reported with BPQ 17/63 (27%), although the frequencies of hotspots for BAQ were of borderline statistical significance over a random distribution (Table 1). Tables of supplemental data (Supporting Information, Tables S1–S6) are provided which lists the mutations observed for each compound by codon number, the nucleotide change observed, the sequence context (5′ and 3′-codons), the type of mutation, and the amino acid substitution that results. In conclusion, under redox cycling conditions, the PAH o-quinones can be highly mutagenic causing a pattern of mutations that closely matches what is seen in lung cancer, and a spectrum of mutations with a statistically significant bias towards mutations at hotspots.
Figure 4.

Mutation spectra induced in p53 by PAH o-quinones. The occurrence of point mutations are plotted against codon number for each PAH o-quinone tested under redox cycling conditions. For comparison, spectra of single base substitutions for lung cancer (2,027 singlet mutations) and all cancer excluding lung cancer (16,117 singlet mutations) are shown. Data was extracted from the p53 IARC database R10 release (50).
Table 1.
Frequency of mutations at lung hotspots by indicated mutagens. For the analysis of hotspots, only base substitution mutations were counted. Hotspots were defined as the 23 mutations that account for 50% of the mutations reported in lung cancer in the IARC data base release R10 (50). In order of frequency, these are codons: 248, 273, 249, 245, 158, 157, 175, 282, 179, 220, 242, 163, 176, 154, 280, 298, 266, 281, 159, 173, 193, 237 and 244. P values indicate the probability that the observed frequency differs from a random distribution of 10.8 % 23 hotspots out of 213 amino acids sequenced. Data for BPQ is from (34) reanalyzed using IARC data base release R10.
| BPQ | BAQ | DMBAQ | BPDE | me-BPDE | |
|---|---|---|---|---|---|
| Observed/Total | 17/63 | 10/56 | 15/59 | 4/27 | 6/41 |
| Frequency (%) | 27 | 18 | 25 | 14.8 | 14.6 |
| Expected (%) | 10.8 | 10.8 | 10.8 | 10.8 | 10.8 |
| P | <0.0001 | 0.09 | <0.0001 | 0.5 | 0.43 |
Mutagenesis of p53 with (±)-anti-BPDE
We initially found it difficult to score (±)-anti-BPDE induced mutations since this compound yielded low numbers of yeast colonies perhaps by interfering with the crucial recombination event. This problem was alleviated if the p53 and carrier DNA were precipitated and washed to remove the mutagens. Concentration dependent mutation of p53 was seen with (±)-anti-BPDE (0–20 μM) irrespective of whether the p53 fragment or plasmid containing the p53 insert was treated. In both instances concentrations as high as 10 μM were necessary to see mutation frequencies similar to those reported for the PAH o-quinones. In addition, at concentrations above 5–10 μM (±)-anti-BPDE, the number of total yeast colonies observed in the assay fell precipitously, suggesting that (±)-anti-BPDE treatment of the plasmid was cytotoxic to the yeast host strain (Figure 5). On a molar basis (±)-anti-BPDE was found to be from 25 to 80 fold less mutagenic than the three quinones.
Figure 5.
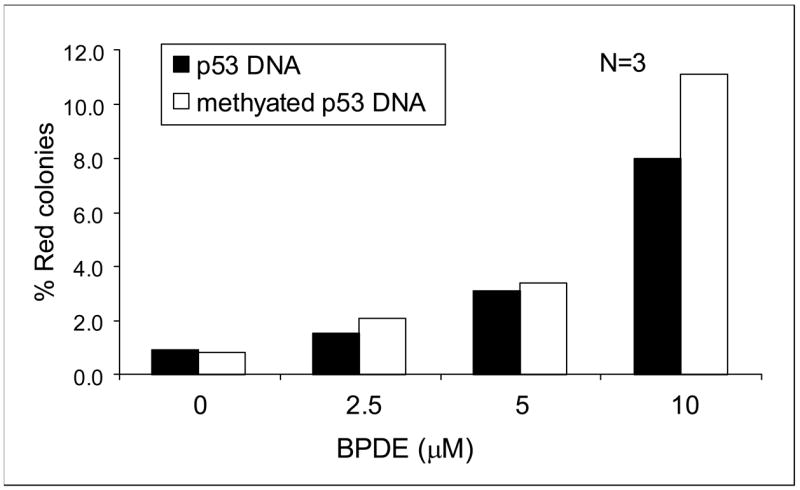
Mutation frequency of (±)-anti-BPDE, on unmethylated vs. methylated DNA.
