

Abstract
The hPMS2 mutation E705K is associated with Turcot syndrome. To elucidate the pathogenesis of hPMS2-E705K, we modeled this mutation in yeast and characterized its expression and effects on mutation avoidance in mammalian cells. We found that while hPMS2-E705K (pms1-E738K in yeast) did not significantly affect hPMS2 (Pms1p in yeast) stability or interaction with MLH1, it could not complement the mutator phenotype in MMR-deficient mouse or yeast cells. Further-more, hPMS2-E705K/pms1-E738K inhibited MMR in wild-type (WT) mammalian cell extracts or yeast cells only when present in excess amounts relative to WT PMS2. Our results strongly suggest that hPMS2-E705K is a recessive loss-of-function allele.
Keywords: Mismatch repair, PMS2, Turcot syndrome
1. Introduction
DNA mismatch repair (MMR) is a highly conserved DNA repair pathway whose primary function is to detect and repair mismatched DNA bases that spontaneously arise during DNA replication and recombination. MMR also participates in the repair of other DNA lesions, acts to suppress recombination between similar but not identical DNA sequences, and plays a role in DNA damage surveillance. Extensive characterization of MMR in bacteria, yeast, and mammals has led to the delineation of two groups of MMR proteins, the MutS and MutL proteins. MutS proteins (e.g., MSH2, MSH6, and MSH3) recognize and bind mismatched or damaged DNA base pairs, while MutL proteins (e.g., MLH1, PMS2, and MLH3) bind MutS proteins and recruit additional proteins for excision of the mismatch and DNA resynthesis (reviewed in [1–6]). In eukaryotes, the primary MutL and MutS complexes are composed of PMS2 (Pms1 in yeast) and MLH1 (MutLα), and MSH2 and MSH6 (MutSα), respectively (reviewed in [1–6]). Defects in the genes coding for these and other MMR proteins result in a mutator phenotype, characterized by higher rates of base pair substitutions and instability in microsatellite sequences (MSI) (reviewed in [1–6]).
The mutator phenotype was associated initially with hereditary nonpolyposis colorectal cancer (HNPCC), a syndrome accounting for ~3% of colorectal cancers and predominantly caused by mutations in MSH2 and MLH1. Until recently, the paucity of germline PMS2 mutations and the absence of intestinal cancer in Pms2−/− mice [7,8] had been widely believed to indicate a minimal role for PMS2 in the initiation of tumorigenesis, even though ample evidence suggested that PMS2-deficiency results in a mutator phenotype [8–10]. However, recent discoveries of new germline hPMS2 mutations have significantly reshaped views about the relationship between PMS2 and cancer [reviewed in 11]. Based on their phenotypic effects, germline hPMS2 defects can now be classified into two groups: (1) compound heterozygosity or homozygosity for recessive PMS2 mutations is associated with Turcot syndrome and often, café-au-lait spots and childhood onset of hematological cancers [12–16] and (2) heterozygosity for a PMS2 mutation that is recessive at the cellular level, followed by mutation or loss of the wild-type (WT) allele is associated with classic autosomal dominant inheritance of HNPCC [17–19].
Interestingly, one common feature of kindreds segregating the first group of PMS2 mutations is that elevated MSI is observed in both tumor and non-neoplastic tissues of the patients, but not in the transmitting parents or in heterozygous family members. The same phenomenon of MSI in normal tissues was described in two reports of Turcot syndrome patients in whom only one PMS2 mutation was originally detected: R134Ter [20] and E705K [21]. The patients carrying hPMS2-R134Ter [20] were later found to carry a recessive mutation in the second PMS2 allele [14]. This finding, coupled with the lack of MSI in the parents’ non-neoplastic tissues and non-dominant functional effects in cultured human cells [22], suggested that hPMS2-R134Ter is a recessive allele and not a dominant allele as originally proposed [23]. In this report, we describe the characterization of hPMS2-E705K through yeast modeling, expression in mouse cells, and in vitro MMR assays. We provide functional evidence that hPMS2-E705K is a recessive allele and unlikely to be the sole etiologic factor for Turcot syndrome in this family.
