

Abstract
Reoviruses have provided insight into the roles played by specific viral genes and the proteins they encode in virus-induced cell death and tissue injury. Apoptosis is a major mechanism of cell death induced by reoviruses. Reovirus-induced apoptosis involves both death-receptor and mitochondrial cell death pathways. Reovirus infection is associated with selective activation of mitogen activated protein kinase (MAPK) cascades including JNK/SAPK. Infection also perturbs transcription factor signaling resulting in the activation of c-Jun and initial activation followed by strain-specific inhibition of NF-κB. Infection results in changes in the expression of genes encoding proteins involved in cell cycle regulation, apoptosis, and DNA damage and repair processes. Apoptosis is a major mechanism of reovirus-induced injury to key target organs including the CNS and heart. Inhibition of apoptosis through the use of caspase or calpain inhibitors, minocycline, or in caspase 3−/− mice all reduce virus-associated tissue injury and enhance survival of infected animals. Reoviruses induce apoptotic cell death (oncolysis) in a wide variety of cancer cells and tumors. The capacity of reoviruses to grow efficiently in transformed cells is enhanced by the presence of an activated Ras signaling pathway likely through mechanisms involving inhibition of antiviral PKR signaling and activation of Ras/RalGEF/p38 pathways. The potential of reovirus-induced oncolysis in therapy of human cancers is currently being investigated in phase I/II clinical trials.
INTRODUCTION
APOPTOSIS IS A PARTICULAR FORM of cell death distinguished from necrosis by the presence of characteristic morphological changes in host cell chromatin and the plasma membrane. Apoptotic cells show condensed nuclei, reduced cytoplasmic volume, and ruffling and/or blebbing of the plasma membrane. Among the biochemical hallmarks of apoptosis are fragmentation of DNA into oligonucleosomal ladders, exteriorization (flipping) of phosphatidyl serine groups from the inner to the outer surface of the plasma membrane, and the activation of specific sets of cysteinyl aspartate proteases (caspases).
Viruses belonging to many different viral families are known to either induce or inhibit apoptosis (4,12,62,75). In the setting of viral infection apoptosis may either represent a part of the host's innate antiviral defense system, or a mechanism utilized by viruses to enhance pathogenesis by facilitating release from cells and dissemination in the host. For viruses that induce apoptosis, it is likely that there are cell-type and organ-specific differences in the pathways involved. Understanding the role of apoptosis in viral pathogenesis and cytopathicity is greatly facilitated by the availability of an experimental system with both in vivo models of disease involving a variety of organ systems, and cell culture models to facilitate detailed investigation of apoptosis-related cell signaling pathways. Experimental reovirus infection has these characteristics, and has become one of the most thoroughly investigated viral models of apoptosis (8,23,25,37,64,94).
REOVIRUS STRUCTURE AND REPLICATION
Reoviruses are non-enveloped viruses with a genome comprised of ten discrete segments of double-stranded RNA (dsRNA) contained within two icosahedrally symmetric concentric protein shells (56). The virion outer shell (capsid) is composed of ∼600 heterodimeric complexes of the σ3 and μ1 proteins. Sixty copies of the λ2 “core spike” protein form twelve pentons located at each of the outer capsid's icosahedral vertices. The inner capsid (core) is composed predominantly of two additional structural proteins, λ1 and σ2, and small numbers of the minor core proteins μ2 and λ3 (68). Trimers of the viral attachment protein σ1 sit in a channel in the outer face of the λ2 pentons (18).
Reovirus replication is purely cytoplasmic (56), although recent studies indicate that virally encoded proteins are transported to the nucleus during infection (42). Viral entry occurs following receptor-mediated endocytosis after virions bind to cell surface molecules including junctional adhesion molecule 1 (JAM1) and sialic acids (9,10,37). Current models suggest that for serotype 3 (T3) reovirus strains capable of binding to both JAM1 and sialic acid, cell attachment is mediated by an initial low affinity binding to sialic acid followed by high-affinity binding to JAM1 in a multi-step adhesion strengthening process (9,37).
Once inside endosomes, virions are progressively uncoated through an acid-pH and cysteine-protease dependent process. Conversion to infectious subvirion particles (ISVPs) involves removal of the major outer capsid protein σ3, cleavage of the major outer capsid protein μ1 into smaller fragments, several of which (δ, ϕ) remain virion-associated, and changes in the conformation of the cell attachment protein σ1 (56).
ISVPs are infectious and can be formed either intracellularly within endosomes of infected cells or extra-cellularly through the action of proteolytic enzymes such as those present in the intestinal lumen. In the intestinal tract extracellularly produced ISVPs, rather than virions, are the major form of infectious particles. When ISVPs infect cells, they no longer require endosomal acidification for processing. Regardless of their site of initial generation, ISVPs are processed further within endosomes to form the non-infectious but transcriptionally active core particle. This conversion involves removal of σ1 and of the virion-associated μ1 fragments and is associated with penetration of the core particle through the endosomal membrane into the host cell cytoplasm. A key event in this process is the exposure of residues on μ1 that facilitate fusion of the ISVP with the endosomal membrane to facilitate delivery of the core particle into the cytoplasm (15,63). Transcription occurs within cores, and involves extrusion of distinct (+)-sense capped primary transcripts corresponding to each of the ten genomic dsRNA (+) strands from the core into the cytoplasm through channels in the λ2 core spike pentons (90). Protein self-assembly is likely to play a critical role in formation of viral capsids and cores, although the exact assembly steps are still not completely resolved. Similarly, the mechanisms that insure that each virion has exactly one copy of each of the ten dsRNA genome segments are not well understood. Following assembly of virus particles, release of mature virions accompanies cell death and disruption of the plasma membrane.
ROLE OF VIRAL GENES AND PROTEINS IN APOPTOSIS
Genetics of strain-specific differences in apoptosis induction
Reovirus strains differ in their capacity to induce apoptosis in a variety of continuous and primary cell lines. This has been most thoroughly investigated in murine L929 fibroblasts, in these cells the T3 prototype strain Dearing (T3D) induces apoptosis much more efficiently than the serotype 1 prototype strain Lang (T1L) (92). Following infection with T3D, cells exhibit all the ultrastructural hallmarks of apoptosis including chromatin condensation and margination, and fragmentation into oligonucleosomal ladders (73,92) (Figs. 1 and 2). In L929 cells viral replication has nearly reached a plateau by 24 h post-infection. The completion of the one-step growth curve precedes the onset of apoptotic changes in nuclear chromatin morphology as identified by staining cells with a DNA intercalating dye (acridine orange) or measuring fragmentation of [3H] thymidine-labeled DNA, which indicate that apoptosis increases progressively at 24−48 h post-infection (92).
FIG. 1.
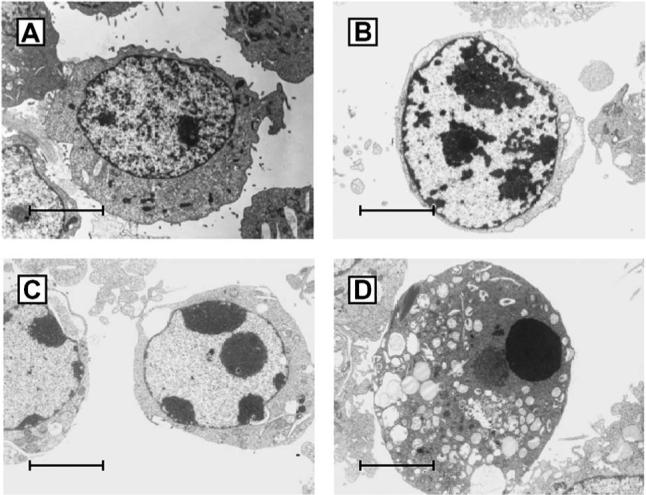
Electron microscopic appearance of uninfected (A) and apoptotic T3D-infected L929 fibroblasts (B–D). Note the progressive margination and compaction of the nuclear chromatin (B,C) and the eventual complete consensation of the nucleus (D). Despite profound changes in nuclear chromatin the cell membrane remains intact. From Tyler et al. (92) with permission.
FIG. 2.
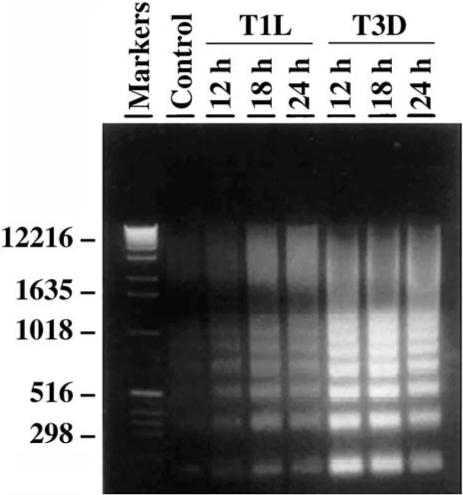
Oligonucleosomal DNA ladders characteristic of apoptosis in reovirus-infected MDCK cells. Oligonucleosomal ladders of fragmented DNA are visible following electrophoresis of extracted total cellular DNA from infected cell lysates through 1.8% TBE/agarose gels stained with ethidium bromide and illuminated with ultraviolet light. From Rodgers et al. (73) with permission.
Reassortant viruses containing different combinations of genes derived from the apoptosis inducing (APO+) T3D strain and the minimally apoptogenic (APO− ) T1L strain were used to identify the S1 viral gene as a determinant of strain-specific differences in apoptosis in L929 (92), MDCK (73) and HeLa cells (28). The M2 gene, encoding the major outer capsid protein μ1, was also identified as a determinant of apoptosis in L929 and MDCK cells, but not HeLa cells. Studies using reassortants generated between another apoptosis-inducing prototypic strain T3 Abney (T3A) and the APOp− T1L strain also identified the S1 and M2 genes as determinants of strain-specific differences in apoptosis (93). The S1 gene is bicistronic and encodes the viral cell attachment protein σ1 and a small non-structural protein, σ1s from over-lapping but out of sequence reading frames. The M2 gene encodes the major outer capsid protein μ1.
Role of the S1 encoded σ1 and σ1s proteins in apoptosis
Both of the two S1 encoded proteins appear to play a role in regulation of apoptosis. A reovirus T3D mutant, variant K, with a single amino acid substitution (lysine for glutamine) at position 419 within the globular head of the σ1 protein (11) has reduced ability to grow and induce apoptosis compared to wild-type virus in both cultured primary hippocampal cortical neurons and in the hippocampus of infected neonatal mice (70). Augmentation of virus-induced apoptosis in variant K infected hippocampal neurons by treatment with Fas-activating antibody partially rescues the defect in viral growth (70). Variant K does not show defective growth or apoptosis induction in mouse cortical neurons, suggesting that cell-type specific factors influence σ1-mediated effects on apoptosis (70).
A T3 reovirus mutant (clone 84-MA) has been isolated which contains a full-length σ1 protein but fails to express σ1s in infected cells due to the presence of a premature stop codon in the σ1s ORF (74). This mutant can still induce apoptosis in L929 cells in culture, indicating that σ1s is dispensable for this process (74). σ1s is a non-structural protein which first appears in infected cells at approximately 8 h post-infection and requires viral transcription. The failure of ribavirin to inhibit reovirus-induced apoptosis in L929 cells and the ability of UV-inactivated replication-incompetent T3D to still induce apoptosis in these cells is consistent with the lack of a requirement for σ1s in apoptosis induction.
Despite the apparent lack of a requirement for σ1s in apoptosis induction in vitro, this protein does appear to play a role in regulating apoptosis in vivo (43). Neonatal mice infected with σ1s null virus (clone 84-MA) still develop apoptosis in the heart and central nervous system (CNS), however the magnitude and onset of this is greatly delayed compared to that seen with σ1s+ wild-type viruses, indicating that σ1s is a regulator of apoptosis in vivo (43).
Avian reovirus proteins and apoptosis
For avian reoviruses the S1 gene encoded σC protein is the cell attachment protein, and is functionally analogous to the S1 gene encoded σ1 protein in mammalian reoviruses. Despite this functional similarity, the avian and mammalian proteins do not have significant amino acid or nucleotide sequence homology. BHK-21 cells transiently transfected with plasmids encoding the σC protein undergo apoptosis as demonstrated by the appearance of oligonucleosomal DNA laddering and histone-associated DNA fragments detectable by ELISA (83). Deletion experiments suggest that the carboxy-terminus of σC is important for induction of apoptosis (83). Interestingly, some C-terminal deletion mutants of σC that lack the capacity to oligomerize into the trimeric form contained in virion particles can still induce apoptosis (83). The finding that transfected protein, which presumably acts at an intracellular site rather than through a cell surface receptor, still induces apoptosis suggests that this is triggered by intracellular protein-protein interactions and not by events triggered by cell surface receptor binding.
Reovirus replication and apoptosis
Replication-incompetent UV-irradiated T3D virions can induce apoptosis, but do so inefficiently compared to non-UV irradiated counterparts (92). Inhibition of viral binding also efficiently inhibits apoptosis (92). These results suggest that an early event occurring between viral receptor engagement and onset of transcriptional activation is required for apoptosis induction—a result which has been subsequently confirmed by studies of the relationship between stages in viral disassembly and apoptosis in reovirus-infected HeLa cells (29). In these studies, inhibition of viral processing into ISVPs in infected HeLa cells by the endosomal acidification inhibitor ammonium chloride or the protease inhibitor E64, blocked apoptosis induction. ISVPs remained able to induce apoptosis in the presence of these inhibitors (29). These results are also consistent with a model suggesting that apoptosis may be triggered by intracellular events during the reovirus replication cycle and not simply by association with cell surface receptors.