As before, the red colonies were isolated and the p53 plasmid rescued for sequencing. We isolated 32 plasmids for sequence analysis. Again >90% of the mutated p53 contained a single point mutation in the DNA binding domain. The spectrum was G to C (63%) > G to A (18%) > G to T (15%). Importantly, 96% of the mutations were at G:C base pairs and this is consistent with the preferential formation of anti-BPDE-N2-dGuo adducts (9–11) (Figure 6). Only 4 of the 27 (14.8%) (p= 0.5) mutations were at hotspot codons, demonstrating a random distribution (Figure 7).
Figure 6.
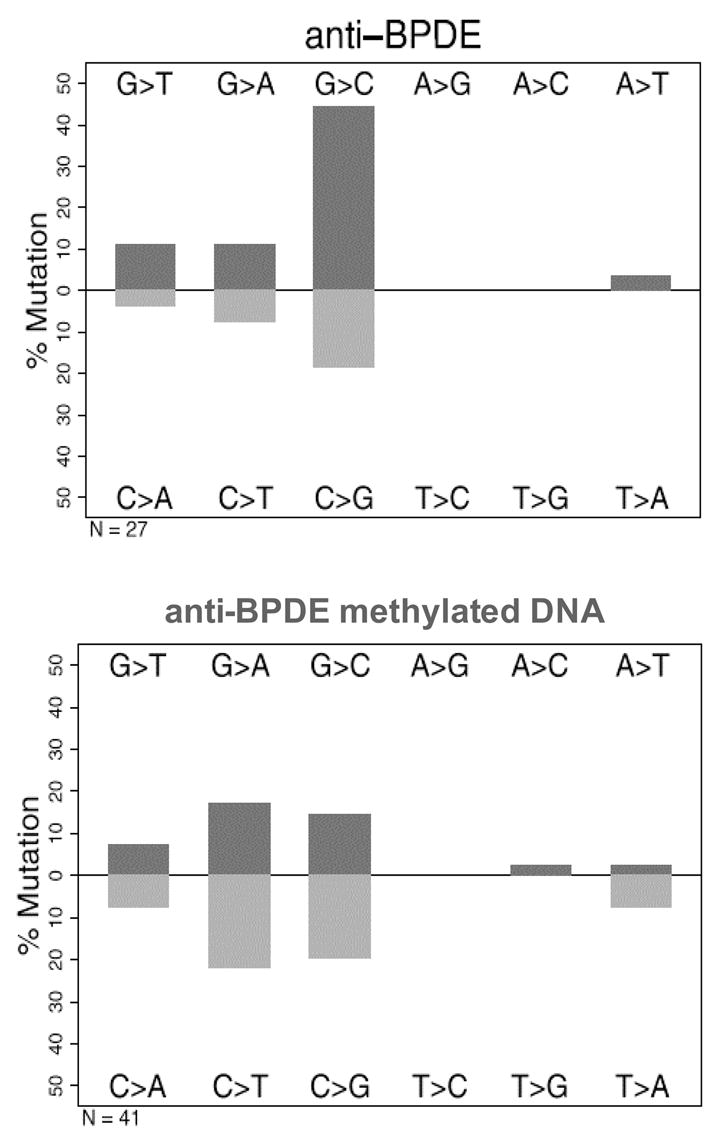
Mutational patterns of (±)-anti-BPDE, induced in unmethylated p53 and methylated p53.
Figure 7.
