

Abstract
Insoluble protein fibrils resulting from the self-assembly of a conformational intermediate are implicated as the causative agent in several severe human amyloid diseases, including Alzheimer’s disease, familial amyloid polyneuropathy, and senile systemic amyloidosis. The latter two diseases are associated with transthyretin (TTR) amyloid fibrils, which appear to form in the acidic partial denaturing environment of the lysosome. Here we demonstrate that flufenamic acid (Flu) inhibits the conformational changes of TTR associated with amyloid fibril formation. The crystal structure of TTR complexed with Flu demonstrates that Flu mediates intersubunit hydrophobic interactions and intersubunit hydrogen bonds that stabilize the normal tetrameric fold of TTR. A small-molecule inhibitor that stabilizes the normal conformation of a protein is desirable as a possible approach to treat amyloid diseases. Molecules such as Flu also provide the means to rigorously test the amyloid hypothesis, i.e., the apparent causative role of amyloid fibrils in amyloid disease.
The accumulation of insoluble protein fibrils in human tissue is a common feature of neurodegenerative disease (1–3). In many cases the protein’s native conformation is destabilized by a genetic mutation or other environmental factors, typically resulting in a β-sheet-rich conformational intermediate (4, 5). These intermediates self-assemble into amyloid fibrils, such as the fibrils found in patients with the transthyretin (TTR)-associated amyloid diseases familial amyloid polyneuropathy (FAP) and senile systemic amyloidosis (SSA) (6, 7). Transthyretin is found in plasma and cerebrospinal fluid and is composed of four identical 127-amino acid β-sheet-rich subunits. The tetrameric structure binds and transports thyroxine (T4) and the retinol binding protein (8, 9). X-ray studies of TTR show two funnel-shaped binding sites for T4, each defined by a dimer–dimer interface (8, 10). In plasma, only 10–15% of TTR has T4 bound to it (Kas of 108 and 106 M−1, negative cooperativity), because thyroxine binding globulin is the major thyroxine carrier (Ka = 6 × 109 M−1) (11). However, in the cerebrospinal fluid, TTR is the main carrier of T4, with 75% of TTR binding T4 (12). Previous biophysical studies demonstrated that the mechanism of TTR amyloid fibril formation requires tetramer dissociation to a monomeric conformational intermediate (6, 7). This conformational intermediate, generated under conditions simulating the pH of a lysosome (5.5 ± 0.5), self-assembles, affording amyloid fibrils (lysosomes are intracellular organelles important for biopolymer degradation and implicated in amyloid fibril formation) (13, 14). Based on fiber diffraction and cryoelectron microscopy studies, the generated amyloid fibril is 130 Å in diameter and is made up of four parallel protofilaments oriented in a square section array, each having an apparent cross-β-helical structure (15).
In heterozygous individuals with FAP, TTR amyloid fibrils deposit in a variety of tissues, but generally not in the brain (16, 17). Amyloid isolated from FAP patients is composed primarily of variant TTR, implying that the variant subunits are less stable in their tetrameric form and therefore more amyloidogenic than wild-type TTR. The age of onset of FAP appears to be directly correlated to both the destabilizing effect of a given mutation on TTR tetramer stability and the increased rate of denaturation to the monomeric amyloidogenic intermediate (Fig. 1, Inset) (18–20). FAP results in several severe and often debilitating conditions by virtue of the neurotoxicity of amyloid and/or its ability to physically interfere with normal organ function (16, 21). Recent studies indicate that amyloidosis caused by the V122I TTR mutation, present in 2.2% of the African American population, is associated with late-onset heart disease in this population (22). Wild-type TTR fibrils are thought to be the disease-causing agent in SSA in patients typically >80 years of age (23). In SSA, it is not clear whether fibril formation accumulates over time or rapidly, after a triggering event. SSA is life-threatening in cases of cardiac infiltration (16).
Figure 1.

Inhibiting amyloid formation. The extent of TTR amyloid fibril formation (37°C) in the absence and presence of various concentrations of Flu probed by OD measurements at 330 nm (shown) and by quantitative Congo red binding (not shown, data virtually identical) (24). The inhibition of wild-type (pH 4.4), V30M (pH 5.0), and L55P (pH 5.0) TTR by variable concentrations of Flu are shown with the errors associated with these measurements. The pHs chosen are those associated with maximal amyloid fibril formation. The table (Inset) provides additional information including pHm, the pH midpoint of the tetramer monomer equilibrium, and the IC50, the concentration of Flu required for reduction of amyloid fibril formation to 50% of its maximum value.