Treatment of HeLa cells with ribavirin, a guanosine nucleoside analog which inhibits the reovirus encoded dsRNA-dependent RNA polymerase and inhibits formation of both single-stranded (ss)–RNA and ds-RNA (66), does not inhibit apoptosis. Temperature sensitive (ts) mutants of T3D (30) with blocks in entry (tsA201), core assembly (tsC447) outer capsid assembly (tsB352, tsG453), and dsRNA synthesis (tsD357, tsE320) and reovirus particles devoid of dsRNA genome (“top component”), are all capable of inducing apoptosis in both HeLa and L929 cells at non-permissive temperatures (29). These studies clearly indicate that reovirus-induced apoptosis does not require viral replication and is triggered at a stage in the replication cycle prior to viral transcription. Interestingly, almost identical results have been found in studies of apoptosis induced by the avian reovirus strain 1133 in chicken embryo fibroblasts (52). Similar to its mammalian counterpart, UV-inactivated avian S1133 retains the ability to induce apoptosis, and apoptosis induced by wild-type virus is inhibited by lysosomotropic agents but not by treatment of cells with ribavirin (52).
Role of reovirus binding to receptors and apoptosis induction
The reovirus σ1 protein serves as the viral cell attachment protein. In virions, the protein forms homotrimers with an externally facing globular head domain and an interior long fibrous tail (18). Most T3 reoviruses, including the apoptosis-inducing prototype strains T3A and T3D, have the capacity to bind to both junctional adhesion molecule–1 (JAM1) (10) and sialic acid (SA) residues on the surface of host cells. Some T3 field isolates (e.g., T3C43, T3C44, and T3C84), although still able to bind to JAM1, fail to bind sialic acids. Serotype 1 (T1) reovirus strains, including the prototype strain type 1 Lang (T1L), also bind to JAM1 and to other as yet uncharacterized cell surface carbohydrate moieties, but not to sialic acid (17). The JAM1 binding region of σ1 involves a cluster of highly conserved amino acid residues in a loop-like structure in the globular head domain (10), while sialic acid binding is mediated by a discrete region in the fibrous tail of σ1 (16,17).
Despite the importance of intracellular events early in the reovirus replication cycle for apoptosis induction, it is important to recognize that initial virion binding to both JAM1 and sialic acid (SA) is essential for optimal expression of apoptosis in infected cells. In both HeLa and L929 cells, non-SA binding T3 strains (clones T3C43, T3C44, T3C84) are still able to induce apoptosis, but do so at a much lower level than their SA+ revertant counterparts (T3C43-MA, T3C44-MA, T3C84-MA) (28). Substitution of a leucine for a proline at amino acid 204 of the σ1 protein in a reovirus monoreassortant with a T3D S1 gene on a T1L genetic background abrogates SA binding and dramatically inhibits apoptosis induction in L929 and HeLa cells (28). These results indicate that the addition of SA binding to JAM1 binding is required for the full induction of apoptosis. This result is supported by studies showing that pre-treatment of HeLa or L929 cells with Arthrobacter ureafaciens neuraminidase, which removes cell surface sialic acids, dramatically reduces apoptosis induced by SA+ T3 strains (28). Apoptosis is also inhibited by pre-incubation of SA+ T3 strains with α-sialyllactose, a trisaccharide containing α-linked terminal sialic acid residues which competitively inhibits binding of T3 viruses to SA, but not to JAM1 (28). The effects of SA binding on apoptosis are not simply due to enhanced viral growth, as in L929 cells both T3SA− and T3SA+ strains grow equivalently (although SA+ strains grow better than their SA− counterparts in HeLa cells) (28). Despite its importance, SA binding in and of itself is insufficient for apoptosis-induction by SA+ T3 strains. In L929 cells, monoclonal antibodies that inhibit viral binding to JAM1 completely block apoptosis by SA+ strains, and have only a modest impact on viral growth (10).
CELLULAR PATHWAYS OF REOVIRUS-INDUCED APOPTOSIS
Death receptor initiated apoptosis signaling pathways
An increasingly more comprehensive picture of the cellular apoptotic pathways activated following reovirus infection has emerged from studies of continuous cell lines, cancer cell lines, and primary neuronal and cardiac myocyte cultures (8,23,25,64) (Fig. 3). Although the basic features of reovirus-induced apoptotic signaling are strikingly similar regardless of cell type, there are variations in the specific details. In all cell lines examined to date the initiating event appears to be activation of cell surface death receptors belonging to the tumor necrosis factor receptor (TNFR) superfamily. In HEK293, L929, and a variety of human cervical (HeLa), lung (H157, A549), and breast (MDA231, ZR75−1) cancer cell lines, apoptosis is initiated by the interaction of TRAIL (TNF-related apoptosis-inducing ligand) with death receptors 4 and 5 (DR4, DR5) (19,20). The supernatant derived from T3 reovirus-infected cells can induce apoptosis in TRAIL-sensitive indicator cells (HeLa), and this apoptosis-inducing activity can be inhibited by treatment of the supernatant with soluble Fc-coupled DR5, but not by anti-reovirus antibody (19). Apoptosis-inducing activity is detectable in the supernatant within 24 h post-infection and increases through 48 h post-infection (19). This suggests that TRAIL is released from infected cells through a yet undefined mechanism, and presumably acts to initiate apoptosis in both virus-infected cells (autocrine pathway) and uninfected neighboring bystander cells (paracrine pathway). Consistent with this model, treatment of infected cells with either antibodies against TRAIL or soluble DR4 or DR5 inhibits T3 reovirus–induced apoptosis (19) (Fig. 4). This activity appears to be specific for TRAIL; as antibodies against TNF or against Fas Ligand and soluble forms of non-TRAIL related death receptors (e.g. TNFR) all fail to inhibit T3 reovirus-induced apoptosis in HEK293 cells (19). Consistent with the key role for TRAIL, stable over-expression of a non-functional decoy receptor for TRAIL (DcR-1) in MDA231 breast cancer cells, inhibits T3 reovirus-induced apoptosis (20). Intriguingly, TRAIL has also been implicated in apoptosis mediated by a diverse group of viruses including measles (95), hepatitis (55), influenza (99), respiratory syncytial virus (50), cytomegalovirus (79), lyssavirus (45), Theiler's virus (44,76), HIV (54), and HTLV (72).
FIG. 3.
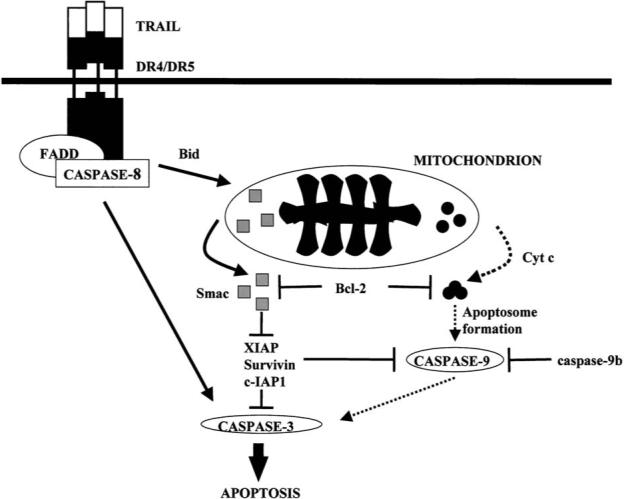
A general outline of caspase pathways activated during reovirus T3 infection of epithelial and human cancer cell lines. Infection results in release of the death inducing ligand TRAIL which binds to cell death receptors DR4 and DR5 which are members of the TNFR superfamily of cell death receptors. Binding of TRAIL to DR4/DR5 activates caspase 8 through the death-inducing signaling complex (DISC). Cleavage of the Bcl-2 family protein Bid plays a key intermediary role in death-receptor initiated activation of mitochondrial apoptosis pathways. Pro-apoptotic factors released from mitochondria following reovirus infection include cytochrome c and Smac/Diablo. Smac/Diablo augments apoptosis by inhibiting the action of cellular inhibitor of apoptosis proteins (IAPs). From Kominsky et al. (49) with permission.
FIG. 4.
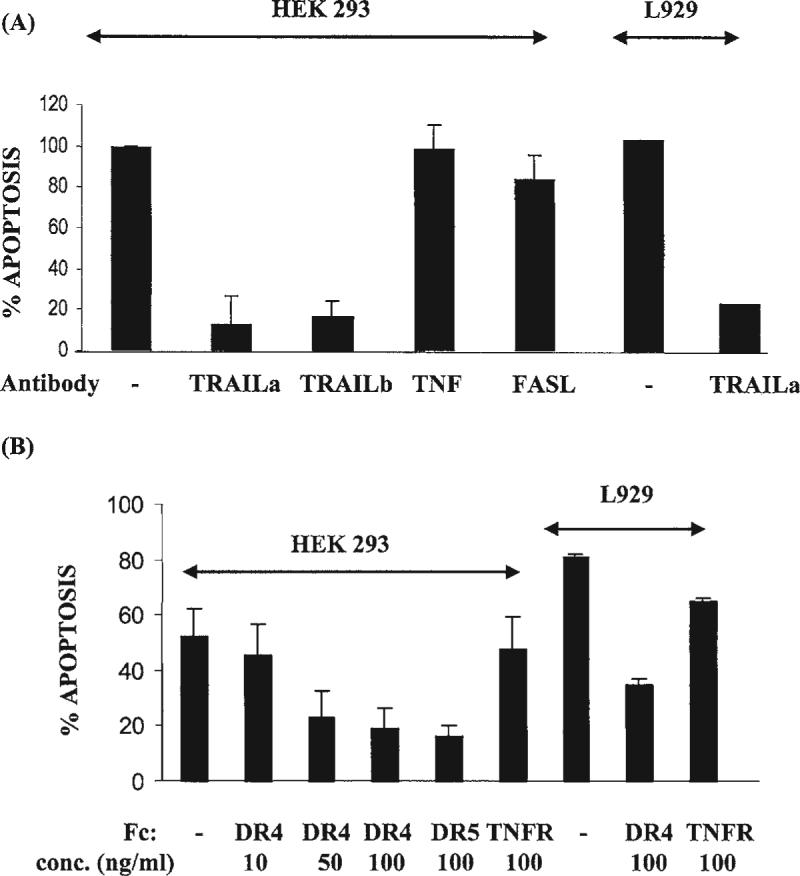
Inhibition of TRAIL binding to reovirus-infected cells with either anti-TRAIL antibodies (A) or soluble Fc-coupled death receptors (B) DR4/DR5 inhibit T3 induced apoptosis in HEK293 and L929 cells. TRAILa and TRAILb are two different polyclonal anti-TRAIL antibodies, TNF and FASL are antibodies against these death ligands. DR4, DR5, and TNFR are Fc-coupled forms of these death receptors. From Clarke et al. (19) with permission.
Death-receptor initiated pathways also play a key role in T3 reovirus–induced apoptosis in primary mouse cortical neuronal cultures (69,70). However, in these cells soluble forms of Fas-receptor and to a lesser extent of soluble forms of TNFR were more effective in inhibiting apoptosis than soluble DR5 (69). Interestingly, the neuroblastoma cell line NB41A3 shows an intermediate phenotype when compared to epithelial and cancer lines (see above) and primary neurons, with apoptosis inhibited by soluble DR5 and TNFR but not by soluble Fas-receptor (69).
Binding of apoptosis-inducing ligands such as TRAIL to their cognate cell surface death receptors results in receptor oligomerization and the apposition of the receptors’ cytoplasmic death effector domains (DEDs). Death receptor oligomerization and DED apposition results in the recruitment of adapter molecules such as FADD (Fas-associated death domain), which also contain DEDs, to the receptor complex. The addition of procaspase 8, the death-receptor associated initiator caspase, completes the components of a “death-inducing signaling complex” (DISC) and leads to the cleavage and activation of caspase 8 (6). Consistent with a model of death receptor-initiated apoptosis, T3 reovirus infection activates caspase 8 in infected epithelial and human cancer cells as well as in primary neuronal cultures (48,69) (Figs. 5 and 6). In addition, T3 reovirus–induced apoptosis is inhibited in HEK293 cells by stable over-expression of a dominant-negative form of FADD (19,49). Treatment with a soluble form of the caspase 8 inhibitory peptide IETD also inhibits T3 reovirus-induced apoptosis in HEK293 and neuronal cells (19,49,69).
FIG. 5.
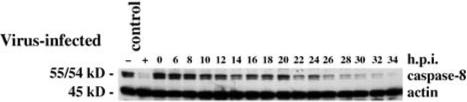
Caspase 8 is activated in T3A-infected HEK293 cells. The immunoblot shows progressive activation-associated disappearance of pro-caspase 8 with an initial phase beginning at ∼8 h post-infection followed by sustained activation after 20 h. From Kominsky et al. (48) with permission.
FIG. 6.
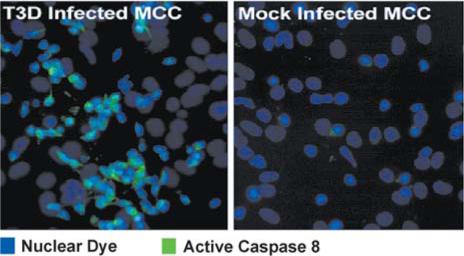
Caspase 8 activation detected using an antibody specific for the activated form of caspase-8 (green staining) in mouse primary cortical neurons at 20 h post-infection with T3D.
Mitochondrial apoptosis signaling pathways
Over-expression of Bc1−2 in MDCK cells markedly reduces T3 reovirus–induced apoptosis (73) (Fig. 7). Over-expression of Bcl-2 also dramatically inhibits effector caspase activation in reovirus-infected HEK293 cells (48). Although the anti-apoptotic actions of Bcl-2 are heterogeneous, the capacity of Bcl-2 to inhibit apoptosis is generally considered a strong indicator that mitochondrial apoptotic pathways are involved (67). This is consistent with results showing that Bcl-2 over-expression blocks reovirus-induced release of a variety of mitochondrial pro-apoptotic factors including cytochrome c, and Smac/ DIABLO (49).
FIG. 7.
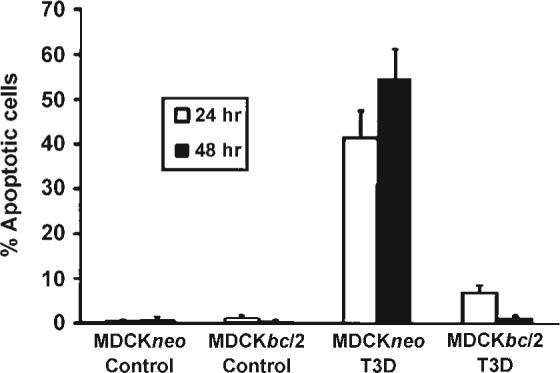
Stable over-expression of Bcl-2 inhibits apoptosis induced by T3D in MDCK cells. MDCKneo is a control plasmid lacking Bcl-2. From Rodgers et al. (73) with permission.