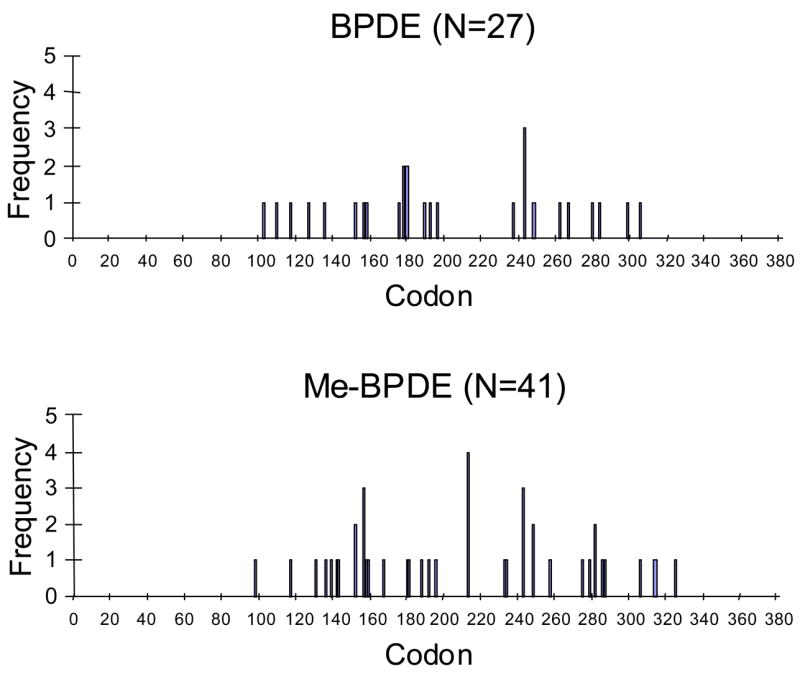
Mutational spectra of (±)-anti-BPDE, induced in unmethylated p53 and methylated p53.
Since methylation of p53 at CpG islands enhances adduct formation at most of the major hotspots (22,23), we tested the mutagenicity of (±)-anti-BPDE with methylated p53 cDNA. We methylated DNA with Sss1 DNA methylase and then confirmed methylation by digesting the DNA with the methylation sensitive restriction endonuclease HpaII. In mutagenesis assays we found that there was essentially no difference in the mutagenic potency of (±)-anti-BPDE irrespective of whether methylated DNA or unmethylated DNA was used (Figure 5). DNA sequencing of 43 mutants showed that the mutation pattern was G to A (37%) > G to C (34%) > G to T (15%). The pattern was somewhat similar to the unmethylated DNA except that there was a significant increase in G>A transitions (p≤ .0001)and the G>C transversions were somewhat reduced (Figure 6). Methylation did not affect the number of mutations at CpG islands (11/30 for methylated DNA versus 6/13 for unmethylated DNA).
Point mutations observed in methylated p53 were plotted against codon number to yield mutational spectrum. Hot-spots were hit 6/41 or 14.6% of the time by (±)-anti-BPDE with the methylated p53 (p=.43), which was again, not significantly different from a random distribution. Tables of supplemental data (Supporting Information, Tables S4–S5) are provided which lists the mutations observed for (±)-anti-BPDE in unmethylated and methylated p53 by codon number, the nucleotide change observed, the sequence context (5′ and 3′-codons), the type of mutation, and the amino acid substitution that results.
Discussion
We have conducted a direct comparison of the mutagenic potential of three representative PAH o-quinones with (±)-anti-BPDE in a yeast mutagenesis assay. The yeast assay provides three points of comparison, mutagenic frequency, pattern of mutations and the spectrum of mutations. The mutagenic frequency measures the potency of the mutagens, the pattern of mutations measures the specific base substitutions and the spectrum of mutations maps the specific codons that are changed. Mutations in p53 are among the most extensively documented in any oncogene and numerous studies have supported that distinct patterns and spectra of mutations occur in lung cancer (18). Neither group of PAH compounds caused mutations that precisely matched those in the p53 data bases, but by all three of these measures, the PAH o-quinones, through their ability to generate reactive oxygen, yielded results that were a closer match to those reported in lung cancer than (±)-anti-BPDE. Three different o-quinones were 25 to 80 times more mutagenic than (±)-anti-BPDE, the pattern of mutations caused by all three were dominated by G>T transversions, whereas those caused by (±)-anti-BPDE were dominated by G>C transitions and finally, the spectrum of mutations caused by all three o-quinones had about twice as many hits at p53 cancer hotspots than (±)-anti-BPDE.