2. Materials and methods
2.1. Yeast strains, vectors, and fluctuation analyses
S. cerevisiae strain W303 (ade2-1 leu2-3,112 his3-11,15c trp1-1 ura3-1 CAN1 RAD5 hom3-10; [24]) was used for gene targeting and over-expression studies. Growth conditions for this strain and others generated in this study have been previously described [24], and transformations were performed according to the polyethylene glycol–lithium acetate method [25]. A 446 bp AatII/SphI fragment of pCoB-yPMS1-E738K (unpublished) was cloned into AatII/SphI-digested pRS416-PMS1 [24], generating pRS416-pms1-E738K. To generate the 2 μ plasmids pRS426-PMS1 and pRS426-pms1-E738K, PvuII fragments of 4.4 kb from plasmids pRS416-PMS1 and pRS416-pms1-E738K were cloned into PvuII-digested pRS426 [26]. The targeting vector pRS406-pms1-E738K was constructed by cloning a 1.5 kb SpeI/SalI fragment from pRS416-pms1-E738K into SpeI/SalI-digested pRS406 [26], and was then linearized with HpaI for integration into the PMS1 genomic locus of W303 cells by a two-step allele replacement method [27]. Construction of the W303 pms1Δ strain was explained previously [24]. Mutations at hom3-10 and CAN1 loci in yeast were measured through fluctuation analyses [28], and rates were determined as previously described [29]. The 95% confidence intervals for mutation rates were calculated with PRISM 2.0a software (GraphPad Software, Inc.).
2.2. Cultured cell lines
The spontaneously-immortalized mouse embryonic fibroblast (MEF) cell lines used in this study were: C18 [Pms2−/−; 30], CAKEM6.7 [WT; 31], MC5 [WT; 8], and MP1 [Mlh1−/−, Pms2−/−; 32]. MEF cultures were grown as previously described [33].
2.3. Mammalian expression vectors
The construction of pCPB-hPMS2 has been described previously [33]. The Quikchange site-directed mutagenesis kit (Stratagene) and mutagenesis primers hPMS2E705K.F 5′-CAGCATGCCACGGACAAGAAGTATAACTTCG-3′ and hPMS2E705K.R 5′-CGAAGTTATACTTCTTGTCCGTGGCATGCTG-3′ were used to introduce a G → A mutation in codon 705 of a full-length hPMS2 cDNA cloned into pBluescript II. The desired point mutation was verified through sequence analysis. EcoRI fragments of 2.7 kb in length and containing either the full-length (including a 3′ UTR) WT or mutated hPMS2 cDNAs were isolated and cloned into the EcoRI sites of pCPB to yield the plasmids pCPB-hPMS2 and pCPB-E705K, respectively. The orientations of the cDNA inserts were determined through restriction mapping, and the entire hPMS2 coding region was sequenced in each plasmid. The construction of the pCNB-hMLH1 vector was described previously [34].
2.4. Transient transfections
Plasmids bearing hMLH1 and hPMS2 cDNAs were transiently transfected into MP1 cells in a manner similar to that described previously [33]. Briefly, various combinations and amounts of purified (Endo-free Maxiprep, Qiagen) hMLH1, hPMS2, hPMS2-E705K, pCPB, and pCNB DNAs were introduced into MP1 cells using Lipofectamine and Plus reagents (Invitrogen). The empty vector DNA was used to equalize the mass of DNA transfected in each well of a 6-well plate to a total of 3 μg, and all reactions were performed in duplicate with two different DNA preparations for each construct. Each experiment was repeated 3–4 times.
2.5. Generation of MEF clones stably expressing hPMS2
The C18 cell line was subcloned by limiting dilution, and metaphase spreads of 6 subclones were prepared to identify subclones with diploid DNA content. The diploid subclone C18.2 was expanded and electroporated with 10 μg of pCPB, pCPBhPMS2, or pCPBE705K. Following selection in puromycin for 7 days, 30 clones were expanded in duplicate 12-well plates, and cells from one 12-well plate were subsequently lysed in 1X Laemmli sample buffer.
2.6. Analyses of hPMS2 expression
Whole cell lysates of stably transfected C18.2 clones were electrophoresed on 8% SDS–PAGE gels and transferred to Immobilon-P membrane (Millipore). Immunoblotting was performed using anti-PMS2 monoclonal antibody A16-4 (1:500; BD Pharmingen) which detects both human and mouse PMS2, and anti-MSH6 monoclonal antibody (1:2500 of clone 44; BD Transduction Laboratories) which was used to control for equal loading of samples. Chemiluminescent signals were developed with an enhanced chemiluminescence detection system (Western Lightning, Perkin-Elmer Life Sciences) and captured on Kodak BioMax Light film. The films were imaged on a UVP EC3 Bio-Chemi Imaging System (UVP Corp.), and the absolute integrated optical density of individual protein bands was determined using the Labworks 4.0 software package (UVP Corp.). To control for loading discrepancies, hPMS2 expression was normalized to endogenous mMSH6 expression.