Direct evidence for involvement of mitochondrial pathways in reovirus-induced apoptosis comes from studies in HEK293 cells indicating that mitochondrial proapoptotic factors including cytochrome c (24,48,49) and Smac/DIABLO (24,49) are released from mitochondria into the cytoplasm of infected cells (Fig. 8). This release is selective, as AIF (apoptosis inducing factor) is not detected in either the cytoplasm or nucleus of infected cells (49). In distinction to the result seen in HEK293 cells, there appears to be only limited and late release of cytochrome c in primary neuronal cultures or NB41A3 cells (69).
FIG. 8.
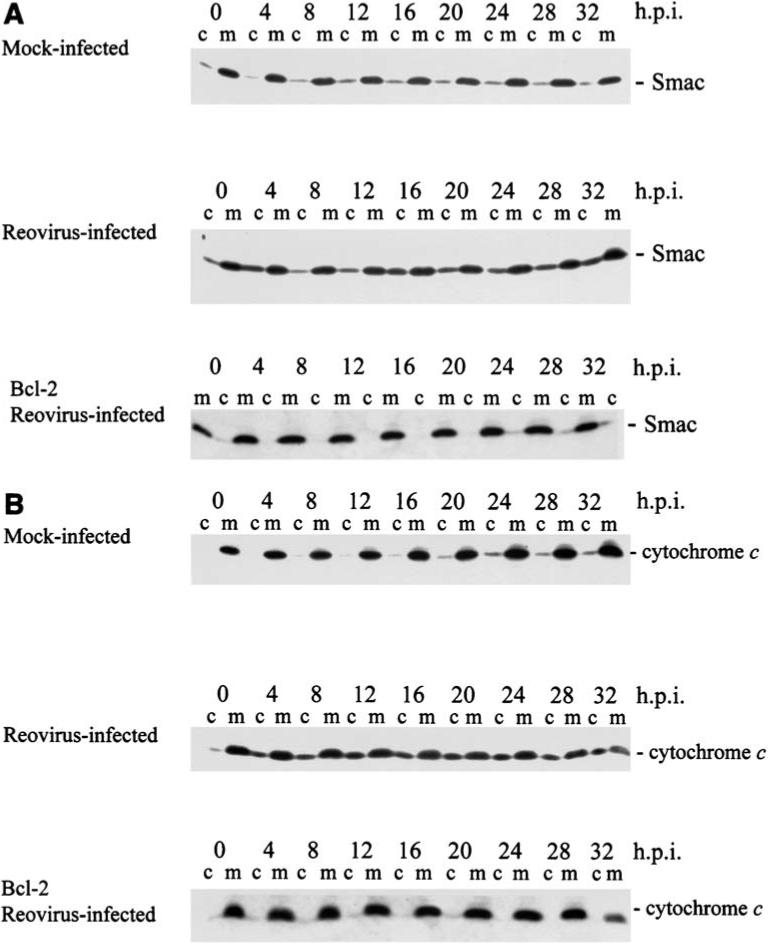
Smac/DIABLO and cytochrome c are released from mitochondria into the cytosol of T3A-infected cells. HEK 293 cells. c, cytosolic fraction; m, mitochondrial fraction of cell lysates; h.p.i., hours post-infection. Both Smac (A) and cytochrome c (B) are released from the mitochondria into the cytosol of reovirus infected cells, and this release is almost completely prevented in cells stably expressing Bcl-2 (bottom immunoblot in each series). From Kominsky et al. (49) with permission.
In some settings, release of mitochondrial apoptotic factors may be associated with profound changes in mitochondrial transmembrane potential (Δψm), likely reflecting the creation of pores in the outer mitochondrial membrane. Reovirus infection is not associated with alterations in mitochondrial Δψm in either HEK293 cells (49) or monkey kidney CV-1 cells (80), suggesting that significant disruption of mitochondrial integrity is not occurring.
Two prominent pathways by which mitochondrial proapoptotic factors facilitate or augment apoptosis are through their activation of caspase 9, and their effects on inhibitor of apoptosis proteins (IAPs). Released cytochrome c forms a complex known as the apoptosome with cytosolic factors including Apaf-1 and procaspase 9 and results in ATP-dependent activation of caspase 9. Caspase 9 activation can be detected in reovirus infected HEK293 cells (48,49). In NB41A3 cells, caspase 9 activation is a late event (<24 h pi) and may occur consequent to activation of caspase 3 rather than as a result of cytochrome c release (69). Even in HEK293 cells, where caspase 9 activation is robust and detectable within 12 h pi, inhibition of this activation by expression of a dominant negative form of caspase 9 (caspase 9b) has no effect on reovirus-induced apoptosis, suggesting that it is not caspase 9 activation, but rather other mitochondrial-dependent events that are critical in reovirus-induced apoptosis (49). Cell permeable caspase 9 inhibitors (LEHD) fail to inhibit reovirus-induced apoptosis in primary neuronal cultures, although both caspase 8 (IETD) and caspase 3 (DEVD) inhibitors effectively inhibit apoptosis (69,70).
The recognition that reovirus infection results in the release of the mitochondrial pro-apoptotic factor Smac/DIABLO (24,49) provides an alternative candidate for the key mitochondrial signaling pathway in reovirus-induced apoptosis. Smac/DIABLO facilitates the activation of caspases by preventing the inhibitory interaction between caspases and IAPs. Selective degradation of specific IAPs, including XIAP, survivin, and cIAP1 (but not cIAP2), occurs in reovirus-infected cells (49) (Fig. 9), and is blocked by over-expression of Bc1−2, which also blocks mitochondrial release of Smac/DIABLO (49). Interestingly, gene expression studies using microarrays suggest that at least one IAP (SMN/NAIP) is up-regulated in T3A infected HEK293 cells by 2.5-fold, a result which is confirmed by RT-PCR and by immunocyto-chemical studies showing increased expression of SMN protein in the hearts of reovirus 8B-infected mice (34).
FIG. 9.
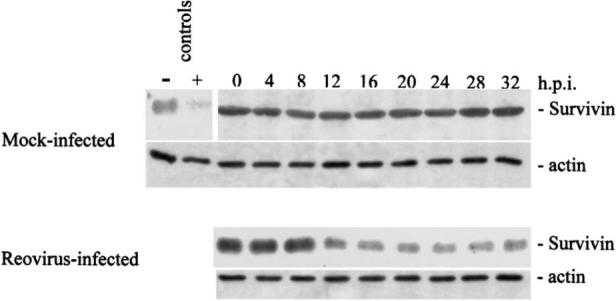
Cellular levels of the inhibitor of apoptosis (IAP) protein survivin are reduced in T3A-infected HEK293 cells. Degradation first appears at 8−12 h post-infection and is inhibited in cells over-expressing Bcl-2 (not shown). From Kominsky et al. (49) with permission.
The pathways linking death-receptor initiated apoptotic signaling to activation of mitochondrial apoptotic pathways in reovirus-infected cells have become increasingly well understood. In reovirus T3-infected cells, caspase 8 activation leads to the cleavage of the Bcl-2 family protein Bid, producing a truncated form of the protein (tBid) (48) which translocates to the mitochondria and facilitate the release of pro-apoptotic mitochondrial factors. The generation of tBid is dependent on death-receptor activation as it can be effectively inhibited by stable over-expression of DN-FADD (48).
The JNK MAPK pathway also plays a key role in activation of mitochondrial apoptotic signaling in reovirus-infected cells (24). HEK293 cells pre-treated with JNK inhibitor 1 show significantly delayed release of both Smac/DIABLO and cytochrome c from mitochondria into the cytosol (24). Although the mechanism of action of JNK in facilitating reovirus-induced activation of the mitochondrial pathway remains unknown, it may be related to the phosphorylation and associated subcellular redistribution of Bcl-2 family proteins.
Effector caspases
The ultimate effect of the activation of death receptor and mitochondrial apoptotic pathways in T3 reovirus-infected cells and tissues is the activation of effector caspases. Caspase 3 activation is detectable in HEK293 cells and in both primary neuronal and cardiac myocyte cultures utilizing immunoblots, fluorescent substrate activity assays, immunohistochemical staining for activated caspase 3, and cleavage of the caspase 3 substrate PARP (35,48,69) (Fig. 10). Caspase 7 activation has also been detected in infected HEK293 cells by immunoblotting (48), although caspase 6, another effector caspase, does not appear to be activated (48). Caspase 3 activation precedes that of caspase 7 and appears at higher levels, suggesting that caspase 3, rather than caspase 7, is the critical effector caspase following T3 reovirus infection (48). In addition to being activated, expression levels of both caspase 3 and 7 are substantially increased (2.6−3.2-fold) in T3A-infected HEK293 cells by 24 h post-infection (34), suggesting that regulation occurs both at the level of gene transcription and protein activation.
FIG. 10.
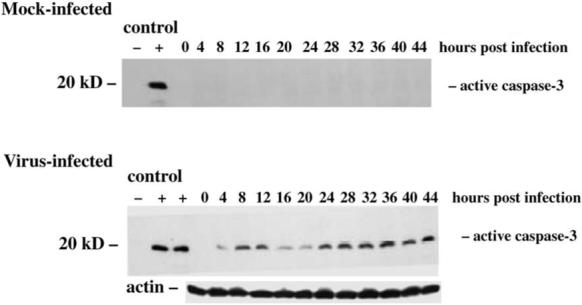
Caspase 3 activation in T3A-infected HEK293 cells detected by immunoblotting using a monoclonal antibody specific for the active form of caspase 3. Note the appearance of an early activation phase at 8−12 h post-infection followed by more sustained activation after 20 h post-infection. From Kominsky et al. (48) with permission.
Caspase 3 activation is detectable in immunoblots from T3 reovirus–infected cells as early as 4 h post-infection, and has a biphasic activation pattern with early initial activation at 4−12 h post-infection followed by a more robust and sustained secondary activation after 24 h post-infection (48). A dual-phase activation pattern is also detectable in fluorogenic substrate assays of caspase 3 activity (48). In these assays, DN-FADD inhibits all caspase 3 activation, whereas Bcl-2 inhibits predominantly the late phase activation. This dual phase activation of caspase 3 would be consistent with an initial transient death-receptor mediated caspase 3 activation phase, followed by a mitochondrially augmented sustained activation phase. As would be predicted from these studies, cell permeable inhibitors of caspase 3 (DEVD) dramatically inhibit T3 reovirus induced apoptosis (48,69).
Calpain
In addition to caspases, the calcium-dependent neutral cysteine protease calpain has been implicated in many models of apoptotic and non-apoptotic cell death (39). Calpains are ubiquitously expressed in the cytosol of many cells, with mu-calpain activated by calcium concentrations in the micromolar range and m-calpain by millimolar concentrations. Both enzymes are specifically inhibited by calpastatin as well as a variety of less specific protease inhibitors. Calpains cleave a variety of proteins at a conformation-dependent rather than sequence specific site. Cellular targets of potential interest in apoptosis signaling include specific cytoskeletal proteins, kinases and phosphatases, caspases, and I-κB the cytosolic inhibitor of NF-κB (39).
T3A infection of L929 cells is associated with increase in calpain activity (1.6-fold) measured by fluorogenic substrate assay in live cells (32). At supra-physiologic multiplicities of infection (MOI 10,000), calpain activation could be detected as early as 30 min post-infection and increased steadily through 2 h post-infection (the last time-point assayed). Calpain activation can also be detected in reovirus 8B infected mouse primary cardiac myocytes as determined by the appearance of calpain-specific 150- and 145-kDa spectrin cleavage products (33). Peak activation (fourfold) occurred at 48 h after infection of cells at MOI 20. Interestingly, gene expression studies using microarrays suggest that calpain gene (Gen-Bank X04366) expression is decreased by 2.6-fold in T3A-infected HEK293 cells at 24 h post-infection, (34) suggesting that activation may be associated with a negative feedback loop that reduces gene expression in infected cells.
Inhibition of calpain activation by pre-treatment of L929 cells with either calpain inhibitor I (aLLN), which blocks the active site, or by PD150606, an α-mercaptoacrylic acid derivative which blocks the Ca2+ binding site; inhibited apoptosis induced by T1L, T3D, and T3A (32). In 8B infected primary cardiac myocytes a similar effect was seen with CX295, a dipeptide α-ketoamide compound that inhibits calpain at the active site (33). Apoptosis was also inhibited in L929 cells treated with calpain inhibitors and infected with replication-incompetent UV-inactivated virus, indicating that the anti-apoptotic effects of calpain inhibition were not due to inhibition of viral replication (32).
The therapeutic efficacy of calpain inhibition was also examined in neonatal mice infected with the myocarditis-inducing reovirus 8B strain (33). Treatment of mice with six daily intraperitoneal injections of CTX 295 (70 mg/kg) beginning 30 min before challenge with 8B (1000 pfu IM) resulted in substantial reduction in the degree of myocardial injury as determined both by a blinded histopathological scoring system and reduction in serum creatine phosphokinase (CPK).
REOVIRUS-INDUCED MODULATION OF TRANSCRIPTION FACTOR ACTIVATION
c-JUN and mitogen-activated protein kinase (MAPK) cascades
Reovirus infection induces changes in several transcription factor systems, notably those involving c-Jun and NF-κB. Increased levels of active, phosphorylated c-Jun are detected in L929 cells infected by the prototypic reovirus strains T1L, T3A, and T3D (21) (Fig. 11). T3A also induces c-Jun phosphorylation in both HEK293 and HeLa cells (24). In L929 cells, phosphorylated c-Jun is first detected at 6−12 h post-infection (pi) and reaches a peak of 30−40-fold over baseline at about 18 h before declining to basal levels by 48 h post-infection (21). The kinetics of c-Jun activation are similar for T3A and T3D. However, T1L shows a slower and less robust activation of c-Jun in comparison to T3 strains (21).
FIG. 11.