Each of the PAH o-quinones were weak direct-acting mutagens yielding low frequencies of mutation at concentrations of 20 μM. However, a significant increase in potency was observed when redox cycling conditions were employed (NADPH plus CuCl2). Under identical redox-cycling conditions, using salmon testis DNA, a significant amount of 8-oxo-dGuo is generated which is formed in a concentration dependent manner (28). Moreover, ROS scavengers attenuate p53 mutagenesis mediated by the PAH o-quinones and eliminate the formation of 8-oxo-dGuo observed under redeox-cycling conditions. Taken together these data suggest that the primary lesion responsible for the G to T transversions observed with the PAH o-quinones is 8-oxo-dGuo. By contrast, micromolar concentrations of (±)-anti-BPDE were required to observe mutations in p53.
The pattern of mutations seen in the mammalian HPRT gene by (±)-anti-BPDE are usually G>T transversions, which occur 70–80% of the time with most of the remainder being A>C transversions (25). One study found that (±)-anti-BPDE also caused G>T transversions in the same yeast assay that we use. However, another study found a preference for G>C transversions in a yeast mutagenesis assay with (±)-anti-BPDE (40). The preference for G to C over G to T transversions may be governed by the trans-lesional by pass polymerase Polζ which has been shown to preferentially incorporate G opposite an (+)-anti-BPDE-N2-dGuo adduct in yeast strains proficient in mutagenesis (40). Thus, while it was somewhat unexpected to observe fewer G>T mutations by (±)-anti-BPDE, it may reflect the repertoire of trans-lesion bypass polymerases that predominate in yeast. Our observation that about 90% of the mutations seen with (±)-anti-BPDE were at GC basepairs is consistent with the formation of (+)-anti-BPDE-N2-dGuo adducts, which are most often observed with this compound. For o-quinones, the probable base lesion is 8-oxo-dGuo, which is readily produced by these compounds under redox cycling conditions (28) and if unrepaired can cause G to T transversions (41).
The transversions in the HPRT gene usually show a strand bias similar to that observed in p53 in lung cancer where the G>T transversions dominate on the non-transcribed strand suggesting that transcription coupled repair can play a dominant role in repair. However, we have not yet observed a significant strand bias with any of the compounds tested in yeast. We hypothesize that the p53 gene is not actively transcribed while it is undergoing recombination into the gapped vector during the assay. Moreover, if the predominant lesion responsible for p53 mutation is 8-oxo-dGuo, strand bias may occur in lung cancer due to the loss of one allele of hOGG1 which preferentially repairs the non-transcribed strand (42).
The mutational spectra with all the PAH o-quinones differed from that observed with (±)anti-BPDE in that the occurrence of mutations was non-random. The p53 structure can be divided into five domains and when the data from the three o-quinones are combined there was clustering of the mutations into regions that form contacts with the DNA and contain hot-spots (domains III, IV and V). But when the spectra are plotted for G>T transversions, even the clustering becomes statistically insignificant. We suggest that the mutational spectra is not faithfully replicated because the biological selection in yeast is not stringent enough.
By contrast, the mutational spectrum with (±)-anti-BPDE appeared to be random, and this was true irrespective of whether the p53 was methylated or not. While there was little effect of methylation on the mutagenic potency of (±)-anti-BPDE, we did find that upon methylation there was a shift in the pattern of mutations form G to C transversions to G to A transitions.