Western analysis of hPMS2 expression in transiently-transfected MP1 cells was performed essentially as described previously [33]. Proteins were detected with primary monoclonal antibodies against hMLH1 (1:200 of clone G168-728; BD Pharmingen), hPMS2 (1:500 of clone A16-4; BD Pharmingen), and α-tubulin (1:1000 of clone B 5-1-2; Sigma). Following incubation with a 1:5000 dilution of a horseradish peroxidase-coupled goat anti-mouse antibody (Jackson Immunoresearch), ECL Plus Western Blotting Detection Reagents (Amersham) were used to visualize proteins. Chemiluminescent signals were captured with the UVP EC3 BioChemi Imaging System (UVP Corp.), and densitometry was performed as described earlier. PMS2 band densities were divided by the band densities for endogenous mouse α-tubulin in each lane to correct for loading discrepancies.
2.7. MSI analyses
Non-isotopic detection of the frequency of MSI at 5–6 of the mouse dinucleotide repeat loci D4Mit27, D19Mit41, D13Mit67, D16Mit4, D17Mit123, and D13Mit139 was performed in up to 12 subclones of each cell line, exactly as described previously [35].
2.8. In vitro MMR assays
The construction of baculovirus expression vectors (pFastBac Dual; Invitrogen) containing full-length hMLH1 and 6× His tagged-hPMS2 cDNAs was described previously [32]. To generate a pDUAL-hPMS2-E705K construct, a 1.6 kb PvuII-AatII fragment of the hPMS2-E705K cDNA was cloned into the PvuII-AatII sites of the pDUAL-hPMS2 vector and verified through restriction analysis and sequencing of the complete coding region. Recombinant hMutLα was produced in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac Baculovirus Expression system (Invitrogen), and purified by nickel affinity and ion exchange chromatography as described previously [32]. The purified proteins were electrophoresed on 8% SDS–PAGE gels, visualized with Coomassie blue stain, and relative amounts were determined with Quantity One software (BioRad).
MP1 and MC2 cell lines were expanded in up to forty 150 mm plates and harvested, and cytoplasmic extracts were prepared as described [36]. The HeLa nuclear extract was a kind gift from Dr. Paul Modrich (Duke Univ. Medical Center, Durham, NC). Circular, double-stranded DNA substrates containing a 5′ nick and either a G/T mismatch or a 1 bp insertion/deletion loop (IDL) were prepared as described earlier [32] and used for in vitro MMR assays with the relevant cytoplasmic or nuclear extracts. The percent repair efficiency represents the sum of the intensity (as determined with Quantity One software, BioRad) of repaired DNA substrates divided by the sum of the intensity of repaired and unrepaired DNA substrates.
3. Results
The glutamic acid residue at codon 705 of hPMS2 lies within a highly conserved region of the MLH1-interacting domain of Pms1p in yeast and PMS2 in mice and humans (Fig. 1), suggesting an important functional role for this residue. To elucidate the effects of the E705K allele on MMR-mediated mutation avoidance, we used both in vivo and in vitro methodologies.
Fig. 1.

Conservation of residue E705 in PMS2 homologs. The non-conservative E → K substitution occurs in hPMS2 residue 705, which is located in a highly conserved region of the MLH1 interaction domain.
3.1. Analyses of mutation rates in yeast expressing pms1-E738K
To determine the effect of the homologous PMS2 mutation pms1-E738K on MMR function in yeast, rates of reversion of a +1 T insertion at the hom3-10 locus [37] were measured in the W303 strain background. Cells with a genomic pms1-E738K mutation expressed as much Pms1p as WT cells (data not shown), and exhibited a mutation rate at the hom3-10 locus comparable to that of a pms1δ strain (Table 1). Thus, the E738K mutation inactivates MMR but does not appear to affect Pms1p stability in yeast. To determine whether pms1-E738K can exert a dominant effect on MMR, WT W303 yeast cells were transformed with either CEN (generally single-copy) or 2 μ (high-copy) plasmids bearing pms1-E738K, and reverse and forward mutation rates were measured at hom3-10 and CAN1 (reports a wide mutational spectrum) loci, respectively. In yeast expressing pms1-E738K from a CEN plasmid, mutation rates were low and comparable to that of WT cells expressing either empty plasmid or PMS1. In contrast, cells expressing pms1-E738K from a 2 μ plasmid exhibited increased mutation rates relative to PMS1 expressors (Table 1). As pms1-E738K exerts dominant effects on MMR in WT cells only when over-expressed, it behaves as a recessive, loss-of-function allele in yeast.
Table 1.