We recently reported that thyroid hormone and derivatives thereof (e.g., 2,4,6-triiodophenol) strongly stabilize the normal native conformation of TTR and inhibit wild-type, V30M, and L55P amyloid formation under partially denaturing conditions simulating lysosomal acidity (24). The ligand-induced inhibition of TTR fibril formation may also operate in vivo, as thyroid-hormone binding to TTR in the cerebrospinal fluid apparently stabilizes TTR against fibril formation in the brain (24). Because thyroid-like molecules are undesirable as amyloid inhibitors (owing to their hormonal activity), we have set out to identify novel small-molecule inhibitors.
EXPERIMENTAL METHODS
Fibril Formation Assay.
Wild-type and variant TTR (L55P and V30M) were purified from an Escherichia coli expression system described previously (18, 19). Concentrated stock solutions of flufenamic acid (Flu) were prepared by dissolving the compounds in dimethyl sulfoxide. The stationary amyloid fibril inhibition assay described previously (7, 24) was used here . Briefly, a series of Eppendorf tubes filled with 200 mM sodium acetate-buffered 100 mM KCl solutions (0.5 ml) containing 5 μl of different concentrations of Flu were prepared at the desired pH. One-half ml of a 0.4 mg/ml TTR stock solution in 10 mM phosphate (pH 7.6), 100 mM KCl, and 1 mM EDTA was then added to the buffered inhibitor solution in each Eppendorf tube to obtain a final TTR concentration of 0.2 mg/ml. In addition, two control samples containing TTR with no inhibitor as well as Flu with no TTR were also evaluated. All solutions were incubated while motionless at 37°C for 72 h, and the extent of fibril formation was measured by OD at 330 nm and by using a quantitative Congo red binding assay; both assays were described in detail previously (7, 24). Preincubation of Flu with TTR before shifting the pH to amyloid-forming conditions exhibited strictly analogous results, implying the on rate for Flu binding to TTR is greater than the rate of denaturation to the amyloidogenic intermediate.
Ultracentrifugation Studies.
The effect of Flu on the quaternary-structure stability of wild-type TTR was evaluated by sedimentation velocity and sedimentation equilibrium ultracentrifugation methods using a temperature-controlled Beckman XL-I analytical ultracentrifuge equipped with an An60Ti rotor and photoelectric scanner. TTR samples in the presence or absence of 10.8 μM Flu were loaded into a double-sector cell and evaluated at 235 nm as described (24).
Isothermal Titration Calorimetry (ITC).
An isothermal titration of a 500 μM solution of Flu (in 10 mM phosphate, pH 7.6/100 mM KCl/1 mM EDTA) into a 20 μM TTR solution (in 10 mM phosphate, pH 7.6/100 mM KCl/1 mM EDTA) using an initial 2-μl injection followed by 24 10-μl injections at 25°C was performed. Integration of the thermogram and subtraction of the blank gives a binding isotherm that fits best to a model of two identical interacting sites demonstrating negative cooperativity. The data was fit by using four variables, K1, ΔH1, K2 and ΔH2 using the ITC data analysis in origin version 2.3 (Microcal Software, Northampton, MD).
Crystallization.
Crystals of TTR were grown by using the hanging-drop vapor diffusion method from solutions of 5 mg/ml protein (in 100 mM KCl/100 mM phosphate, pH 7.4/1 M ammonium sulfate) equilibrated against 2 M ammonium sulfate. To prepare the TTR-drug complex, crystals were soaked for 5 wk with a 10-fold molar excess of Flu to ensure full saturation of both binding sites.
Structure Determination.