While there was a significant bias towards mutations at hotspots with the PAH o-quinones, there was a lack of correlation between the mutational spectra from the yeast based assays and the spectra seen in lung cancers. This may be due to the fact that the yeast assays are too sensitive and score almost any loss of function in p53. The most common mutations observed in cancers are point mutations that generate a full length protein more stable than the wild type protein. Significantly, these mutants have dominant negative effects when tested in transcription assays and some of the most potent dominant negatives are hotspot mutants. Selection for dominant negative mutants may occur during tumorigenesis since patients with dominant negative mutations have an earlier onset of glioblastoma and a poorer prognosis (43). Additionally, while several studies suggest that diol-epoxide carcinogens target hotspots, there are very few lung cancer specific hotspots (eg 157 is lung specific, but even this site has been questioned as discussed below), as almost all of the hotspot mutations occur in most other tumors as well. Together, these data suggest that during tumorigenesis there is a strong selection for dominant negative p53 mutants. However, most yeast screens do not distinguish between dominant negative and recessive mutations. In one yeast study when dominant-negative p53 mutations alone were scored, there was a clustering of mutations around the hot-spot codons, especially in domains IV and V (44). Thus by employing a selection for dominant negative mutations in mutagenesis studies, it should be possible to recapitulate both the pattern and spectra of mutations found in p53 using a yeast based assay.
Another component of hot-spot bias may be seen if some types of mutations are nonconservative and therefore more likely to cause a change in function. This could occur if the codon changes caused by the G to T transversions are more prone to cause non-conservative changes than those caused by G to A or G to C mutations, since they would be more likely to be scored in the transcription assay. For example, codon 157 only appears specific for lung cancer because G to T transversions at this site cause non-conservative substitutions while G to A and G to C mutations cause conservative substitutions (45). Carcinogens that cause distinctive patterns may, in some cases, appear to show a bias towards hotspot codons without specifically targeting them.
Our study provides additional support for the quinone route of carcinogenesis as an alternative mechanism to anti-diol epoxide formation of (+)-anti-BPDE N2-dGuo lesions. Our model of carcinogenesis would proceed as follows. PAH derived from tobacco smoke would be metabolically activated by AKRs to redox-active PAH-o-quinones in lung cells. The ROS produced during the redox-cycling of the o-quinones then overwhelms endogenous protective mechanisms to produce 8-oxo-dGuo lesions. The 8-oxo-dGuo, if left unrepaired, gives rise to G to T transversions. The lesions are not completely random due site specific intercalation of the quinones with DNA. The final spectrum is probably largely driven by genetic selection for the p53 mutations that provide the greatest growth advantage.
Whether this mechanism occurs will depend upon the expression levels of AKRs involved in the metabolic activation of PAH in lung cells. A consistent theme is emerging which suggests that this is the case. The AKR isoforms most involved in the formation of PAH o-quinones in lung cells would be AKR1A1 (aldehyde reductase) and AKR1C1-AKR1C3 (hydroxysteroid dehydrogenases) (46,47). Differential display of 317/372 NSCLC patients showed that AKR1C1/AKR1C2 were over expressed by up to 50-fold in tumor tissues versus the adjacent normal tissue and this was indicative of poor prognosis (30). In addition three independent studies showed that AKR1C1 was overexpressed in NSCLC and SCLC (31), bronchial epithelial cells of NSCLC (32), or oral cancer cells (33) by Affymetrix array. Expression levels were among the highest of all genes examined and correlated with smoking history. These studies were validated in human lung adenocarcinoma cells (A549) which were shown to overexpress AKR1C1-AKR1C3 based on RT-PCR, Northern analysis and functional enzyme assay (48). Additionally, these cells were able to convert PAH trans-dihydrodiols to PAH-o-quinones when lysates were supplemented with the NADP+ cofactor (48). Finally, 1-hydroxychavicol the active ingredient of Acrea Quid (ingested with chewing tobacco) was found to act as a monofunctional inducer in buccal cells and induce AKR1C1. Treatment of these cells with BP led to a reduction in stable (+)-anti-BPDE-N2-dGuo adducts and an increase in 8-oxo-dGuo (49), consistent with the proposed mechanism.