Effects of genomic and plasmid-borne pms1-E738K mutations on mutation rates in the W303 strain of S. cerevisiae
| Genotype (genome) |
hom3-10 |
Genotype(plasmid)a |
hom3-10 |
CAN1 |
|||
|---|---|---|---|---|---|---|---|
| Mutation rate (×10−8) | Fold increase | Mutation rate (×10−8)a,b | Fold increase | Mutation rate (×10−8)a,b | Fold increase | ||
| PMS1 | 1.4 | 1 | Empty vector | 0.9 (0.7–1.1) | 1 | 24 (11–37) | 1 |
| pms1Δ | 873 | 642 | PMS1 (CEN) | 1.1 (0.6–1.6) | 1.2 | 30 (23–37) | 1.3 |
| pms1-E738K | 971 | 714 | pms1-738 (CEN) | 1.7 (0.9–2.5) | 1.9 | 36 (18–53) | 1.5 |
| PMS1 (2 μ) | 3.3 (1.4–5.2) | 3.7 | 27 (23–30) | 1.1 | |||
| pms1-738 (2 μ) | 213 (165–262) | 237 | 184 (54–314) | 7.7 | |||
In WT W303 yeast cells.
95% confidence intervals are indicated in parentheses.
3.2. Expression and functional analyses of WT and mutant hPMS2 in MMR-deficient MEFs
To evaluate the expression of hPMS2-E705K in mammalian cells, we used a transient transfection assay that allows rapid assessments of MLH1 and PMS2 stability [33]. As shown in Fig. 2A, hPMS2 and hPMS2-E705K were expressed at nearly equivalent levels in hMutLα-deficient MP1 cells in the presence of hMLH1, which is known to stabilize hPMS2 through direct interaction [33]. To specifically determine whether hPMS2-E705K was less stable than its wild-type counterpart, MP1 cells were transiently-transfected with 0.25–2 μg of hPMS2 or hPMS2-E705K cDNA alone (Fig. 2B). Quantitation of the PMS2 signals on immunoblots revealed that there was no significant difference in stability between WT and mutant hPMS2 in these cells (Fig. 2C).
Fig. 2.

Expression of hPMS2-E705K in transiently transfected mouse cells. (A) Representative immunoblot of whole cell lysates (WCLs) of MP1 cells transfected with hMLH1 (1 μg). hPMS2 (0.25 μg,), and/or hPMS2-E705K (0.25 μg) cDNAs, together with pCPB (1.25–1.5 μg) and pCNB (0.5–1.5 μg) empty vector DNAs to bring the total amount of DNA transfected to 3 μg. (B) Representative immunoblot of WCLs of MP1 cells transfected with 0.25–2 μg of hPMS2 or hPMS2-E705K cDNAs. (C) Quantitative analysis of PMS2 expression in MP1 cells transfected with increasing amounts of hPMS2 versus hPMS2-E705K cDNAs. Each point on the graph represents the average of 3–4 experiments consisting of duplicate transfections. Linear regression was performed on each data series.
Next, we tested the ability of hPMS2-E705K to complement the mutator phenotype of Pms2−/− MEFs (C18 cells). It was expected that hPMS2 would heterodimerize with mMLH1 in MEFs to yield functional MutLα complexes because human and mouse PMS2 are highly conserved and functional transspecies MutLα have been reported previously [34,38]. The mutator phenotypes of a vector-transfected clone and two clones expressing nearly equivalent amounts of WT hPMS2 or hPMS2-E705K (Fig. 3) were analyzed by determining the mutation frequency at 5–6 microsatellite loci. While mutation levels were very low in the WT hPMS2-transfected clone, the microsatellite mutation frequencies in vector and hPMS2-E705K-transfected cells were comparably elevated (Table 2), indicating that hPMS2-E705K cannot complement the mutator phenotype of C18 cells, even when expressed at levels comparable to WT hPMS2.
Fig. 3.
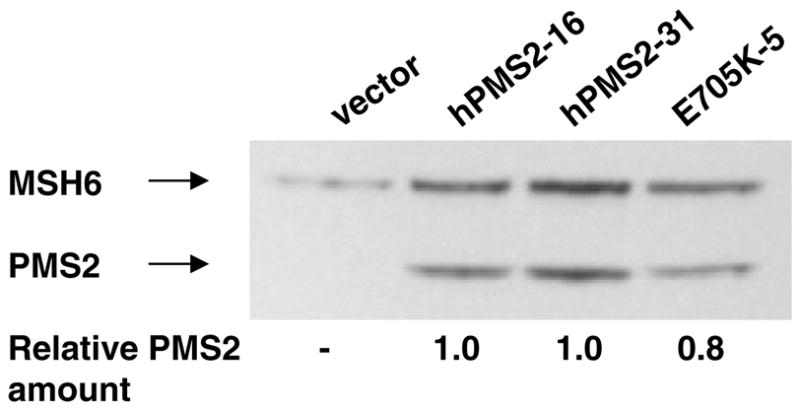
Expression of hPMS2 and hPMS2-E705K in stably transfected mouse cells. Whole cell lysates prepared from a subset of C18.2 clones expressing nearly equivalent amounts of hPMS2 and hPMS2-E705K were electrophoresed, immunoblotted, and quantitated via densitometry as described in Section 2. All PMS2 signals were normalized to endogenous mMSH6 expression in each lane, and relative amounts of PMS2 were calculated by dividing all PMS2 signals by the signal detected in clone hPMS2-16 (lane 2).