X-ray data for the apo- and complexed crystal forms of TTR were collected on a DIP2030 imaging plate system (MAC Science, Yokohama, Japan). The crystals were isomorphous with unit-cell dimensions of a = 43.2 Å, b = 85.3 Å, and c = 64.5 Å. They belong to the space group P21212 and contain one-half of the homotetramer in the asymmetric unit. Data were reduced with denzo and scalepack (25). The protein coordinates from the TTR–bromoflavone complex (Protein Data Bank accession no. 1THC) were used as a starting model for the initial refinement of the native structure using molecular dynamics and energy minimization by using the program xplor (26). Simulated annealing and individual temperature factor refinement provided a model which was used to phase a “complex−native” difference-Fourier map. In the resulting map, Flu could be located with peak heights of 10 σrms in both binding pockets of the TTR tetramer. Flu was found to bind in two propeller-like conformations. The two minimum-energy conformations of the drug (conformation trans: Φ1(C1-C6-N-C1′) = 14° and Φ2(C6-N-C1′-C2′) = 16°; conformation cis: Φ1(C1-C6-N-C1′) = 14°/Φ2(C6-N-C1′−C2′) = −168°) calculated by the program insight (biosym insight II, Molecular Simulations, Waltham, MA) were in good agreement with the initial |Fo, complex| − |Fo, apo| Φapo maps and were used as an initial model in the crystallographic refinement. The molecular conformations representing a mirror image of these two conformations with identical formation energies did not match the observed density maps. Because of the twofold crystallographic axis along the binding channel, a statistical disorder model had to be applied, giving rise to four binding conformations of Flu in each of the two binding sites of TTR (cis, cis′, trans, trans′). After several cycles of simulated annealing and subsequent positional and temperature factor refinement, 56 water molecules were placed into difference-Fourier maps. Both molecular conformations (trans Φ1/Φ2 = 10°/34°; cis Φ1/Φ2 = 21°/−151°) were in good agreement with unbiased annealed 2 |Fo| − |Fc| Φcalc omit maps, phased in absence of the drug. All four binding conformations of the drug are shown as an Inset to Fig. 3. Because of the lack of interpretable electron densities in the final map, the nine N-terminal and two C-terminal residues were not included in the final model. A summary of the crystallographic analysis can be found in Table 1.
Figure 3.
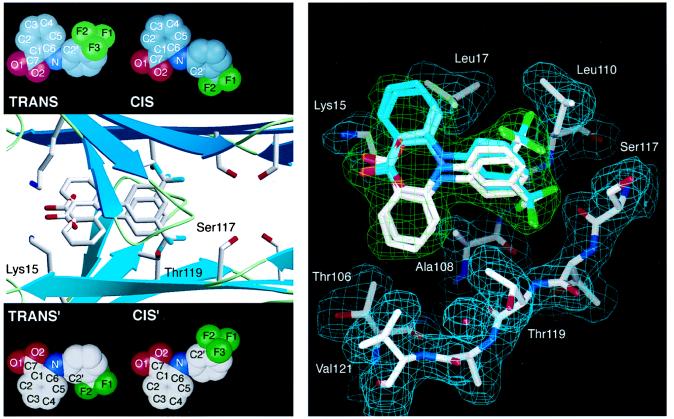
(Left) Flu–TTR interactions. Ribbon diagram of the tetrameric TTR with Flu showing a closeup of one of the two funnel-shaped binding cavities computed by the program setor (32). The binding site is formed by two adjacent TTR monomers related by a twofold axis of symmetry (from left to right in the paper plane). Residues Ser-117, Thr-119, and Lys-15, which are displaced on binding of Flu, are highlighted. For clarity, only the trans conformation of Flu is shown with its symmetry mate. Insets show all four observed binding conformations (the molecular conformations cis and trans with the symmetry related binding modes cis′ and trans′). (Right) TTR conformational changes mediated by Flu binding. A stick model of the Flu binding site as computed by o (33). Only residues from one subunit of TTR that align the binding cavity are displayed, with their 2 |Fo| − |Fc| Φcalc density in cyan. Flu is shown in all four of its binding modes. Because of the twofold-symmetry axis (from left to right in paper plane) both molecular conformations of Flu fit in two symmetry-related binding modes into the final 2 |Fo| − |Fc| Φcalc in green. Oxygen atoms are red, nitrogen atoms are blue, fluorine atoms are green, and carbon atoms are shown in white and cyan, respectively. The protein–ligand interactions are described in the text.
Table 1.