Acknowledgments
This work was supported by grants R01 CA39504 and P01 CA92537 awarded to T.M.P and by grant R01 GM48241 and pilot project support from 1P30 ES013508-01 to J.F. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH. We thank the NCI Chemical Carcinogen Standard Reference Repository for (±)-anti-BPDE.
Footnotes
Abbreviations: (±) anti-BPDE, anti-benzo[a]pyrene diol epoxide; BPQ, benzo[a]pyrene-7,8-dione; BAQ, benz[a]anthracene-3,4-dione; DMBAQ, dimethylbenz[a]anthracene-3,4-dione; MNNG, N-methyl-N′-nitro-N-nitroso-guanidine; PAH, polycyclic aromatic hydrocarbons.
Supporting Information Available:
Tables of supplemental data S1–S6. These tables list the mutations observed for each PAH o-quinone, BPDE and MNNG in unmethylated or methylated p53 cDNA by codon number, the nucleotide change observed, the sequence context (5′ and 3′-codons), the type of mutation, and the amino acid substitution. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LA, Schrag D, Jamison PA, Jemal A, Wu XC, Friedman C, Harlan L, Warren J, Anderson RM, Pickle LW. Annual report to the nation on the status of cancer, 1975–2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- 2.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. G.H.A Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 3.Gelboin HV, T’so POP. Benzo[a]pyrene metabolism, activation and carcinogenesis: Role and regulation of mixed function oxidases and related enzymes. Physiol Rev. 1980;60:1007–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 4.Sutter TR, Tang YM, Hayes CL, Wo Y-Y, Jabs PW, Li X, Yin H, Cody CW, Greenlee WF. cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem. 1994;269:13092–13099. [PubMed] [Google Scholar]
- 5.Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P450 1B1. Cancer Res. 1996;56:2979–2984. [PubMed] [Google Scholar]
- 6.Penning TM, Burczynski ME, Hung C-F, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 7.Cavalieri EL, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 8.Jennette KW, Jeffery AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Nucleoside adducts from the in vitro reaction of benzo[a]pyrene-7,8-dihydrodiol-9,10-oxide or benzo[a]pyrene-4,5-oxide with nucleic acids. Biochemistry. 1977;16:932–938. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]
- 9.Jeffrey AM, Jennette KW, Blobstein SH, Weinstein IB, Beland FA, Harvey RG, Kasai H, Miura I, Nakanishi K. Benzo[a]pyrene-nucleic acid derivative found in vivo: Structure of a benzo[a]pyrene-tetrahydrodiol epoxide-guanosine adduct. J Amer Chem Soc. 1976;98:5714–5715. doi: 10.1021/ja00434a060. [DOI] [PubMed] [Google Scholar]
- 10.Koreeda M, Moore PD, Wislocki PG, Levin W, Conney AH, Yagi H, Jerina DM. Binding of benzo[a]pyrene-7,8-diol-9,10-epoxides to DNA, RNA and protein of mouse skin occurs with high stereoselectivity. Science. 1978;199:778–781. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- 11.Osborne MR, Beland FA, Harvey RG, Brookes P. The reaction of (±)-7α,8β-dihydroxy-9β,10β-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene with DNA. Int J Cancer. 1976;18:362–368. doi: 10.1002/ijc.2910180315. [DOI] [PubMed] [Google Scholar]
- 12.Shou M, Harvey RG, Penning TM. Reactivity of benzo[a]pyrene-7,8-dione with DNA. Evidence for the formation of deoxyguanosine adducts. Carcinogenesis. 1993;14:475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- 13.McCoull KD, Rindgen D, Blair IA, Penning TM. Synthesis and characterization of depurinating adducts of polycyclic aromatic hydrocarbon o-quinones. Proc Am Assoc Cancer Res. 1998;39:637. doi: 10.1021/tx980182z. [DOI] [PubMed] [Google Scholar]
- 14.Balu N, Padgett WT, Lambert GR, Swank AE, Richard AM, Nesnow S. Identification and characterization of novel stable deoxyguanosine and deoxyadenosine adducts of benzo[a]pyrene-7,8-quinone from reactions at physiological pH. Chem Res Toxicol. 2004;17:827–838. doi: 10.1021/tx034207s. [DOI] [PubMed] [Google Scholar]