Table 2.
Frequency of MSI in Pms2-deficient MEFs stably transfected with hPMS2 or hPMS2-E705K
| Cell line | MSI frequency (%) |
|---|---|
| Vector | 13.3 |
| hPMS2-31 | 4.6 |
| E705K-5 | 18.3 |
3.3. Characterization of MMR in MP1 cytoplasmic extracts supplemented with hMutLα-E705K
Our earlier studies had shown that WT but not mutant recombinant hMutLα can complement cytoplasmic extracts of MP1 cells [32]. A similar strategy was used to characterize the mutation avoidance activity of hPMS2-E705K relative to WT PMS2. As shown in Fig. 4A, the amount of hPMS2-E705K is similar to that in both WT hMutLα and a non-functional mutant form of hMutLα (hMutLα-mE34A/pE41A, [32]) with mutations affecting the ATPase activities of both hMLH1 and hPMS2. Thus E705K has little, if any, effects on hPMS2 stability. Furthermore, the stoichiometry of MLH1 to PMS2 was comparable in WT and mutant hMutLα complexes, indicating that the mutation has minimal effects on heterodimerization of hMLH1 and hPMS2 in insect cells. Consistent with this conclusion, yeast two-hybrid analysis revealed that Mlh1p:Pms1p-E738K interaction was not significantly reduced relative to WT Mlh1p:Pms1p interaction (data not shown). Taken together, these data suggest that residue 705/738 has minimal effects on hPMS2/Pms1p stability or heterodimerization with hMLH1/Mlh1p.
Fig. 4.
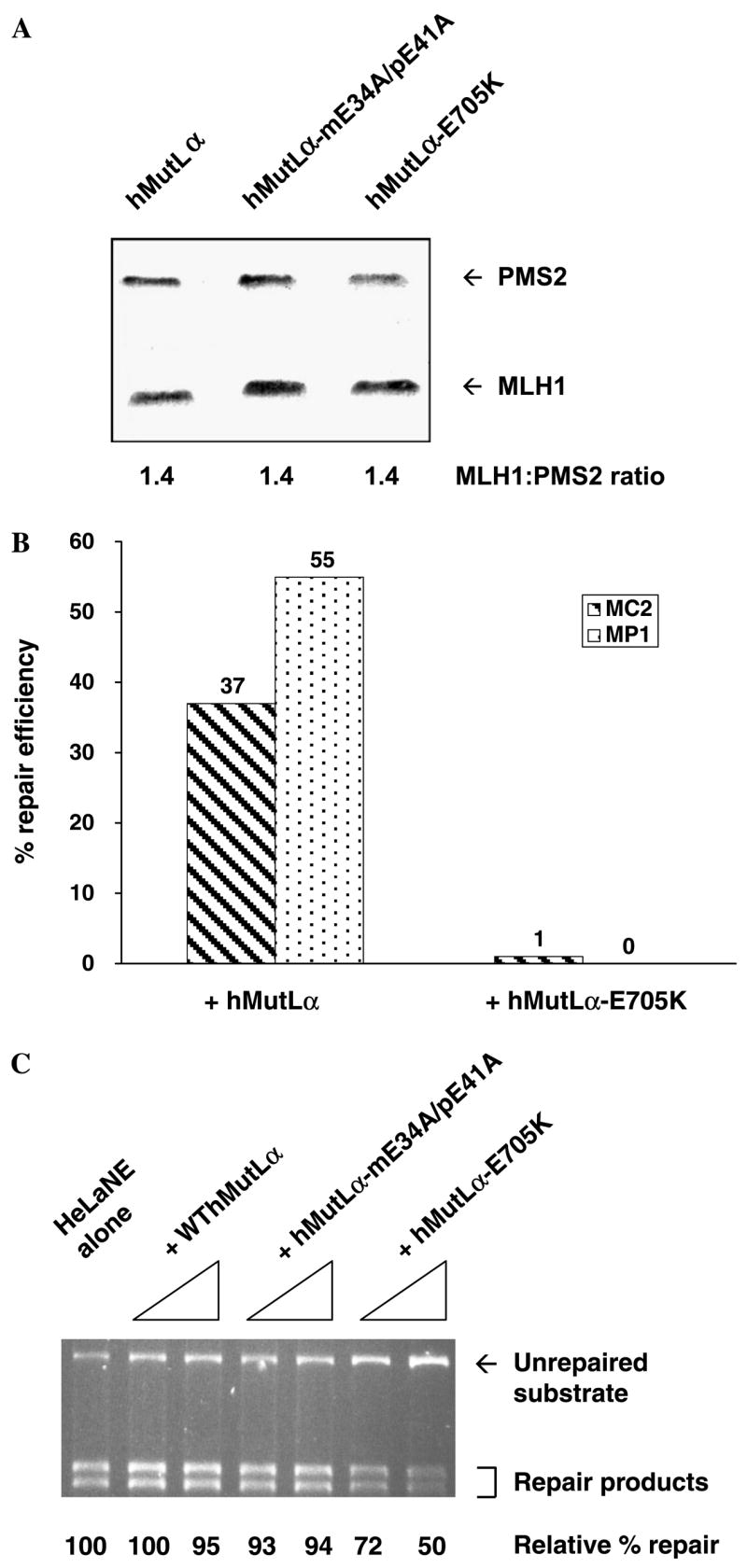
In vitro analyses of the ability of hMutLα-E705K to complement MMR deficiency or dominantly inhibit MMR.(A) Coomassie-stained SDS–PAGE gel containing recombinant wild-type and mutant hMutLα that was purified from baculovirus-transduced insect cells through nickel affinity and ion exchange chromatography. hMutLα-mE34A/pE41A, a non-functional hMutLα complex with wild-type MLH1/PMS2 stoichiometry [31], was included for comparison purposes. (B) The 5′ nick-directed repair of a 1 bp IDL was determined in cytoplasmic extracts of MutLa-deficient MEFs supplemented as follows: 350 ng of wild-type or 700 ng of mutant MutLα was added to MC2 extract, whereas 100 ng of either complex was added to MP1 extract. (C) To a HeLa nuclear extract, 200–400 ng of wild-type or mutant hMutLα was added and its ability to perform 5′ nick-directed repair of a G/T mismatch was determined through agarose gel electrophoresis and densitometry.