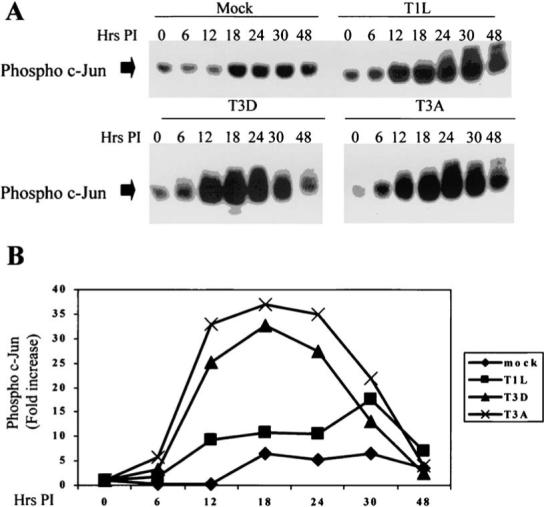
Phosphorylated c-Jun is increased in reovirus-infected L929 cells as detected in immunoblots of infected cell lysates using a phospho-c-Jun specific antibody. Both T3A and T3D induce more robust activation of c-Jun than T1L. B is a graphical representation of the data shown in the immunoblots in A. From Clarke et al. (21) with permission.
In L929 cells, a strong correlation exists between the capacity of the reovirus prototype strains to activate c-Jun and to induce apoptosis (R2 = 0.93 − 0.96 at 12 and 18 h pi), while a similar, but less robust correlation is observed using T1L × T3D reassortant viruses (R2 0.3, p 0.035) (21). This suggests that the effects of c-Jun activation are pro-apoptotic. However, later studies, using an adenovirus vector (TAM67) expressing a dominant-negative form of c-Jun (DN-C-Jun) suggest that it is not c-Jun activation per se but likely an upstream event in the mitogen activated protein kinase (MAPK) pathway leading to c-Jun activation that is the key pro-apoptotic event (24).
JNK and ERK but not p38 MAPK are activated in reovirus infected cells by T3A and T3D but not by T1L, and contribute to reovirus-induced c-Jun activation (21). In HEK293 cells, inhibition of either JNK or ERK activity partially reduces T3A-induced c-Jun phosphorylation and the combination of both inhibitors completely blocks c-Jun phosphorylation (24). Inhibition of p38 and ERK activation with pharmacologic inhibitors does not effect reovirus-induced apoptosis (21,24), in contrast to the results seen with JNK inhibition (24), suggesting that JNK but not p38 or ERK, contribute to reovirus-induced apoptosis (see below). The kinetics of JNK activation parallels that of activation of c-Jun (21). Following infection with T3D, JNK activation is detectable as early as 10 hrs pi and increases steadily through 24 h (Fig. 12). T1L does not significantly induce JNK activity in these cells. Studies using reassortant T1L × T3D viruses indicate that the same reovirus genes that determine strain-specific differences in apoptosis (S1, M2) also determine differences in the capacity of reoviruses to activate JNK (21). There is a stronger correlation between the capacity of T1L × T3D reassortants to activate JNK and to induce apoptosis (R2 = 0.61, p = 0.003), than for c-Jun activation (R2 = 0.3, p = 0.035), suggesting that it is JNK rather than c-Jun activation that may be the key factor in modulating reovirus induced apoptosis.
FIG. 12.
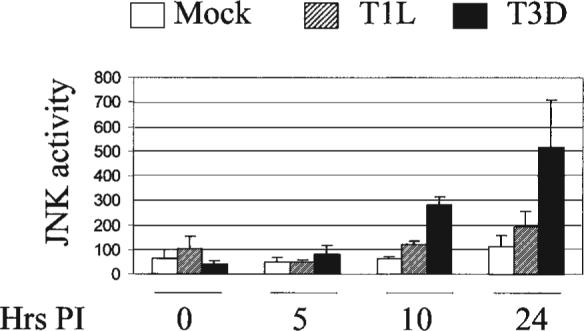
Reovirus infection activates JNK in infected L929 cells. JNK activity was determined using an in vitro kinase assay measuring c-Jun phosphorylation. T3D infection results in significantly higher JNK activity in infected cells than T1L infection. From Clarke et al. (21) with permission.
Additional evidence suggesting that JNK activation rather than c-Jun activation was the key factor in reovirus-induced apoptosis comes from studies with MAPK inhibitors. JNK inhibitor 1 (Alexis) is a cell permeable peptide inhibitor that contains the minimal inhibitory sequence of JNK inhibitory protein 1 (JIP-1/IB1) and inhibits downstream signaling events by inhibiting the interaction between JNK and its substrates. JNK inhibitor II (1,9-pyrazoloanthrone) (Calbiochem) is a cell permeable, selective and reversible competitive inhibitor of JNK. Pre-treatment of either HeLa or HEK293 cells with these inhibitors significantly reduces T3A-induced apoptosis at 24−48 h post infection (57−67% reduction), although this effect could be overcome with high multiplicity of infection (MOI = 100) at the late (48 h) time-point (24). Similar results were seen when caspase 3 activity was examined in T3A-infected HeLa cells. In HEK293 cells, inhibitors of MAPK p38 (SB203580) and ERK (PD98059) had no effect on apoptosis, while JNK inhibitors decreased both caspase-3 induction and apoptosis. In contrast to the specificity of JNK inhibition on apoptosis in HEK293 cells, in HeLa cells the p38 inhibitor did inhibit apoptosis, suggesting that cell-type specific variations exist in MAPKs involved in reovirus-induced apoptosis. The effects of MAP kinase inhibitors on apoptosis and viral growth are distinct. Although JNK inhibition blocks apoptosis it does not affect viral yield in HEK293 cells (24) nor viral protein synthesis in Ras-transformed NIH 3T3 cells (59). Conversely, p38 inhibition does inhibit viral protein synthesis, at least in Ras-transformed NIH 3T3 cells (59).
The exact pathways by which JNK is activated in reovirus-infected cells have not been elucidated. JNK is phosphorylated by JNK kinase (JNKK), which in turn can be activated by the MAP kinase kinase kinase (MAP3K) MEKK1. MEKK1 preferentially activates the JNK pathway and also influences the activity of ERK but does not affect the p38-MAPK pathway. Embryonal stromal (ES) cells lacking MEKK1 show reduced T3A-induced JNK activation (103). MEKK1 −/− mouse embryo fibroblasts show almost complete inhibition of T3A-induced apoptosis and caspase 3 activation (24), as do HEK293 cells expressing a kinase-dead mutant MEKK1 (24). These results suggest that MEKK1-dependent JNK activation may play a key role in reovirus-induced apoptosis.
The mechanism by which reovirus infection could potentially activate MEKK1 remains unknown. It has been shown that viral engagement of the JAM1 receptor and sialic acid co-receptor are critical for T3D-induced activation of NF-κB (28,29), but it is unknown if receptor engagement or early disassembly events are also critical for activation of MAPK pathways. MEKK1 can also be activated through pathways downstream of death receptors (see below). However inhibition of reovirus-induced TRAIL-mediated engagement of death receptors 4 and 5 (DR4, DR5) in L929 cells with soluble DR4/DR5 receptors (19,21), although dramatically inhibiting apoptosis, does not inhibit reovirus-induced phosphorylation of c-Jun, suggesting that activation of the DR-pathway is not the critical event in JNK activation (21).
Ras signaling pathways
The presence of an active Epidermal Growth Factor Receptor (EGF-R) on mouse fibroblasts enhances the efficiency of reovirus infection (87). This activity appears dependent on functional EGF-R tyrosine kinase activity. Studies in NIH-3T3 fibroblasts indicates that a similar enhanced efficiency of infection occurs in cells transfected with v-erbB oncogene constructs with active tyrosine kinase (TK) activity (88). Activation of EGF-R by ligand binding results in receptor autophosphorylation and subsequent recruitment of adapter molecules including Grb2 and the guanine nucleotide–exchange factor Sos. Grb2/Sos in turn activates the Ras-GTP signaling pathway. Transfection of NIH-3T3 cells with either Sos or Ras also enhances susceptibility to reovirus infection (89). Activated Ras-GTP is involved in a wide variety of kinase signaling pathways including those involving PI3 kinase and Raf kinase, and may inhibit virus-induced PKR responses perhaps through a Ras-inducible PKR kinase inhibitor (RIKI). In NIH-3T3 cells reovirus replication appears to be normally restricted by reovirus-induced activation of PKR (89), suggesting that this may be a key antiviral pathway that is inactivated by activated Ras.
Recent studies suggest that the RalGEF signaling pathway may also play an important role in the enhanced replication efficiency of reovirus in Ras-transformed cells (59). Ras and RalGEFs have been associated with activation of NF-κB, cyclin D, and activation of both JNK and p38 MAP kinases. Although JNK activation has been associated with reovirus-induced apoptosis (21,24), inhibition of JNK with SB600125 did not inhibit reovirus protein synthesis in NIH 3T3 cells with activated Ras (59), nor does JNK inhibition reduce viral yield in HEK293 cells (24). Interestingly, inhibition of p38 with SB203580 did inhibit reovirus protein synthesis in NIH 3T3 cells (59) despite the absence of effects of this inhibitor on reovirus-induced apoptosis in HEK293 cells (24). These data suggest that cell signaling pathways involved in apoptosis and enhanced viral growth may be dissociable, consistent with results suggesting a lack of correlation between viral growth and apoptosis induction.
Oncolysis
Reovirus-induced killing of tumor cells, a property often referred to as “oncolysis,” is due to induction of apoptosis in target cells (20). Tumors and tumor cells with an activated Ras pathway appear particularly susceptible to reovirus-induced cell death (26,59,89). Reovirus-induced oncolysis has been demonstrated in an extensive variety of tumor types both in vitro and in vivo, including gliomas (98), breast cancers (20,58,101), lung cancers (20), ovarian cancers (41), colon cancers (41), lymphoid malignancies (2), medulloblastoma (100), and bladder cancer (47). Based on these animal studies, a proprietary reovirus preparation (“reolysin,” Oncolytics Biotech Inc., Calgary, Canada) has recently been tested in Phase I human clinical trials involving direct inoculation of virus into subcutaneous tumors, prostate cancers, and recurrent malignant gliomas. A trial of intravenous administration of reolysin in patients with advanced stage primary or metastatic solid tumors that have failed other chemotherapies has also recently been instituted (see www.oncolytics-biotech.com).
NF-κB
Reoviruses share with many other viruses the capacity to perturb regulation of the transcription factor NF-κB (14). All reovirus strains tested to date induce an initial early phase of NF-κB activation (22,27), with T3A and T3D strains having the capacity to subsequently inhibit NF-κB activation (22). Under resting conditions, NF-κB is complexed with its inhibitor, IκB, and retained in the cytoplasm. The canonical pathway of NF-κB activation involves the phosphorylation, ubiquitination, and proteosomal degradation of IκB, which exposes a nuclear localization signal on NF-κB, allowing it to translocate to the nucleus where it activates expression of genes containing promoters with NF-κB consensus binding sequences.
T3-induced NF-κB activation can be detected as early as 2−4 h post-infection, peaking at 4 h in HEK293 cells and at 8−10 h post-infection in HeLa cells as determined by electrophoretic mobility shift assays (EMSAs) (22,27). Nuclear extracts from T3-infected HeLa cells contain both the p50 and p65 subunits of the NF-κB RelA heterodimer (27). In T3D-infected HeLa, NF-κB-dependent expression of a luciferase reporter gene is detected by 12 h pi, reaches a maximum at 18 h pi, and has declined to baseline by 48 h pi (27) (Fig. 13). Results are essentially similar in T3A-infected HEK293 cells with luciferase activity detected at 6 h, peaking at 12 h, and returning to baseline by 24 h pi (22). In HeLa cells, this initial phase of T3-induced NF-κB activation requires viral binding to sialic acid residues, as a T3SA− strain failed to activate NF-κB when compared to its isogenic SA+ counterpart. Furthermore, pre-treatment of cells with neuraminidase, to remove cell surface sialic acid, abrogates NF-κB activation by SA+ virus (28). The pathways by which T1 strains, such as the prototype T1L activate NF-κB (22) differ from those utilized by T3 strains as T1 strains fail to bind sialic acid.
FIG. 13.
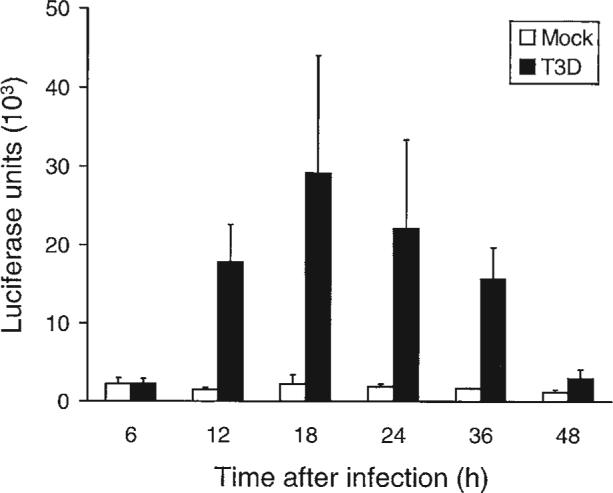
T3D-induced expression of an NF-κB–dependent luciferase reporter gene in HeLa cells. From Connolly et al. (27) with permission.
NF-κB activation in HeLa cells does not require viral replication, as it is not inhibited by ribavirin (29). However, receptor binding alone is apparently insufficient to induce NF-κB activation, as it is inhibited in cells treated with lysosomotropic agents (ammonium chloride, E64) that prevent virion disassembly (29). These results indicate that it is a step in the disassembly process rather than just receptor engagement that triggers NF-κB activation.
The initial phase of NF-κB activation is followed by a later second phase of reovirus-induced inhibition of NF-κB activation (22). In HEK293 cells, both etoposide and TNFα are potent inducers of NF-κB activation. Cells infected with T3A and then stimulated with either etoposide or TNFα show markedly reduced levels of NF-κB activation compared to uninfected cells (Fig. 14). This inhibitory effect is associated with failure to degrade the cytoplasmic inhibitor of NF-κB, IκB. This effect is detectable as earlier as 4 h pi, and reaches a maximum by 12 hrs pi, by which time IκB levels are comparable to those seen in unstimulated control cells.