- 15.Devanesan PD, RamaKrishna NVS, Todorovic R, Rogan EG, Cavalieri EL, Jeong H, Jankowiak R, Small GJ. Idenitification and quantitation of Benzo[a]pyrene-DNA adducts formed by rat liver microsomes in vitro. Chem Res Toxicol. 1992;5:302–309. doi: 10.1021/tx00026a024. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Devanesan PD, Higginbotham S, Ariese F, Jankowiak R, Small GJ, Rogan EG, Cavalieri EL. Expanded analysis of benzo[a]pyrene-DNA adducts formed in vitro and in mouse skin: their significance in tumor initiation. Chem Res Toxicol. 1996;9:897–903. doi: 10.1021/tx960004a. [DOI] [PubMed] [Google Scholar]
- 17.Beroud C, Soussi T. p53 gene mutation: software and database. Nucleic Acids Res. 1998;26:200–204. doi: 10.1093/nar/26.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 19.Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat. 2003;21:229–239. doi: 10.1002/humu.10177. [DOI] [PubMed] [Google Scholar]
- 20.Hussain SP, Amstad P, Raja K, Sawyer M, Hofseth L, Shields P, Hewer A, Phillips DH, Ryberg D, Huagen A, Harris CC. Mutability of p53 hotspot codons to benzo[a]pyrene diol epoxide (BPDE) and the frequency of p53 mutations in nontumorous human lung. Cancer Res. 2001;61:6350–6355. [PubMed] [Google Scholar]
- 21.Denissenko MF, Pao A, Tang M-S, Pfeifer GP. Preferential formation of Benzo[a]pyrene adducts at lung cancer mutation hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 22.Denissenko MF, Chen JX, Tang M-S, Pfeifer GP. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc Natl Acad Sci USA. 1997;94:3893–3898. doi: 10.1073/pnas.94.8.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoon JH, Smith LE, Feng Z, Tang M, Lee CS, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 2001;61:7110–7117. [PubMed] [Google Scholar]
- 24.Yoon JH, Lee CS, Pfeifer GP. Simulated sunlight and benzo[a]pyrene diol epoxide induced mutagenesis in the human p53 gene evaluated by the yeast functional assay: lack of correspondence to tumor mutation spectra. Carcinogenesis. 2003;24:113–119. doi: 10.1093/carcin/24.1.113. [DOI] [PubMed] [Google Scholar]
- 25.Wei SJ, Chang RL, Bhachech N, Cui XX, Merkler KA, Wong CQ, Hennig E, Yagi H, Jerina DM, Conney AH. Dose-dependent differences in the profile of mutations induced by (+)-7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in Chinese hamster V-79 cells. Cancer Res. 1993;53:3294–3301. [PubMed] [Google Scholar]
- 26.Lu R, Nash HM, Verdine GL. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr Biol. 1997;7:397–407. doi: 10.1016/s0960-9822(06)00187-4. [DOI] [PubMed] [Google Scholar]
- 27.Penning TM, Ohnishi ST, Ohnishi T, Harvey RG. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase. Chem Res Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- 28.Park JH, Gopishetty S, Szewczuk LM, Troxel AB, Harvey RG, Penning TM. Formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) by PAH o-quinones: involvement of reactive oxygen species and copper(II)/copper(I) redox cycling. Chem Res Toxicol. 2005;18:1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JH, Troxel AB, Harvey RG, Penning TM. Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem Res Toxicol. 2006;19:719–728. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu NY, Ho H-C, Chow K-C, Lin T-Y, Shih C-S, Wang L-S, Tsai C-M. Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res. 2001;61:2727–2731. [PubMed] [Google Scholar]
- 31.Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, Taniguchi H, Ishikawa S, Ito H, Yamamoto S, Iwanari H, Hironaka M, Ishikawa Y, Niki T, Sohara Y, Kodama T, Nishimura M, Fukayama M, Dosaka-Akita H, Aburatani H. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res. 2005;11:1776–1785. doi: 10.1158/1078-0432.CCR-04-1238. [DOI] [PubMed] [Google Scholar]