Next, either hMutLα or hMutLα-E705K was added to cytoplasmic extracts of the MutLα-deficient MEF cell lines MC2 and MP1. MC2 cells are MutLα-deficent because in the absence of MLH1, PMS2 is unstable [e.g., 34]. Whereas hMutLα exhibited efficient 5′ nick-directed repair of a 1 bp IDL, hMutLα-E705K showed little to no activity in repairing the same mismatch, even when present at twice the amount of hMutLα in MC2 extract (Fig. 4B). The same mismatch correction defect was observed in HCT116 (human colorectal carcinoma cells deficient in MLH1) cytoplasmic extracts supplemented with hMutLα-E705K versus hMutLα (data not shown).
To determine if hPMS2-E705K can dominantly inhibit mammalian MMR, purified hMutLα-E705K was added to a HeLa nuclear extract and in vitro MMR assays were performed. In this extract endogenous hMutLα is not limiting, as it contains ~100 ng (560 fmol) of endogenous hMutLα, which is twice that required for maximal activity in reconstituted in vitro MMR reactions [39]. Thus, we reasoned that only the addition of excess hMutLα (e.g., 200 ng and 400 ng) would be likely to provoke changes in MMR activity. As shown in Fig. 4C, the repair of a 5′ nicked substrate with a G/T mismatch was inhibited by up to 50% when hMutLα-E705K was added. Addition of equivalent amounts of WT hMutLα or hMutLαmE34A/pE41A did not negatively affect MMR in the same extract, excluding the possibility of artifact-induced MMR inhibition (Fig. 4C). Furthermore, the failure of hMutLα-mE34A/pE41A to dominantly inhibit MMR indicates that the mere presence of a non-functional mutant hMutLα complex is not sufficient to explain the negative effects exhibited by hMutLα-E705K. These results are interesting in light of a recent report in which hMutLα was shown to lose single-stranded endonucleolytic activity as a result of either the E705K or mE34A/pE41A substitutions [40]. Finally, the inhibitory effects of excess hMutLα-E705K were also observed for the 5′ nick-directed repair of a 1 bp IDL in HeLa nuclear extract and when defined ratios of purified WT hMutLα and hMutLα-E705K were added to MMR-deficient cytoplasmic extracts (data not shown). Consistent with the results of the yeast mutator assays, then, the in vitro MMR data demonstrate that hPMS2-E705K exerted dominant effects on MMR only when present in excess over WT hPMS2.