FIG. 14.
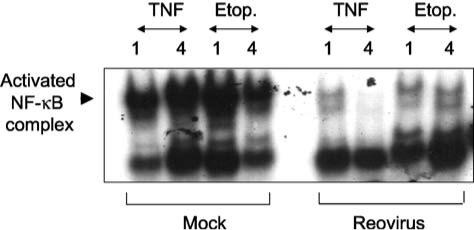
T3A prevents TNF and etoposide-induced activation of NF-κB in HEK293 cells. Cells were infected with T3A and then 12 h later treated with TNF (100 ng/mL) or etoposide (100 μM). Nuclear extracts were prepared 1 or 4 h post-stimulus treatment as indicated in the figure. NF-κB activation was measured by electrophoretic mobility shift assay (EMSA) using an oligonucleotide probe with NF-κB binding sequences. Note the marked reduction in the size of the shifted (activated) NF-κB complex in the reovirus infected lanes compared to mock-infected controls. From Clarke et al. (22) with permission.
Reovirus-induced NF-κB inhibition in HEK293 cells requires viral replication. Following treatment with etoposide, cells infected with T3A and the reovirus replication inhibitor ribavirin show a normal pattern of NF-κB activation and associated degradation of the inhibitor, IκB (22). This suggests a model in which an early step (ribavirin insensitive) in viral replication results in NF-κB activation, whereas a later step (ribavirin sensitive) is required for the subsequent inhibition of NF-κB activation. Ribavirin treatment also inhibits the ability of reovirus to induce apoptosis in HEK293 cells (22), suggesting that in these cells the second inhibitory phase may be more critical to apoptosis induction than the initial NF-κB activation. This phenomenon may be cell type specific, as in contrast to the results seen in HEK293 cells, ribavirin does not inhibit apoptosis in HeLa cells (22,29).
Reovirus infection sensitizes many epithelial cells and human cancer cell lines, to killing by the apoptosis inducing ligand TRAIL (19,20). Cell lines vary in their sensitivity to TRAIL, and reovirus infection can make some previously TRAIL-resistant lines sensitive to killing by TRAIL (19,20). The capacity of reovirus to sensitize HEK293 cells to TRAIL killing was blocked by ribavirin treatment, suggesting that the TRAIL sensitization phenomenon required the late phase of NF-κB inhibition. Further evidence in support of this concept came from studies in HEK293 cells stably expressing a dominant negative IκB-ΔN2 that acts to inhibit activation of NF-κB. These cells show enhanced sensitivity to apoptotic killing by TRAIL, similar to that seen in wild-type cells in which NF-κB inhibition occurred as a consequence of reovirus infection.
Although reovirus-induced regulation of NF-κB is still a subject of active investigation, a unitary hypothesis would suggest that at an early stage following viral infection (prior to onset of viral RNA synthesis and therefore insensitive to ribavirin inhibition) virus induces the activation of NF-κB. This activation exerts a pro-apoptotic influence, presumably through NF-κB mediated regulation of expression of as yet unidentified pro-apoptotic genes. At a later step in replication, following RNA synthesis and therefore sensitive to inhibition by ribavirin, infection induces an inhibitory state that may be designed to prevent expression of NF-κB–dependent apoptosis-inhibitory genes (51).
REOVIRUS-INDUCED CHANGES IN EXPRESSION OF APOPTOSIS-RELATED HOST CELL GENES
Reovirus-induced alterations in activation of transcription factors, including c-Jun and NF-κB, suggests that virus-induced changes in host cell gene expression are likely to play an important role during viral pathogenesis in general and in apoptosis in particular. Reovirus T1L and T3A-induced changes in gene expression following infection of HEK293 cells have been examined using oligonucleotide microarrays (34,65). Using microarrays (Affymetrix HU95A) containing probes for over 12,000 human genes, T3A was found to induce altered expression (twofold or greater change) compared to mock infection of 18 genes at 6 h pi, 86 at 12 h pi, and 309 at 24 h pi (34). T1L produced more modest changes, inducing alteration in expression compared to mock of only 59 genes at 24 h pi. All of the changes induced by T3A at 6 h pi involved up-regulation of expression, although at 12 and 24 h there were both up- and down-regulated genes (34). Categorization of these genes into functional groupings indicates that a significant number of affected genes encode proteins involved in apoptotic signaling including mitochondrial, endoplasmic reticulum, and death receptor signaling as well as proteases including calpain and caspases (Tables 1 and 2). An additional group of affected genes encode proteins involved in DNA damage repair pathways (34). Of 24 genes involved in apoptotic signaling pathways whose expression was altered following infection with the APO+ T3A strain, only five were also found to be altered following infection with the APO− T1L strain (34). Similarly, of 14 identified genes involved in DNA damage and repair pathways whose expression was altered by APO+ T3A, none were altered following infection with APO− T1L. Interestingly, the majority of T3A induced changes in expression involved up-regulation of apoptotic signaling genes (19 of 24 at 24 h pi) and down-regulation of DNA repair genes (11 of 14 at 12 or 24 h) (34). Taken together the patterns of gene expression suggest that infection of cells with APO+ reovirus strains enhances expression of genes involved in ER stress and both death receptor and mitochondrial apoptotic signaling, and shifts the balance toward pro-apoptotic Bcl-2 family proteins (34). This was combined with a down-regulation by the APO+ virus of genes encoding proteins involved in cellular DNA repair, which would be predicted to impair the cell's ability to repair DNA damage and thereby promote apoptosis induction.
Table 1.
Reovirus-Induced Alteration in Expression of Genes Encoding Proteins Known to Regulate Apoptotic Signaling
|
Change in expression (n-fold)bat the indicated time (h) after infection with: |
|||||
|---|---|---|---|---|---|
| GenBank accession no.a |
T3A |
T1L |
|||
| Gene | 6 | 12 | 24 | 24 | |
| Mitochondrial signaling | |||||
| Pim-2 proto-oncogene homologue | U77735 | −2.2 ± 0.1 | |||
| MCL1 | L08246 | 2.0 ± 0.0 | 2.2 ± 0.0 | ||
| BAC 15E1-cytochrome C oxidase polypeptide | AL021546 | 2.1 ± 0.0 | |||
| Par-4 | U63809 | 2.1 ± 0.0 | |||
| HSP-70 (heat shock protein 70 testis variant) | D85730 | 2.2 ± 0.1 | |||
| BNIP-1 (BCL-2 interacting protein) | U15172 | 2.3 ± 0.2 | |||
| SMN/Btfp44/NAIP (survival motor neuron/neuronal apoptosis inhibitor protein) | U80017 | 2.5 ± 0.1 | |||
| DRAK-2 | AB011421 | 2.8 ± 0.2 | |||
| SIP-1 | AF027150 | 3.0 ± 0.2 | |||
| DP5 | D83699 | 5.5 ± 1.1 | |||
| ER stress-induced signaling | |||||
| ORP150 | U65785 | −2.4 ± 0.2 | |||
| GADD 34 | U83981 | 6.8 ± 0.2 | 3.7 ± 0.2 | 2.9 ± 0.2 | |
| GADD45 | M60974 | 3.3 ± 0.2 | 4.9 ± 0.1 | 4.4 ± 0.1 | |
| Death receptor signaling | |||||
| Bcl-10 | AJ006288 | 5.6 ± 1.1 | |||
| PML-2 | M79463 | 3.4 ± 0.3 | |||
| Ceramide glucosyltransferase | D50840 | 4.0 ± 1.2 | |||
| Sp100 | M60618 | 6.5 ± 0.3 | |||
| 5.8 ± 0.6 | |||||
| Proteases | |||||
| Calpain (calcium-activated neutral protease) | X04366 | −2.6 ± 0.1 | |||
| Beta-4 adducin | U43959 | −2.1 ± 0.1 | |||
| Caspase 7 (lice-2 beta cysteine protease | U67319 | 2.6 ± 0.2 | |||
| Caspase 3 (CPP32) | U13737 | 3.2 ± 0.2 | 2.8 ± 0.1 | ||
| Undefined | |||||
| Frizzled related protein | AF056087 | −2.5 ± 0.1 | −3.3 ± 0.5 | ||
| TCBP (T cluster binding protein) | D64015 | 3.3 ± 0.2 | |||
| Cug-BP/hNAb50 RNA binding protein | U63289 | 6.6 ± 1.1 | |||
GenBank accession number corresponds to sequence from which the Affymetrix U95A probe set was designed.
Data are means ± standard errors of the means.
Table 2.
Reovirus-Induced Alteration in Expression of Genes Encoding Proteins Known to be Involved in DNA Repair
|
Change in expression (n-fold)bat the indicated time (h) after infection with: |
|||||
|---|---|---|---|---|---|
| GenBank accession no.a | T3A |
TIL 24 | |||
| Gene | 6 | 12 | 24 | ||
| DNA ligase 1 | M36067 | −8.2 ± 1.1 | |||
| PARPL | AF057160 | −6.3 ± 0.7 | |||
| XP-C repair complementing protein (p125) | D21089 | −3.4 ± 0.1 | |||
| DNA polymerase gamma | U60325 | −1.9 ± 0.1 | −2.9 ± 0.1 | ||
| ERCC5 | L20046 | −2.7 ± 0.1 | |||
| DNA polymerase alpha | L24559 | −2.5 ± 0.2 | |||
| HLP (helicase-like protein) | U09877 | −2.4 ± 0.1 | |||
| GTBP | U28946 | −2.0 ± 0.1 | −2.1 ± 0.0 | ||
| DDB2 (p48 subunit) | U18300 | −2.0 ± 0.0 | |||
| RAD 54 homologue | X97795 | −2.0 ± 0.1 | |||
| Mi2 autoantigen | X86691 | −1.3 ± 0.1 | −2.0 ± 0.1 | ||
| MMS2 | AF049140 | 2.1 ± 0.0 | |||
| Rad-51-interacting protein | AF006259 | 2.6 ± 0.2 | |||
| Rec-1 | AF084513 | 2.4 ± 0.4 | |||
GenBank accession number corresponds to sequence from which the Affymetrix U95A probe set was designed.
Data are means ± standard errors of the means.
REOVIRUS APOPTOSIS IN VIVO
Central nervous system (CNS)
In addition to inducing apoptosis in a variety of continuous cell lines, reovirus-induced apoptosis also occurs in vivo in both the central nervous system (CNS) (60,61,69,70,71) and heart (33,35), in addition to both primary neuronal cultures (69,70) and primary cardiac myocytes (33,35). Following intracerebral (ic) inoculation of reovirus T3D into 1-day-old Swiss Webster (Tac:(SW)fBR) mice neurons undergo morphological changes characteristic of apoptosis including cytoplasmic shrinkage, chromatin condensation, and nuclear pyknosis and fragmentation. Cells staining positive by TUNEL (terminal deoxynucleotidyl transferase (TdT)–mediated dUTP nick end–labeling) can be detected as early as 3 days pi and progressively increasing in both number and extent until the death of animals at ∼day 8−10 (60,61). DNA extracted from whole brains at 8−9 days pi showed a characteristic laddering pattern consistent with oligonucleosomal DNA fragmentation pathognomonic of apoptosis (60,61). Staining for the activated form of the effector caspase, caspase 3, co-localizes with TUNEL staining and is widespread in areas of virus injury by day 7 pi (69,70).
Within the CNS, the most significantly involved areas include the cingulate gyrus (notably layer V), thalamus, and hippocampus (CA1–CA3). The involved cells appear to be predominantly neurons as determined by their morphology (60,61), consistent with results in primary neuronal cultures derived from E20-P0 Swiss Webster mice in which cell-type specific markers confirm that it is neurons (neuron nuclear protein, NeuN-positive cells) rather than glial cells (GFAP positive) than are infected by T3D and that undergo apoptosis (70).
In the CNS, there is an excellent correlation between the areas of viral injury detected histopathologically, the sites of viral infection detected by immunocytochemistry for viral antigen, and regions of apoptosis detected by either TUNEL staining or staining for activated caspase 3 (60,61,69,70) (Fig. 15). Double staining of tissue sections for both viral antigen and apoptosis suggests that apoptosis occurs both in productively infected cells (direct apoptosis) and in uninfected cells in close proximity to infected cells (bystander apoptosis) (60,61). A similar phenomenon can be observed in primary neuronal cultures. In T3D-infected mouse cortical neuronal cultures at 48 h pi, 38% of cells are both antigen and TUNEL positive, and 12% TUNEL positive but antigen negative, suggesting that direct apoptosis accounts for three-fourths of the apoptotic neurons, and bystander apoptosis for the remainder. Essentially similar results were seen in the NB41A3 neuroblastoma cell line (69).
FIG. 15.
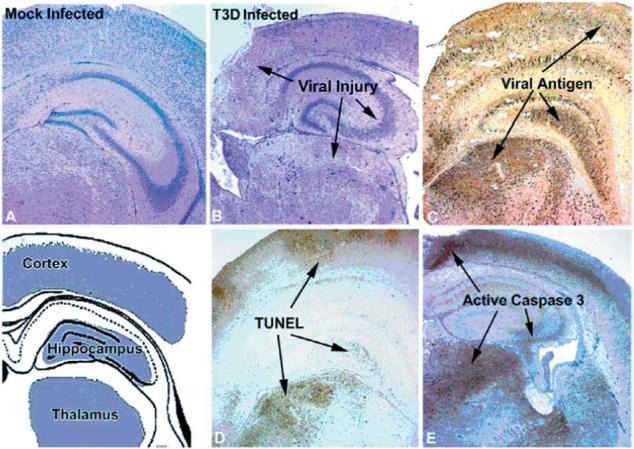
Correlation between viral injury, viral antigen, apoptosis, and caspase-3 activation in the brains of neonatal mice 7 days after intracerebral inoculation with T3D. From Richardson-Burns et al. (69) with permission.