Summary of crystallographic analysis
| Apo | TTR∗Flu | |
|---|---|---|
| Resolution, Å | 20–2.0 | 20–2.0 |
| Reflections, measured/unique | 64,327/16,218 | 86,716/16,664 |
| Completeness, % overall/outer shell | 96.7/84.0 | 99.6/99.4 |
| Rsym, % overall/outer shell | 5.2/11.7 | 6.3/28.5 |
| I/σ1 | 13.9 | 10.6 |
| Refinement statistics | ||
| Resolution, Å | 6.0–2.0 | 8.0–2.0 |
| Rfactor/Rfree, % | 18.3/24.4 | 18.9/25.1 |
| rmsd bond length, Å | 0.015 | 0.016 |
| rmsd bond angles, degrees | 2.011 | 2.037 |
Rsym = Σ |I − 〈I〉|/Σ 〈I〉, where I is the observed intensity, and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections. Rfactor Σ |Fo − Fc|/ΣFo where Fo and Fc are observed and calculated structure factor amplitudes, respectively. Rfree is calculated for a randomly chosen subset of 10% of the reflections. rmsd is the root mean squared deviation from ideal geometry.
RESULTS AND DISCUSSION
Compounds whose structures complement the known binding site of TTR were screened as possible inhibitors by using established amyloid fibril assays including a light-scattering assay and a quantitative Congo red fibril-formation assay (7, 27). Active compounds were further evaluated by analytical ultracentrifugation to demonstrate that the normal tetrameric conformation of TTR was adopted in the presence of the inhibitors under acidic conditions that, in the absence of inhibitor, would normally result in tetramer dissociation and the conformational changes leading to amyloid formation (24). Screening allowed us to identify several bisarylamine inhibitors including the nonsteroidal antiinflammatory drug Flu, which binds and stabilizes the nonamyloidogenic tetramer. Incubation of physiological concentrations of TTR (3.6 μM) at pH 4.4 (the optimal pH for wild-type amyloid fibril formation) results in the conversion of the majority of TTR (≈60%) into amyloid after 72 h in the absence, but not in the presence of 3 equivalents (10.8 μM) of Flu (Fig. 1). In the presence of Flu, the sedimentation equilibrium and velocity data fit to a single ideal species model corresponding to nonamyloidogenic tetrameric TTR (54 kDa), demonstrating that the ligand inhibits fibril formation by stabilizing the normal fold against the pathogenic conformational changes (24). Two equivalents of Flu bind to the wild-type TTR tetramer (pH 7.6) with negative cooperativity (Kd1 = 30 ± 14 nM, Kd2 = 255 ± 97 nM) as measured by ITC. The observed dissociation constants for the FAP variants V30M (Kd1 = 41 ± 10 nM, Kd2 = 320 ± 125 nM), and L55P TTR (Kd1 = 74 ± 16 nM, Kd2 = 682 ± 137 nM) are slightly higher. However, the Kds are low enough in every case to saturate both binding sites in TTR when the Flu concentration equals 10.8 μM, i.e., three times the physiological TTR concentration (3.6 μM), ensuring that there is enough drug to bind to both binding sites. It is not yet clear whether one or both binding sites in TTR must be occupied to achieve complete amyloid fibril inhibition. The conversion of 30% of a 3.6-μM solution of TTR into amyloid fibrils (70% inhibition, 3.6 μM Flu, pH 4.4) when TTR has one of the two ligand-binding sites occupied suggest that complete inhibition requires occupancy of both sites. However, this interpretation assumes that the dissociation constant does not increase appreciably with a decrease in pH, which remains to be demonstrated. ![]()
To further understand the structural basis for the efficacy of Flu as a fibril inhibitor, co-crystals of Flu bound to TTR were produced, and the crystal structure of the binary complex was solved by using molecular-replacement methods and was refined to a resolution of 2.0 Å (Table 1). As in the TTR–T4 complex, two molecules of Flu were bound deep into the central hormone-binding funnel of the TTR tetramer, Fig. 2. In each binding site, four different binding conformations of Flu were found with approximately equal occupancy (Fig. 3).
Figure 2.