- 32.Woenckhaus M, Klein-Hitpass L, Grepmeier U, Merk J, Pfeifer M, Wild P, Bettstetter M, Wuensch P, Blaszyk H, Hartmann A, Hofstaedter F, Dietmaier W. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J Pathol. 2006 doi: 10.1002/path.2039. In press. [DOI] [PubMed] [Google Scholar]
- 33.Nagaraj NS, Beckers S, Mensah JK, Waigel S, Vigneswaran N, Zacharias W. Cigarette smoke condensate induces cytochromes P450 and aldo-keto reductases in oral cancer cells. Toxicol Lett. 2006;165:182–194. doi: 10.1016/j.toxlet.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu D, Berlin JA, Penning TM, Field J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem Res Toxicol. 2002;15:832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- 35.Yu D, Penning TM, Field JM. Benzo[a]pyrene-7,8-dione is more mutagenic than anti-BPDE on p53 and is dependent upon the generation of ROS. Polycyclic Aromatic Compounds. 2002;22:881–891. [Google Scholar]
- 36.Boiteux S, Gellon L, Guibourt N. Repair of 8-oxoguanine in Saccharomyces cerevisiae: interplay of DNA repair and replication mechanisms. Free Radic Biol Med. 2002;32:1244–1253. doi: 10.1016/s0891-5849(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 37.Harvey RG, Dai Q, Ran C, Penning TM. Synthesis of the o-quinones and other oxidized metabolites of polycyclic aromatic hydrocarbons implicated in carcinogenesis. J Org Chem. 2004;69:2024–2032. doi: 10.1021/jo030348n. [DOI] [PubMed] [Google Scholar]
- 38.Ishioka C, Frebourg T, Yan Y-X, Vidal M, Friend SH, Schmidt S, Iggo R. Screening patients for heterozygous p53 mutations using a functional assay in yeast. Nat genet. 1993;5:124–129. doi: 10.1038/ng1093-124. [DOI] [PubMed] [Google Scholar]
- 39.Guthrie C, Fink GR, editors. Methods in Enzymology. Vol. 194. Academic Press, Inc; New York: 1998. Guide to yeast genetics and molecular biology. [PubMed] [Google Scholar]
- 40.Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, Wang Z. Mutagenesis of benzo[a]pyrene diol-epoxide in yeast: requirement for DNA polymerase ξ and involvement of DNA polmerase η. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 41.Breen AP, Murphy JP. Reactions of oxyl radicals with DNA. Free Radic Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 42.Le Page F, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc Natl Acad Sci USA. 2000;97:8397–8402. doi: 10.1073/pnas.140137297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marutani M, Tonoki H, Tada M, Takahashi M, Kashiwazaki H, Hida Y, Hamada J, Asaka M, Moriuchi T. Dominant-negative mutations of the tumor suppressor p53 relating to early onset of glioblastoma multiforme. Cancer Res. 1999;59:4765–4769. [PubMed] [Google Scholar]
- 44.Brachmann RK, Vidal M, Boeke JD. Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc Natl Acad Sci USA. 1996;93:4091–4095. doi: 10.1073/pnas.93.9.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodin SN, Rodin AS. Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci USA. 2000;97:12244–12249. doi: 10.1073/pnas.180320897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry. 1998;37:6781–6790. doi: 10.1021/bi972725u. [DOI] [PubMed] [Google Scholar]
- 47.Palackal NT, Burczynski ME, Harvey RG, Penning TM. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: potential role in polycyclic aromatic hydrocarbon activation. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- 48.Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J Biol Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- 49.Tang DW, Chang KW, Chi CW, Liu TY. Hydroxychavicol modulates benzo[a]pyrene-induced genotoxicity through induction of dihydrodiol dehydrogenase. Toxicol Lett. 2004;152:235–243. doi: 10.1016/j.toxlet.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]