The co-localization of viral antigen, apoptosis, and tissue injury strongly suggests that apoptosis is an important mechanism of reovirus-induced CNA injury. Additional support for this idea comes from studies of an attenuated reovirus T3D variant, variant K (84). Variant K was selected from T3D stocks based on its capacity to resist neutralization by a monoclonal antibody (9BG5) directed against the viral σ1 protein (84). Nucleotide sequence analysis indicates that variant K differs from its T3D parent by a single amino acid substitution at amino acid position 419 (K419L) within the globular head domain of the S1 gene encoded sigma 1 (11). Variant K is of particular interest, as the S1 gene is a determinant of strain-specific differences in the ability of reovirus strains to induce apoptosis in a variety of cell lines. After intracerebral inoculation variant K has attenuated neurovirulence associated with reduced growth and a restricted pattern of tissue injury compared to T3D (84,85). Studies with a reassortant virus containing the Variant K S1 gene on a T1L (“1HAK”) background confirm that it is the S1 gene mutation that accounts for these properties (46). Both variant K and T3D produce injury in the hippocampus, but only T3D produces significant injury in the thalamus, frontoparietal cortex and cingulate gyrus (70,85). Variant K infected mice can survive infection and clear virus, although focal hippocampal lesions are still detectable in mice sacrificed >80 days pi (70).
Following IC inoculation, variant K and T3D show similar patterns of regional growth, apoptosis, and caspase 3 activation in the hippocampus (70). By contrast, the growth of variant K is markedly reduced in other brain regions, and apoptosis is not detectable. These results have been replicated in primary neuronal cultures derived from hippocampus and cortex. Variant K and T3D grow to identical titers hippocampal cultures and produce equivalent amounts of apoptosis. By contrast, in cortical cultures T3D grows significantly better and produces significantly more apoptosis and higher levels of caspase 3 activation than variant K (70).
Studies in non-neuronal cells indicate that the capacity of reovirus strains to grow in these cells does not correlate with its capacity to induce apoptosis. For example, both T3D and T1L grow to equivalent titers in L929 cells, yet T3D induces significantly more apoptosis than T1L (92). In MDCK cells, T1L grows significantly better than T3D, yet T3D induces significantly higher levels of apoptosis (73). This suggests that it might be the reduced capacity of variant K to induce apoptosis in cortical neurons that results in its impaired growth in these cells, rather than the impaired growth inhibiting apoptosis. Support for this hypothesis comes from studies in which inhibiting apoptosis reduced the growth of T3D in primary cortical neurons and augmenting apoptosis enhanced growth of variant K (70). T3D-infected cortical neurons were treated with the cell-permeable pancaspase inhibitor ZVAD-fmk, which reduces apoptosis and decreases viral titer in addition to viral yield to levels at or below those seen in variant K infected cultures. Similar results are seen with the caspase 3 inhibitor DEVD-fmk, but not with an irrelevant caspase inhibitor used as a negative control (YVAD-fmk an inhibitor of caspase 1, a caspase not involved in reovirus-induced apoptosis). These results clearly indicate that inhibition of apoptosis could inhibit viral growth in neuronal cultures. Conversely, treatment of variant K infected cortical neurons at 18 h pi with an antibody that augments apoptosis by binding to and activating the death receptor FAS, enhances apoptosis to levels similar to those seen following T3D infection and significantly augments viral titer and yield, although not to levels seen with T3D. Treatment of variant K infected cortical neuronal cultures with caspase inhibitors does not further reduce either apoptosis or viral yield, nor does treatment of T3D-infected cultures with FAS activating antibody further increase apoptosis or viral yield in T3D-infected cultures.
The variant K studies provide support for the importance of σ1 as a mediator of apoptosis, as at least in cortical neurons, variant K has attenuated apoptosis inducing capacity. However, variant K remained fully capable of inducing apoptosis and activating caspase 3 in hippocampal neurons, indicating that apoptotic signaling pathways and requirements for induction differ within distinct populations of neurons. These studies also provide evidence that inhibiting apoptosis may decrease viral yield and augmenting apoptosis may increase viral yield in the CNS.
σ1 is not the only reovirus protein that can influence CNS apoptosis in vivo. The reovirus S1 gene is bicistronic, encoding the viral attachment protein σ1 and a small non-structural protein, σ1s, from overlapping but out-of-sequence reading frames. The reovirus T3 σ1s null mutant virus clone 84MA (C84MA) has attenuated neurovirulence after intracerebral inoculation (43). The σ1s null virus grows to equivalent titer in the brain as σ1s+ control viruses, indicating that its attenuated neurovirulence is not simply the result of reduced viral growth. By contrast, mice infected with σ1s null virus show significantly delayed onset of caspase 3 activation and apoptosis compared to mice injected with control serotype 3 σ1s+ viruses (43). This suggests that σ1s serves to modulate the efficiency with which T3 reoviruses induce CNS apoptosis, with the σ1s null virus showing less severe and less extensive apoptosis than its σ1s+ counterparts. The fact that mice infected with σ1s null virus eventually developed significant apoptosis indicated that lack of this protein only delayed but did not prevent apoptosis induction in the CNS. In σ1s null virus infected mice, the extent of CNS tissue injury and apoptosis were both delayed and paralleled each other in extent and severity, providing additional evidence that apoptosis is the major mechanism of tissue injury following T3 infection of the CNS.
Given the importance of apoptosis in the development of reovirus-induced CNS tissue injury, it is of great interest to determine whether inhibition of apoptosis could provide a novel strategy for antiviral therapy by influencing the pathogenesis of reovirus-induced CNS disease (71). The antibiotic minocycline, a synthetic tetracycline derivative, is known to be neuroprotective in several models of neurodegenerative disease, traumatic CNS injury, and CNS hypoxic-ischemic injury (5,38,97,104). The mechanisms by which minocycline exerts its neuroprotective effects have not been definitively established, but can include inhibition of mitochondrially mediated apoptosis pathways through decreasing the release of mitochondrial pro-apoptotic factors including cytochrome c and Smac/DIABLO (78,104), and through up-regulation of Bcl-2 and IAPs (78,96). In addition, minocycline may inhibit microglial-mediated excitotoxic pathways that contribute to neuronal cell death (91). Minocycline delays but does not prevent CNS injury and death in a reovirus model of encephalitis, consistent with its effects in other models of neurodegeneration. In the reovirus studies, neonatal mice were injected intracerebrally with 3000 pfu of T3D (∼300 × LD50) and then treated beginning at 48 hrs post infection with minocycline (35 mg/kg ip daily). Minocycline treated mice survived an average of three days longer than untreated controls (mean day of death 11.6 ± 0.9 vs. 8.6 ± 0.7, p < 0.01) (71). A survival effect was even noted when the challenge dose was increased to 300,000 pfu (30,000 × LD50). The prolonged survival in treated mice was associated with reduction and delay in onset of the extent of CNS viral injury in thalamus, hippocampus, and cingulate gyrus. Reduction in injury correlated with a decrease in the number of apoptotic neurons in each of these brain regions (71). This effect is likely due to an anti-apoptotic action of minocycline, as microglial activation and astrogliosis is not prominent at times when tissue injury has been dramatically reduced by minocycline (71). Consistent with earlier results with other apoptosis inhibitors, minocycline slowed the kinetics of viral replication in the CNS, although peak titers eventually reached those achieved in untreated animals (71). A similar beneficial effect of minocycline treatment has recently been described in a Sindbis-virus model of spinal motor neuron death (31).
The identification of mammalian Toll-like receptors (TLRs) was followed by the recognition that binding of microbial ligands to these receptors could trigger the activation of NF-κB, and the subsequent up-regulation of specific cytokines and related co-stimulatory molecules (1,13). TLR3 is of potential interest in the context of reovirus infection, as it is the only TLR that binds double-stranded RNA, and this leads to NF-κB activation and enhanced production of type 1 interferons (α/β) (3,53). TLR3 −/− mice have been utilized to study the potential role of this TLR in viral pathogenesis (36). The extent and distribution of reoviral injury, antigen, apoptosis, and total brain titer are identical in wild-type (TLR 3+/+ ) and TLR 3 −/− mice, suggesting that this TLR does not play a critical role in either reovirus-induced CNS apoptosis or pathogenesis (36).
Heart
Reoviruses provide an important experimental model system for studying viral myocarditis (81). The most extensively characterized myocarditic strain is a reassortant virus, 8B, which was initially isolated following simultaneous inoculation of mice with reovirus strains T1L and T3D. When injected intramuscularly (im) into neonatal mice, 8B is efficiently myocarditic even at low doses, producing extensive cardiac tissue injury, that is the result of direct viral injury to myocardiocytes rather than immune-mediated mechanisms (81).
The myocardial injury induced by 8B is the result of apoptosis. At 7 days pi (im) sections from infected mice show extensive areas of TUNEL-positive nuclei which co-localize with regions of viral injury seen histologically, and viral infection as identified by antigen staining (33) (Fig. 16). DNA extracted from infected hearts shows characteristic oligonucleosomal DNA laddering (33). The kinetics of development of apoptosis and myocardial injury in the heart has been examined in detail (35). Apoptosis, as determined by TUNEL and staining for activated caspase 3, viral growth, and evidence of histologic injury develop in parallel. Injury and apoptosis are typically detectable as early as 4 days pi, and increase steadily in severity and extent thereafter. By day 7, the degree of involvement is extensive, involving 18% (TUNEL) to 23% (activated caspase 3) of the cross-sectional myocardial area.
FIG. 16.
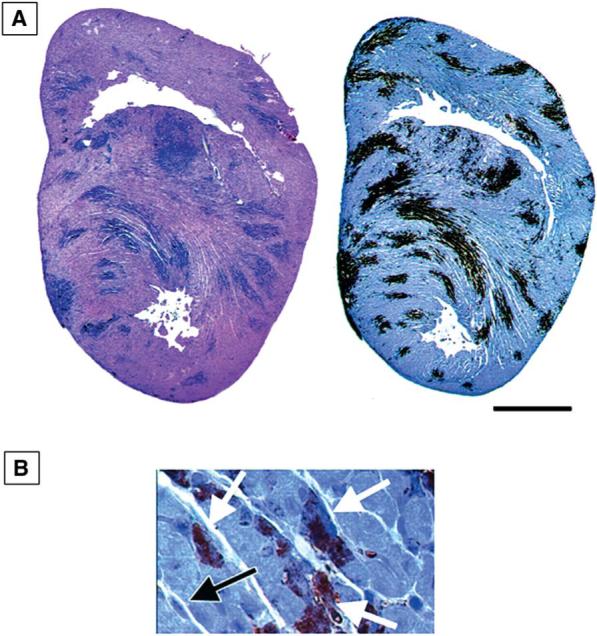
Apoptosis in the hearts of neonatal mice 7 days after infection with the myocarditic reovirus strain 8B. (A) TUNEL staining (left) and staining for active caspase 3 (right). (B) Positive staining for active caspase 3 in cardiac myocytes (white arrows). A negative fibroblast is also shown (black arrow). From DeBiasi et al. (35) with permission.
Apoptosis is also clearly demonstrable using TUNEL, Annexin PI and activated caspase 3 staining in 8B-infected primary cardiac myocyte cultures derived from neonatal mice (35). The “flipping” of phosphatidylserine residues from the inner to the outer surface of the cell membrane, a marker of apoptosis-related changes in cell membranes, can be detected by binding of Annexin V. Using flow cytometric analysis of annexin V positivity, nearly 50% of virus-infected cells are positive at 48 h pi, increasing to >90% by 72 h. Although the absolute numbers are lower (35−30% positive cells), there is also a significant increase at 48 h pi in the percentage of both TUNEL and activated caspase 3–positive cells in virus compared to mock infected cultures at 48 h pi (35).
The reovirus myocarditis model is a valuable system for testing the effects of apoptosis inhibition on viral pathogenesis (33,35). A variety of apoptosis inhibitors have been shown to reduce reovirus-induced cell death in vitro. This suggests that apoptosis inhibitors might also reduce the extent of virus-induced tissue injury in vivo, providing a potentially novel strategy for antiviral therapy. Proof of this principle came initially from studies of calpain inhibitor treatment of 8B-induced myocarditis. Calpain was initially shown to be activated following reovirus infection of L929 cells in vitro, and inhibition of this activation inhibits reovirus induced apoptosis (32). Calpain was subsequently shown to be activated in 8B-infected primary cardiac myocytes as detected by the appearance of calpain-specific spectrin cleavage products in infected cells (33). To test the effects of calpain inhibition on 8B-induced myocardial apoptosis in vivo, neonatal mice were treated with six daily intraperitoneal (ip) inoculations of the calpain inhibitor CX295 (Cortex Pharmaceuticals) beginning at 30 min prior to infection with 8B. CX295 is a specific inhibitor of calpain, and does not inhibit caspase 2 or 3 at doses tested (100 μM) and was shown to inhibit reovirus-induced calpain activation in 8B-infected mouse primary cardiac myocytes (33). 8B-infected CX295-treated mice showed a significant reduction in the severity and extent of cardiac lesions using a standardized lesion scoring scale (3.0 ± 0.1 to 0.6 ± 0.1, p < 0.0001) (33). A significant reduction in serum CPK levels was also seen in treated compared to control mice. CPK is an intracellular enzyme found in cardiac myocytes that is released into serum in response to tissue injury. Staining of cardiac tissue sections from treated mice showed marked reduction in apoptosis as determined both by morphology of cardiac myocytes and almost complete absence of TUNEL staining (33).
Further evidence for the significance of apoptosis in the pathogenesis of 8B-induced reovirus myocarditis comes from studies using peptide caspase inhibitors (35). 8B-infected mice were treated at days 3−6 pi with daily ip inoculations of the irreversible broad spectrum peptide pancaspase inhibitors Q-VD-OPH or Z-VAD(OMe)-FMK (Enzyme Systems). Compared to controls, Q-VD-OPH reduced myocardial lesion score by 60% and cross-sectional lesion area by 89% (Fig. 17). The effects with Z-VAD(OMe)-FMK were similar but more modest (39% reduction in lesion score, 55% in cross-sectional lesion area). In Q-VD-OPH–treated mice, histological sections were also examined for apoptosis, and treated mice showed a significant decrease in both TUNEL and staining for activated caspase 3 compared to controls.