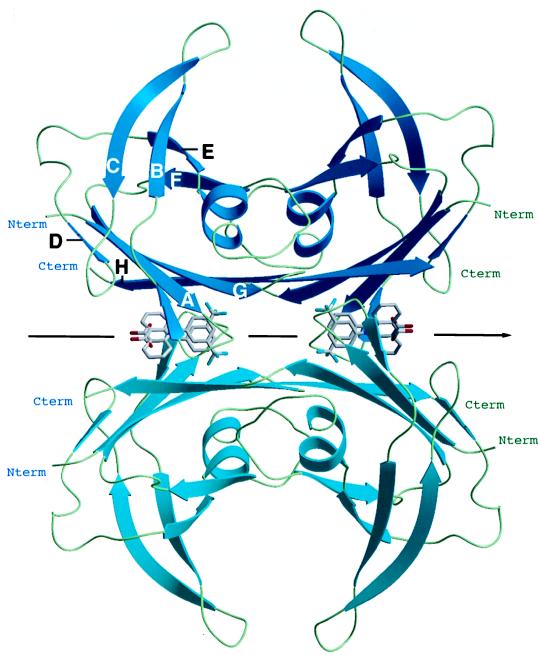
Structure of the Flu–TTR complex. Ribbon diagram of tetrameric TTR with Flu as computed with the program setor (32). The two molecules of Flu, binding into the T4-binding channels of TTR, are shown as a stick model. Because of the twofold axes along the binding channel (pointing from left to right in the paper plane) a second symmetry-related binding mode is present for both molecules (statistic disorder). For clarity, a second molecular conformation of the Flu that is also in agreement with the crystallographic data is not displayed. Each monomer of TTR consists of 127 amino acid residues that form two stacked sheets, each containing four antiparallel β-strands (sheet I: HGAD; sheet II: FEBC). The subunits are linked by hydrogen bonds within the β-strands H and F with the equivalent strands H′ and F′ in the second subunit. These interactions extend the two four-stranded β-sheets into eight-stranded β-sheets in the dimer. Two of these dimers are sandwiched on top of one another to assemble the tetrameric molecule through interactions of the AB and GH loops. The opposition of the convex faces of the β-sheets I and I′ (because of the right-hand twist of their β-strands) gives the funnel-like depressions, representing the two Flu-binding channels.
The refined structure of the TTR–Flu binary complex clearly defines the molecular interactions that allow the drug to bind to TTR. In all four binding modes, Flu interacts with residues of two adjacent TTR molecules, thus mediating several intersubunit interactions. The CF3 substituent occupies the innermost halogen-binding pocket, interacting with Ser-117, Thr-119, Leu-110, and Ala-108, providing van der Waals contacts (Fig. 3). For complex interaction distances see Table 2. The biphenyl system of Flu is found in a low-energy propeller-like conformation in a hydrophobic patch of the T4 binding site between the residues Leu-17, Thr-106, Ala-108, Thr-119, and Val-121. Finally, the carboxylate group on the outer phenyl ring is placed at the entrance of the funnel-shaped binding pocket, forming electrostatic interactions with the side chains of the Lys-15 residues from opposing subunits.
Table 2.
Interaction distances in the complex of TTR with Flu
| Ligand atom | Protein atom | Distance, Å
|
|
|---|---|---|---|
| cis | trans | ||
| F1 | Ser-117 O | 3.22 | 3.38* |
| Ser-117 Cβ | 2.92 | 2.83* | |
| Leu-110 Cβ | 3.30 | 3.52* | |
| F2 | Thr-119 Cβ | 3.23 | 3.12* |
| Leu-110 Cδ2 | 3.11* | 3.18 | |
| F3 | Ser-117 O | 3.57 | 3.46* |
| Ala-109 C | 3.83 | 3.72* | |
| Ala-108 O | 3.31 | 3.43* | |
| Ala-108 Cβ | 3.68 | 3.87* | |
| C4′ | Leu-110 Cβ | 3.94 | |
| C6′ | Ala-108 Cβ | 3.49* | 3.59 |
| C3 | Val-121 Cγ2 | 3.95 | |
| Thr-106 Cγ2 | 3.76 | 3.49 | |
| Ala-108 Cβ | 3.77 | ||
| C4 | Ala-108 Cβ | 3.23 | 3.39 |
| Val-121 Cγ2 | 3.88 | 3.57 | |
| Thr-119 Cγ2 | 3.69 | 3.91 | |
| Leu-17 Cδ1 | 3.33* | 3.55* | |
| C5 | Ala-108 Cβ | 3.37 | 3.18 |
| Leu-17 Cδ1 | 2.87* | 2.96* | |
| Thr-119 Cγ2 | 3.60 | 3.51 | |
| C6 | Leu-17 Cδ1 | 3.34* | 3.31* |
| O1 | Lys-15 N | 2.88 | 2.96 |
| Lys-15 N | 3.72* | 3.57* | |
The values for both molecular conformations of Flu have been averaged over the two symmetry-related monomers and are listed in Å. Both molecular conformations differ in their dihedral angles along the aryl–N–aryl bonds (trans Φ1/Φ2 = 10°/34°; cis Φ1/Φ2 = 21°/−151°). Distances marked with an asterisk reflect interactions of the drug with the second of the two monomers forming the binding cavity.