4. Discussion
In this report, we describe the use of three different model systems to characterize the expression and mutation avoidance function of hPMS2-E705K, an allele found in a Turcot syndrome patient [21]. When introduced into yeast, insect, and mouse cells, this mutant Pms1p/hPMS2 was expressed at or near-equivalent levels to its WT counterpart and appeared to heterodimerize with MLH1 with an efficiency comparable to that of WT Pms1p/hPMS2. We demonstrated that the orthologous mutation, pms1-E738K, inactivated the mutation avoidance function of MMR in yeast. Similarly, hPMS2-E705K did not complement the mutator phenotype of Pms2-deficient MEFs. In vivo, Pms2−/− MEFs transfected with hPMS2-E705K exhibited high microsatellite instability, and in vitro purified hMutLα-E705K failed to restore MMR activity in MMR-deficient MEF cytoplasmic extracts. Finally, we found that (1) pms1-E738K exerted dominant effects on MMR in WT yeast only when it was over-expressed, and (2) purified hMutLα-E705K could inhibit MMR-proficient HeLa nuclear extracts only when added in excessive amounts. Taken together, our results strongly suggest that hPMS2-E705K is a recessive loss-of-function allele rather than a dominant allele.
Assuming that hPMS2-E705K is a recessive allele, two alternative explanations for the pathogenesis of Turcot syndrome in the E705K patient [21] can be made. First, a second and possibly different PMS2 mutation in the patient may have escaped detection because of the confounding effect of paralogous genes on PMS2 mutation identification [e.g., 14,17] and thus could account for the presence of the phenotype in the E705K patient but not the parents. Indeed, the use of sequencing techniques designed to circumvent PMS2 pseudogene sequences has led to the identification of compound heterozygosity or homozygosity for recessive PMS2 mutations in families with no history of colorectal cancer and probands with café-au-lait spots and hematological or brain cancers:, 1221delG/2361delCTTC [12], 1169ins20/1169ins20 [13], R134Ter/2184delTC [14], R802Ter/R802Ter [14,16], and Y181Ter/Y181Ter [16]. These findings, coupled with recent reports of heterozygosity for recessive PMS2 mutations associated with autosomal dominant inheritance of HNPCC [17–19], suggest that the population frequency of recessive PMS2 mutations may be as high as the MSH2 mutant allele frequency, albeit with lower penetrance [18].
Second, the E705K patient may have carried a second recessive, but undetected, mutation in another MMR or DNA repair gene, or in a gene involved in assuring DNA replication fidelity. Heterozygosity for recessive mutations in two different mutation avoidance genes could result in a mutator phenotype through either synergistic effects on mutation rates [41] or “unlinked non-complementation” [42].
Until recently, the significance of the evolutionary conservation of residue 705 and surrounding residues was unknown. After the completion of our studies, Kadyrov and colleagues reported that residue 705 of hPMS2 is critical for a latent, single-stranded endonucleolytic activity in hMutLα that is stimulated in in vitro reconstituted MMR reactions [40]. The introduction of the E705K substitution in hPMS2 inactivated the endonucleolytic activity of hMutLα, suggesting that residue 705 is part of a highly conserved amino acid motif in the active site of the latent endonuclease. The same single-stranded endonucleolytic activity has also been observed in yeast MutLα. In this report we extend the above findings by demonstrating that residue 705/738 is critical in vivo for the mutation avoidance function of human PMS2 and yeast Pms1p. Thus, the hPMS2-E705K-associated inactivation of endonucleolytic activity in hMutLα [40] likely explains the loss-of-function phenotype that we observed in both mammalian and yeast cells.
Acknowledgments
We gratefully acknowledge Sandy Dudley for expert technical assistance, and Dr. Andrew Buermeyer for helpful comments. This work was supported by grants to S.M. Deschênes (NIH F32 CA79200-03; University Research and Creativity Grants, Sacred Heart Univ.), N. Erdeniz (NIH GM045413), G. Tomer (Human Frontier Science Program Fellowship), and R.M. Liskay (NIH GM032741).