FIG. 17.
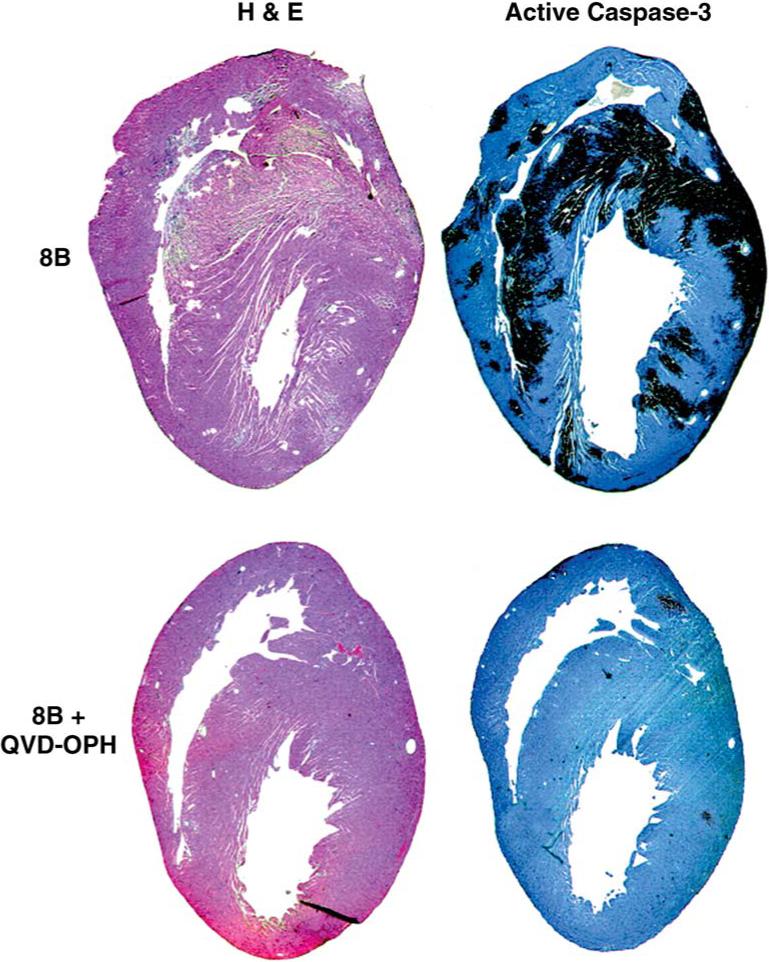
Treatment of neonatal mice with the pancaspase inhibitor Q-VD-OPH inhibits reovirus 8B–induced apoptosis and myocardial injury. Day 7 post-infection. From DeBiasi et al. (35). with permission.
The studies with both calpain and caspase inhibitors indicated that apoptosis was a major mechanism of myocardial injury in 8B-infected mice. This result was confirmed in studies using transgenic C57B6 caspase-3–deficient mice (gift of Richard Flavell, Yale University) (35). 8B-infected caspase 3 −/− deficient mice showed reduced cardiac lesion scores compared to both syngeneic caspase 3 +/− heterozygotes and wild-type caspase 3 +/+ homozygotes. The caspase 3 −/− mice had a 58% reduction in lesion score and a 56% reduction in cross-sectional lesion area compared to infected wild-type caspase 3 +/+ mice (35). Heterozygous caspase 3 +/− mice showed an intermediate phenotype. As expected, staining for activated caspase 3 was not detectable in caspase-3 −/− mice (although present in hearts of infected wild-type caspase 3 +/+ mice). Apoptosis was reduced in the hearts of infected caspase 3 −/− mice as assayed by TUNEL (35). Long-term survival is not seen in 8B infected caspase 3 +/+ mice (100% mortality within 9 days pi after im inoculation of 2 day-old mice with 104 PFU of 8B). By contrast, 50% of caspase 3 −/− survived at least 21 days pi, and 37.5% >50 days (Fig. 18). These long-term survivors did not show cardiac lesions when sacrificed (day 54 pi) (35).
FIG. 18.
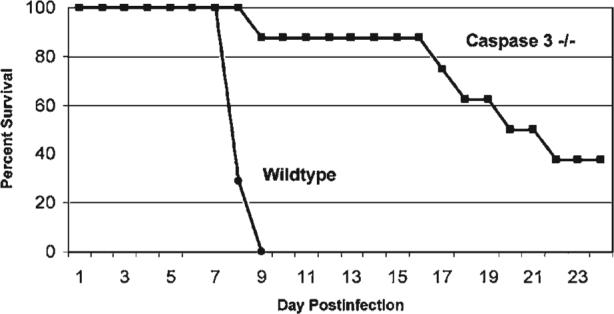
Survival of caspase 3 −/− (deficient) mice and their syngeneic caspase 3 +/+ (wild-type) counterparts following infection with the myocarditic reovirus strain 8B. The caspase 3–deficient mice show enhanced survival and markedly reduced cardiac lesions after infection. From DeBiasi et al. (35) with permission.
Studies in the CNS of an attenuated T3 mutant virus had suggested that defects in apoptosis were associated with decreased viral yield and peak titer (70). Hearts of mice treated with calpain or pancaspase inhibitors showed modest (2−5-fold, 0.3−0.7 log10) reductions in viral titer compared to controls. Significantly more dramatic effects were sign in caspase 3 −/− mice in which a 80-fold (1.9 log10) reduction in heart titer was seen compared to caspase 3 +/+ mice (35).
An additional signaling pathway of paramount importance in reovirus-induced myocarditis is the IFN-β signaling pathway (77,81,82). Myocarditis can be induced by a normally non-myocarditic reovirus strain in mice depleted of IFN-α/β. In addition, non-myocarditic reovirus strains differ from their myocarditic counterparts by both inducing higher levels of IFN-β and by increased sensitivity to its action (82). IFN-β induction in infected cardiac myocytes appears to require activation of the transcription factor interferon regulatory factor 3 (IRF3), as over-expression of a dominant negative IRF-3 in these cells blocks virus-induced IFN-β (57). Although the actions of IFN-β are pleiotropic, a major candidate for mediating its antiviral activity during reovirus infection is the ds-RNA activated protein kinase, PKR. After binding to dsRNA, PKR becomes activated and phosphorylates the eukaryotic initiation factor eIF2α , which inhibits host cell protein translation. Activated PKR may also act as an I-κB kinase, with phosphorylated I-κB undergoing ubiquitination and degradation allowing cytosolic NF-κB to translocate to the nucleus and activate genes with κB-responsive promoter elements, a group that includes the IFN-β gene. As might be expected from this model, loss of PKR enhances the virulence of both myocarditic and non-myocarditic reovirus strains (86). Perhaps not surprisingly, both avian and mammalian strains of reovirus encode proteins that inhibit the activation of PKR (40,102). Reovirus infection of cardiac myocytes also induces IRF-1, likely through an indirect mechanism that requires prior induction of the IFN-α/β response, and mice lacking IRF-1 show more extensive cardiac lesions than wild-type mice (7).
CONCLUSION
Reovirus infection has served as an experimental model system to understand how specific viral genes and the proteins they encode induce cell death in vitro and tissue injury in vivo. Recent studies indicate that apoptosis is a major mechanism by which reovirus T3 strains kill target cells. Apoptosis is triggered by early events in the viral replication cycle consequent to viral binding to JAM1 and sialic acid receptors, and not dependent on viral RNA synthesis. In a wide variety of continuous and primary cell lines reovirus-induced apoptosis is initiated by death-receptor activation. Activation of death-receptor signaling pathways results in activation of the death receptor associated initiator caspase 8, which is followed by cleavage of the Bcl-2 family protein Bid. Cleaved Bid translocates to the mitochondria where it is involved with other Bcl-2 family proteins in the activation of intrinsic mitochondrial-associated apoptotic signaling pathways. Activation of pro-apoptotic mitochondrial signaling pathways are also influenced by reovirus-induced activation of JNK which acts to phosphorylate specific Bcl-2 family proteins and may influence their redistribution from cytosol to mitochondria. Pro-apoptotic factors released from mitochondria following reovirus infection include cytochrome c and Smac/DIABLO. The mitochondrial-associated initiator caspase 9 is activated following reovirus infection in some but not all cells. Recent studies suggest that Smac/DIABLO-associated degradation of cellular inhibitor of apoptosis proteins plays a critical role in the augmentation of apoptosis signaling pathways in many reovirus-infected cells. Activation of extrinsic and intrinsic cell death pathways leads ultimately to the activation of effector caspases including caspase 3. Inhibition of caspase cascades at multiple levels can inhibit reovirus-induced cell death in cell culture.
Apoptosis is not limited to reovirus-infected cells in culture, but also appears to be the major mechanism of reovirus-induced tissue injury in experimental models of reovirus-induced encephalitis and myocarditis. Apoptosis and activation of caspases can be detected in both the heart and CNS of reovirus-infected mice, and correlates with the localization of viral antigen and tissue injury. Inhibition of virus-induced apoptosis through use of peptide inhibitors of caspases, calpain inhibitors, or treatment of animals with minocycline all ameliorate reovirus-induced tissue injury and prolong survival of infected animals. Even more dramatic reductions in virus-induced tissue injury and beneficial effects on survival are seen in caspase 3 −/− mice.
In addition to triggering activation of apoptosis signaling pathways, reovirus infection also results in the selective activation of mitogen activated protein kinase cascades including those involving JNK/SAPK. There is an excellent correlation between the capacity of reovirus strains to activate JNK and their capacity to induce apoptosis, and JNK inhibition results in inhibition of apoptosis. In many cells including a wide variety of cancer cell lines, the efficiency of reovirus infection is enhanced by the presence of activated Ras signaling pathways. This effect may be mediated by inhibition of the antiviral effects of PKR and by activation of Ras/RelGEF/p38 signaling. Interestingly, the signaling pathways involved in apoptosis and viral growth may be dissociable, as inhibition of JNK signaling can inhibit apoptosis without effecting efficiency of viral growth. Conversely, inhibition of p38 MAPK signaling may inhibit viral replication without affecting apoptosis.
Reovirus infection also results in selective changes in the regulation of specific transcription factors. Both ERK and JNK activation contribute to the subsequent activation of c-Jun. Reovirus infection also results in early activation of NF-κB, which is followed in the case of infection with T3 strains by subsequent inhibition of NF-κB activation. These alterations in transcription factor regulation play a key role in regulating reovirus-induced apoptosis, suggesting that virus-induced alterations in gene expression play a key role in regulating apoptosis. Oligonucleotide microarrays have been utilized to gain a more comprehensive picture of reovirus-induced alterations in gene expression. These studies reveal significant alterations in genes encoding proteins involved in cell cycle regulation, apoptosis, and DNA damage and repair processes.
ACKNOWLEDGMENTS
Work described in this paper has been supported by MERIT and REAP grants from the Department of Veterans Affairs (K.L.T.), Public Health Service/NIH grants R01 NS050138 (K.L.T.), 5K08AI52261 (R.L.D.), the U.S. Army Medical Research and Materiel Command Grant DAMD17-98-1-8614 (K.L.T.), the Ovarian Cancer Research Fund (P.C.), and the Reuler-Lewin Family Professorship of Neurology (K.L.T.).