The binding of Flu also induces conformational changes in TTR that appear to have a stabilizing effect on the TTR tetramer. Specifically, the side chains of Ser-117 and Thr-119 rearrange to facilitate additional hydrogen bonding between the subunits of the tetramer (Fig. 3). In the ligand-free tetramer, the side chains of all four Ser-117 residues are pointing toward the two T4 binding cavities interacting with bulk H2O. Binding of Flu, however, causes the side chains of the Ser-117 residues of all four monomers to rotate about 120°, leading to the formation of two nonsolvated hydrogen bonds between the Ser-117 residues on adjacent subunits (four new hydrogen bonds per tetramer). A similar ligand-induced conformational change of the Thr-119 side chain leads to the formation of a hydrogen bond to an ordered water molecule, which in turn is hydrogen bonded to the carbonyl oxygen of Asp-18 of an adjacent subunit of the tetramer. In summary, Flu binding appears to initiate intersubunit hydrogen bonding and hydrophobic interactions, the latter being mediated by the arylamine substructure of Flu. These intermediates stabilize the normal fold of TTR against the pH-mediated dissociation and conformational changes associated with amyloid fibril formation.
These data demonstrate that Flu binds with high affinity to wild-type TTR and with modest affinity to L55P and V30M TTR, stabilizing the normally folded state against the pH-induced conformational changes that lead to amyloid fibril formation. Because TTR transports only 10–15% of T4 in plasma (thyroid-binding globulin being the main T4 carrier), inhibitor binding to TTR should not perturb normal thyroid metabolism. This hypothesis is further supported by the small effect that the TTR knockout has on T4 metabolism in mice (28). Unlike other inhibitors we have identified that bind to a variety of plasma proteins (e.g., albumin), Flu exhibits strong binding selectivity for TTR (29). In addition, Flu exhibits nonsteroidal antiinflammatory activity, which is very interesting considering the promising effect exhibited by some nonsteroidal antiinflammatory drugs in delaying the onset of Alzheimer’s disease (30). However, the role of inflammation in SSA and FAP pathology is less clear. Based on the structural data outlined herein and emerging crystallographic data with other inhibitors, it is likely that success will be realized in designing novel compounds that bind to TTR with high affinity and prevent amyloid formation by stabilization of the normally folded form of this potentially amyloidogenic protein.
A small-molecule therapeutic strategy is highly desirable in comparison to the surgical strategy currently used to treat eligible FAP patients, where the FAP-associated TTR gene is replaced by the wild-type gene via liver transplantation (31). Currently, there is no effective treatment for SSA, which should be amenable to management by using the approach described within. Perhaps most importantly, a small-molecule inhibitor strategy makes it practical to further evaluate the amyloid hypothesis. Even though there is an overwhelming amount of circumstantial evidence favoring the amyloid hypothesis, it remains to be demonstrated that the inhibition of fibrils will prevent disease onset.
Acknowledgments
We thank the Lita Annenberg Hazen family, the Skaggs family, the Regina M. Ackermann family, the Welch Foundation, and the Deutsche Forschungsgemeinschaft for a postdoctoral fellowship (T.K.) and the San Diego Achievement Rewards for College Scientists foundation for a predoctoral fellowship (H.P.). This work was supported by National Institutes of Health Grant R01 DK46335.