REFERENCES
- 1.Akira S, Takeda K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–4153. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 3.Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 4.Amiesen JC, Pleskoff O, Lelievre JD, et al. Subversion of cell survival and cell death: viruses as enemies, tools, teachers, and allies. Cell Death Diff. 2003;10(Suppl 1):S3–S6. doi: 10.1038/sj.cdd.4401117. [DOI] [PubMed] [Google Scholar]
- 5.Arvin KL, Han BH, Du Y, et al. Minocycline markedly protects the neonatal brain against hypoxicischemic injury. Ann. Neurol. 2002;52:54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 6.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 7.Azzam-Smoak K, Noah DL, Stewart MJ, et al. Interferon regulatory factor–1, interferon-beta, and reovirus-induced myocarditis. Virology. 2002;298:20–29. doi: 10.1006/viro.2002.1470. [DOI] [PubMed] [Google Scholar]
- 8.Barton ES, Chappell JD, Connolly JL, et al. Reovirus receptors and apoptosis. Virology. 2001;290:173–180. doi: 10.1006/viro.2001.1160. [DOI] [PubMed] [Google Scholar]
- 9.Barton ES, Connolly JL, Forrest JC, et al. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 10.Barton ES, Forrest JC, Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 11.Bassel-Duby R, Spriggs DR, Tyler KL, et al. Identification of attenuating mutations on the reovirus type 3 S1 double-stranded RNA segment with a rapid sequencing technique. J. Virol. 1986;60:64–67. doi: 10.1128/jvi.60.1.64-67.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benedict CA, Norris PS, Ware CF. To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 2002;3:1013–1018. doi: 10.1038/ni1102-1013. [DOI] [PubMed] [Google Scholar]
- 13.Boehme KW, Compton T. Innate sensing of viruses by toll-like receptors. J. Virol. 2004;78:7867–7873. doi: 10.1128/JVI.78.15.7867-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowie AG, Zhan J, Marshall WL. Viral appropriation of apoptotic and NF-kappaB signaling pathways. J. Cell. Biochem. 2004;91:1099–1108. doi: 10.1002/jcb.20026. [DOI] [PubMed] [Google Scholar]
- 15.Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chappell JD, Gunn VL, Wetzel JD, et al. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein σ1. J. Virol. 1997;71:1834–1841. doi: 10.1128/jvi.71.3.1834-1841.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappell JD, Duong J, Wright BW, et al. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 2000;74:8472–8479. doi: 10.1128/jvi.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chappell JD, Prota A, Dermody TS, et al. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke P, Meintzer SM, Gibson S, et al. Reo-virus-induced apoptosis is mediated by TRAIL. J. Virol. 2000;74:8135–8139. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke P, Meintzer SM, Spalding AC, et al. Caspase 8–dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene. 2001;20:6910–6919. doi: 10.1038/sj.onc.1204842. [DOI] [PubMed] [Google Scholar]
- 21.Clarke P, Meintzer SM, Widmann C, et al. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 2001;75:11275–11283. doi: 10.1128/JVI.75.23.11275-11283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke P, Meintzer SM, Moffitt LA, et al. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor–related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J. Biol. Chem. 2003;278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- 23.Clarke P, Tyler KL. Reovirus-induced apoptosis: a minireview. Apoptosis. 2003;8:141–150. doi: 10.1023/a:1022966508671. [DOI] [PubMed] [Google Scholar]
- 24.Clarke P, Meinzter SM, Wang Y, et al. JNK regulates the release of pro-apoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 2004;78:13132–13138. doi: 10.1128/JVI.78.23.13132-13138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke P, Richardson-Burns SM, DeBiasi RL, et al. Mechanisms of apoptosis during reovirus infections. Curr. Topics Microbiol. Immunol. 2005;289:1–24. doi: 10.1007/3-540-27320-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffey MC, Strong JE, Forsyth PA, et al. Reovirus therapy of tumors with an activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 27.Connolly JL, Rodgers SE, Clarke P, et al. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 2001;75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connolly JL, Dermody TS. Virion disassembly is required for apoptosis induced by reovirus. J. Virol. 2002;76:1632–1641. doi: 10.1128/JVI.76.4.1632-1641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coombs KM. Temperature-sensitive mutants of reovirus. Curr. Top. Microbiol. Immunol. 1998;233:69–107. doi: 10.1007/978-3-642-72092-5_4. [DOI] [PubMed] [Google Scholar]
- 31.Darman J, Backovic S, Dike S, et al. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J. Neurosci. 2004;24:7566–7575. doi: 10.1523/JNEUROSCI.2002-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeBiasi RL, Squier MKT, Pike B, et al. Reo-virus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J. Virol. 1999;73:695–701. doi: 10.1128/jvi.73.1.695-701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeBiasi RL, Edelstein C, Sherry B, et al. Calapin inhibition protects against virus-induced myocardial injury. J. Virol. 2001;75:351–361. doi: 10.1128/JVI.75.1.351-361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeBiasi RL, Clarke P, Meintzer S, et al. Reo-virus-induced alteration in expression of apoptosis and DNA repair genes with potential roles in viral pathogenesis. J. Virol. 2003;77:8934–8947. doi: 10.1128/JVI.77.16.8934-8947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeBiasi RL, Robinson BA, Sherry B, et al. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J. Virol. 2004;78:11040–11050. doi: 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edelmann KH, Richardson-Burns SM, Alexopoulou A, et al. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 37.Forrest JC, Dermody TS. Reovirus receptors and pathogenesis. J. Virol. 2003;77:9109–9115. doi: 10.1128/JVI.77.17.9109-9115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 39.Goll DE, Thompson VF, Li H, et al. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez-Lopez C, Martinez-Castas J, Esteban M, et al. Evidence that avian reovirus protein sigmaA is an inhibitor of the double-stranded RNA-dependent protein kinase. J. Gen. Virol. 2003;84:1629–1639. doi: 10.1099/vir.0.19004-0. [DOI] [PubMed] [Google Scholar]
- 41.Hirasawa K, Nishikawa SG, Norman KL, et al. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–1701. [PubMed] [Google Scholar]
- 42.Hoyt CC, Bouchard RJ, Tyler KL. Novel nuclear herniations induced by nuclear localization of a viral protein. J. Virol. 2004;78:6360–6369. doi: 10.1128/JVI.78.12.6360-6369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoyt CC, Richardson-Burns SM, Goody RJ, et al. Reovirus non-structural protein σ1s is a determinant of virulence and the efficiency of apoptosis induction in the heart and CNS. J. Virol. 2005;79:2743–2753. doi: 10.1128/JVI.79.5.2743-2753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jelachich ML, Lipton HL. Theiler's murine encephalomyelitis virus induces apoptosis in gamma interferon–activated M1 differentiated myelomonocytic cells through a mechanism involving tumor necrosis factor–alpha (TNF-alpha) and TNF-alpha–related apoptosis-inducing ligand. J. Virol. 2001;75:5930–5938. doi: 10.1128/JVI.75.13.5930-5938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kassis R, Larrous F, Estaquier J, et al. Lyssavirus matrix protein induces apoptosis by a TRAIL-dependent mechanism involving caspase-8 activation. J. Virol. 2004;78:6543–6555. doi: 10.1128/JVI.78.12.6543-6555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaye KM, Spriggs DR, Bassel-Duby R, et al. Genetic basis for altered pathogenesis of an immune-selected antigenic variant of reovirus type 3 (Dearing). J. Virol. 1986;59:90–97. doi: 10.1128/jvi.59.1.90-97.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kilani RT, Tamimi Y, Panel EG, et al. Selective reovirus killing of bladder cancer in a co-culture spheroid model. Virus Res. 2003;93:1–12. doi: 10.1016/s0168-1702(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 48.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Diff. 2002;9:926–933. doi: 10.1038/sj.cdd.4401045. [DOI] [PubMed] [Google Scholar]
- 49.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 2002;76:11414–11424. doi: 10.1128/JVI.76.22.11414-11424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotelkin A, Prikhod'ko EA, Cohen JI, et al. Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J. Virol. 2003;77:9156–9172. doi: 10.1128/JVI.77.17.9156-9172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kucharczak J, Simmons MF, Fan Y, et al. To be, or not to be: NF-kappaB is the answer-the role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 52.Labrada L, Bodelon G, Vinuela J, et al. Avian reoviruses cause apoptosis in cultured cells: viral uncoating, but not viral gene expression, is required for apoptosis induction. J. Virol. 2002;76:7932–7941. doi: 10.1128/JVI.76.16.7932-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsumoto M, Funami K, Oshiumi H, et al. Toll-like receptor 3: a link between toll-like receptor, interferon, and viruses. Microbiol. Immunol. 2004;48:147–154. doi: 10.1111/j.1348-0421.2004.tb03500.x. [DOI] [PubMed] [Google Scholar]
- 54.Miura Y, Misawa N, Kawano Y, et al. Tumor necrosis factor–related apoptosis-inducing ligand induces neuronal death in a murine model of HIV central nervous system infection. Proc. Natl. Acad. Sci. USA. 2003;100:2777–2782. doi: 10.1073/pnas.2628048100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mundt B, Kuhnel F, Zender L, et al. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J. 2003;17:94–96. doi: 10.1096/fj.02-0537fje. [DOI] [PubMed] [Google Scholar]
- 56.Nibert ML, Schiff L. Reoviruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven; Philadelphia: 2001. pp. 1679–1728. [Google Scholar]
- 57.Noah DL, Blum MA, Sherry B. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J. Virol. 1999;73:10208–10213. doi: 10.1128/jvi.73.12.10208-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Norman KL, Coffey MC, Hirasawa K, et al. Reovirus oncolysis of human breast cancer. Hum. Gene Ther. 2002;13:641–652. doi: 10.1089/10430340252837233. [DOI] [PubMed] [Google Scholar]
- 59.Norman KL, Hirasawa K, Yang AD, et al. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oberhaus SM, Smith RL, Clayton GH, et al. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J. Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oberhaus SM, Dermody TS, Tyler KL. Apoptosis and the cytopathic effects of reovirus. Curr. Top. Microbiol. Immunol. 1998;233/II:23–49. doi: 10.1007/978-3-642-72095-6_2. [DOI] [PubMed] [Google Scholar]
- 62.O'Brien V. Viruses and apoptosis. J. Gen. Virol. 1998;79:1833–1845. doi: 10.1099/0022-1317-79-8-1833. [DOI] [PubMed] [Google Scholar]
- 63.Odegard AL, Chandran K, Zhang X, et al. Putative autocleavage of outer capsid protein μl, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Donnell SM, Hansberger MW, Dermody TS. Viral and cellular determinants of apoptosis induced by mammalian reovirus. Int. Rev. Immunol. 2003;22:477–503. doi: 10.1080/08830180305212. [DOI] [PubMed] [Google Scholar]
- 65.Poggioli GJ, DeBiasi RL, Bickel R, et al. Reovirus-induced alterations in gene expression related to cell cycle regulation. J. Virol. 2002;76:2585–2594. doi: 10.1128/JVI.76.6.2585-2594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rankin JT, Jr., Eppes SB, Antczak JB, et al. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology. 1989;168:147–158. doi: 10.1016/0042-6822(89)90413-3. [DOI] [PubMed] [Google Scholar]
- 67.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 68.Reinisch KM, Nibert ML, Harrison SC. Structure of the reovirus core at 3.6 A resolution. Nature. 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 69.Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with activation of death receptors. J. Neurovirol. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- 70.Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J. Virol. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richardson-Burns SM, Tyler KL. Minocycline delays disease onset and mortality in a murine model of viral encephalitis. Exp. Neurol. 2005;192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 72.Rivera-Walsh I, Waterfield M, Xiao G, et al. NF-kappaB signaling pathway governs TRAIL gene expression and human T-cell leukemia virus–I Tax-induced T-cell death. J. Biol. Chem. 2001;276:40385–40388. doi: 10.1074/jbc.C100501200. [DOI] [PubMed] [Google Scholar]
- 73.Rodgers SE, Barton ES, Oberhaus SM, et al. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodgers SE, Connolly JL, Chappell JD, et al. Reovirus growth in cell culture does not require the full complement of viral proteins: identification of a sigma 1s-null mutant. J. Virol. 1998;72:8597–8604. doi: 10.1128/jvi.72.11.8597-8604.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- 76.Rubio N, Martin-Clemente B, Lipton HL. High-neurovirulence GDVII virus induces apoptosis in murine astrocytes through tumor necrosis factor (TNF)–receptor and TNF-related apoptosis-inducing ligand. Virology. 2003;311:366–375. doi: 10.1016/S0042-6822(03)00157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Samuel CE. Reoviruses and the interferon system. Curr. Top. Microbiol. Immunol. 1998;233:125–145. doi: 10.1007/978-3-642-72095-6_7. [DOI] [PubMed] [Google Scholar]
- 78.Scarabelli TM, Stephanou A, Pasini E, et al. Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to Smac/DIABLO, and reduces the mitochondrial leakage of cytochrome c and Smac/DIABLO. J. Am. Coll. Cardiol. 2004;43:865–874. doi: 10.1016/j.jacc.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 79.Sedger LM, Shows DM, Blanton RA, et al. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 1999;163:920–926. [PubMed] [Google Scholar]
- 80.Sharpe AH, Chen LB, Fields BN. The interaction of mammalian reoviruses with the cytoskeleton of monkey kidney CV-1 cells. Virology. 1982;120:399–411. doi: 10.1016/0042-6822(82)90040-x. [DOI] [PubMed] [Google Scholar]
- 81.Sherry B. Pathogenesis of reovirus myocarditis. Curr. Top. Microbiol. Immunol. 1998;233/II:51–66. doi: 10.1007/978-3-642-72095-6_3. [DOI] [PubMed] [Google Scholar]
- 82.Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J. Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shih WL, Hsu HW, Liao MH, et al. Avian reovirus σC protein induces apoptosis in cultured cells. Virology. 2004;321:65–74. doi: 10.1016/j.virol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 84.Spriggs DR, Fields BN. Attenuated reovirus type 3 strains generated by selection of hemagglutinin antigenic variants. Nature. 1982;297:68–70. doi: 10.1038/297068a0. [DOI] [PubMed] [Google Scholar]
- 85.Spriggs DR, Bronson RT, Fields BN. Hemagglutinin variants of reovirus type 3 have altered central nervous system tropism. Science. 1983;220:505–507. doi: 10.1126/science.6301010. [DOI] [PubMed] [Google Scholar]
- 86.Stewart MJ, Blum MA, Sherry B. PKR's protective role in viral myocarditis. Virology. 2003;314:92–100. doi: 10.1016/s0042-6822(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 87.Strong JE, Tang D, Lee PW. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology. 1993;197:405–411. doi: 10.1006/viro.1993.1602. [DOI] [PubMed] [Google Scholar]
- 88.Strong JE, Lee PW. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J. Virol. 1996;70:612–616. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Strong JE, Coffey MC, Tang D, et al. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tao Y, Farsetta DL, Nibert ML, et al. RNA synthesis in a cage—structural studies of reovirus polymerase lambda3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 91.Tikka T, Fiebich BL, Goldsteins G, et al. Minocycline, a tretracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglial. J. Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tyler KL, Squier MKT, Rodgers SE, et al. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein σ1. J. Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyler KL, Squier MKT, Brown AL, et al. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J. Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tyler KL, Clarke P, DeBiasi RL, et al. Reoviruses and the host cell. Trends Microbiol. 2001;9:560–564. doi: 10.1016/s0966-842x(01)02103-5. [DOI] [PubMed] [Google Scholar]
- 95.Vidalain PO, Azocar O, Lamouille B, et al. Measles virus induces functional TRAIL production by human dendritic cells. J. Virol. 2000;74:556–559. doi: 10.1128/jvi.74.1.556-559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, Wei Q, Wang CY, et al. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J. Biol. Chem. 2004;279:19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 97.Wang X, Zhu S, Drozda M, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc. Natl. Acad. Sci. USA. 2003;100:10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilcox ME, Yang W, Senger D, et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J. Natl. Cancer Inst. 2001;93:903–912. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 99.Wurzer WJ, Ehrhardt C, Pleschka S, et al. NF-kappaB–dependent induction of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004;279:30931–30937. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- 100.Yang WQ, Senger D, Muzik H, et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63:6162–6172. [PubMed] [Google Scholar]
- 101.Yang WQ, Saenger DL, Lun XQ, et al. Reovirus as an experimental therapeutic for brain and leptomeningeal metastases from breast cancer. Gene Ther. 2004;11:1579–1589. doi: 10.1038/sj.gt.3302319. [DOI] [PubMed] [Google Scholar]
- 102.Yue Z, Shatkin AJ. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology. 1997;234:364–371. doi: 10.1006/viro.1997.8664. [DOI] [PubMed] [Google Scholar]
- 103.Yujiri T, Ware M, Widmann C, et al. MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-kappaB activation. Proc. Natl. Acad. Sci. USA. 2000;97:7272–7277. doi: 10.1073/pnas.130176697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu S, Starvrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]