ABBREVIATIONS
- FAP
familial amyloid polyneuropathy
- TTR
transthyretin
- SSA
senile systemic amyloidosis
- Flu
flufenamic acid
- T4
thyroxine
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 [PDB ID codes 1BMZ (apo⋅TTR) and 1BM7 (TTR⋅Flu)].
References
- 1. Selkoe D J. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 2.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hosenbank R, Bates J P, Davies S W, Lehrach H, Wanker E E. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 3.Pepys M B, Hawkins P N. Nature (London) 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- 4.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 5.Kelly J W. Structure. 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 6.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 7.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 8.Blake C C F, Geisow M J, Oatley S J. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 9.Monaco H L, Rizzi M, Coda A. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 10.Blake C C F, Geisow M J, Swan I D A, Rerat C, Rerat B. J Mol Biol. 1974;88:1–12. doi: 10.1016/0022-2836(74)90291-5. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson S F, Rask L, Peterson P. J Biol Chem. 1975;250:8554–8563. [PubMed] [Google Scholar]
- 12.Herbert J, Wilcox J N, Pham K T, Fremeau R T, Zeviani M, Dwork A, Sopprano D R, Makover A, Goodman D S, Zimmerman, et al. Neurology. 1986;36:900–911. doi: 10.1212/wnl.36.7.900. [DOI] [PubMed] [Google Scholar]
- 13.Badman M K, Pryce R A, Charge S B P, Morris J F, Clark A. Cell Tissue Res. 1998;291:285–294. doi: 10.1007/s004410050998. [DOI] [PubMed] [Google Scholar]
- 14.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 15.Blake C, Serpell L. Structure (London) 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson D R, Buxbaum J N. Adv Hum Genet. 1991;20:69–123. doi: 10.1007/978-1-4684-5958-6_2. [DOI] [PubMed] [Google Scholar]
- 17.Sipe J D. Crit Rev Clin Lab Sci. 1994;31:325–354. doi: 10.3109/10408369409084679. [DOI] [PubMed] [Google Scholar]
- 18.McCutchen S, Colon W, Kelly J W. Biochemistry. 1993;32:12119–12127. doi: 10.1021/bi00096a024. [DOI] [PubMed] [Google Scholar]
- 19.McCutchen S L, Lai Z, Miroy G, Kelly J W, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 20.Lai Z, McCulloch J, Kelly J W. Biochemistry. 1997;36:10230–10239. doi: 10.1021/bi963195p. [DOI] [PubMed] [Google Scholar]
- 21.Pepys M B. In: Immunological Diseases. Samter M, editor. Vol. 1. Brown, Boston: Little; 1988. pp. 631–674. [Google Scholar]
- 22.Jacobson D R, Pastore R D, Yaghoubian R, Kane I, Gallo G, Buck F S, Buxbaum J N. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 23.Cornwell G C, Sletten K, Johansson B, Westermark P. Biochem Biophys Res Comm. 1988;154:648–653. doi: 10.1016/0006-291x(88)90188-x. [DOI] [PubMed] [Google Scholar]
- 24.Miroy G J, Lai Z, Lashuel H, Peterson S A, Strang C, Kelly J W. Proc Natl Acad Sci USA. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend: Data Collection and Processing. Sawyer L, Isaacs N, Bailey S, editors. Science and Engineering Research Council Darsbury Laboratory; 1993. [Google Scholar]
- 26.Brünger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 27.Baures P W, Peterson S A, Kelly J W. Bioorg Med Chem Lett. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 28.Palha J A, Episkopou Y, Maeda S, Shimada K, Gottesman M E, Saraiva M J. J Biol Chem. 1994;269:33135–33139. [PubMed] [Google Scholar]
- 29.Munro S L, Lim C-F, Hall J G, Barlow J W, Craik D J, Topliss D J, Stockigt J R. J Clin Endocrinol Metab. 1989;68:1141–1147. doi: 10.1210/jcem-68-6-1141. [DOI] [PubMed] [Google Scholar]
- 30.McGeer P L, Schulzer M, McGeer E G. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 31.Coelho T. Curr Opin Neurol. 1996;9:355–359. [PubMed] [Google Scholar]
- 32.Evans S V. J Mol Graphics. 1993;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 33.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr. 1991;A 47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]