Footnotes
Publisher's Disclaimer: This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues that you know, and providing a copy to your institution's administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution's website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial
References
- 1.Buermeyer AB, Deschénes SM, Baker SM, Liskay RM. Mammalian DNA mismatch repair. Annu Rev Genet. 1999;33:533–564. doi: 10.1146/annurev.genet.33.1.533. [DOI] [PubMed] [Google Scholar]
- 2.Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- 3.Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 4.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signaling. DNA Repair. 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 6.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 7.Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 8.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, et al. Pmsl and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- 9.Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proc Natl Acad Sci USA. 1997;94:3122–3127. doi: 10.1073/pnas.94.7.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao X, Buermever AB, Narayanan L, Tran D, Baker SM, Prolla TA, et al. Different mutator phenotypes in Mlhl-versus Pms2-deficient mice. Proc Natl Acad Sci USA. 1999;96:6850–6855. doi: 10.1073/pnas.96.12.6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gryfe R, Gallinger S. Germline PMS2 mutations: one hit or two? Gastroenterology. 2005;128:1506–1509. doi: 10.1053/j.gastro.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 12.de Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, Iannelli A, et al. Evidence for a recessive inheritance of Turcot’s syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene. 2000;19:1719–1723. doi: 10.1038/sj.onc.1203447. [DOI] [PubMed] [Google Scholar]
- 13.Trimbath JD, Petersen GM, Erdman SH, Ferre M, Luce MC, Giardiello FM. Cafe-au-lait spots and early onset colorectal neoplasia (a variant of HNPCC?) Fam Cancer. 2001;1:101–105. doi: 10.1023/a:1013881832014. [DOI] [PubMed] [Google Scholar]
- 14.De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet. 2004;74:954–964. doi: 10.1086/420796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agostini M, Tibiletti MG, Lucci-Cordisco E, Chiaravalli A, Morris H, Furlan D, et al. Two PMS2 mutations in a Turcot syndrome family with small bowel cancers. Am J Gastroenterol. 2005;100:1886–1891. doi: 10.1111/j.1572-0241.2005.50441.x. [DOI] [PubMed] [Google Scholar]
- 16.De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, et al. PMS2 mutations in childhood cancer. J Natl Cancer Inst. 2006;98:358–361. doi: 10.1093/jnci/djj073. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, et al. Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res. 2004;64:4721–4727. doi: 10.1158/0008-5472.CAN-03-2879. [DOI] [PubMed] [Google Scholar]
- 18.Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JQ, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128:1160–1171. doi: 10.1053/j.gastro.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 19.Worthley DL, Walsh MD, Barker M, Ruszkiewicz A, Bennett G, Phillips K, et al. Familial mutations in PMS2 can cause autosomal dominant hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;128:1431–1436. doi: 10.1053/j.gastro.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- 21.Miyaki M, Nishio J, Konishi J, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, et al. Drastic instability of tumors and normal tissues in Turcot syndrome. Oncogene. 1997;15:2877–2881. doi: 10.1038/sj.onc.1201668. [DOI] [PubMed] [Google Scholar]
- 22.Yamada NA, Castro A, Farber RA. Variation in the extent of micro satellite instability in human cell lines with defects in different mismatch repair genes. Mutagenesis. 2003;18:277–282. doi: 10.1093/mutage/18.3.277. [DOI] [PubMed] [Google Scholar]
- 23.Nicolaides NC, Littman SJ, Modrich P, Kinzler KW, Vogelstein B. A naturally occurring hPMS2 mutation can confer a dominant negative mutator phenotype. Mol Cell Biol. 1998;18:1635–1641. doi: 10.1128/mcb.18.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdeniz N, Dudley S, Gealv R, Jinks-Robertson S, Liskav RM. Novel PMS1 alleles preferentially affect the repair of primer strand loops during DNA replication. Mol Cell Biol. 2005;25:9221–9231. doi: 10.1128/MCB.25.21.9221-9231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 26.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran PT, Liskay RM. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLα. Mol Cell Biol. 2000;20:6390–6398. doi: 10.1128/mcb.20.17.6390-6398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang Q, Prolla TA, Liskay RM. Functional domains of the Saccharomyces cerevisiae Mlhlp and Pmslp DNA mismatch repair proteins and their relevance to human hereditary nonpolyposis colorectal cancer-associated mutations. Mol Cell Biol. 1997;17:4465–4473. doi: 10.1128/mcb.17.8.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fritzell JA, Narayanan L, Baker SM, Bronner CE, Andrew SE, Prolla TA, et al. Role of DNA mismatch repair in the cytotoxicity of ionizing radiation. Cancer Res. 1997;57:5143–5147. [PubMed] [Google Scholar]
- 31.Farber RA, Liskay RM. Karvotypic analysis of a near-diploid established mouse cell line Cytogenet. Cell Genet. 1974;13:384–396. doi: 10.1159/000130288. [DOI] [PubMed] [Google Scholar]
- 32.Tomer G, Buermeyer AB, Nguyen MM, Liskay RM. Contribution of human mlhl and pms2 ATPase activities to DNA mismatch repair. J Biol Chem. 2002;277:21801–21809. doi: 10.1074/jbc.M111342200. [DOI] [PubMed] [Google Scholar]
- 33.Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen G, Huo X, et al. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair. 2006;5:347–361. doi: 10.1016/j.dnarep.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Buermever AB, Wilson-Van Patten C, Baker SM, Liskav RM. The human MLH1 cDNA complements DNA mismatch repair defects in Mlh1-deficient mouse embryonic fibroblasts. Cancer Res. 1999;59:538–541. [PubMed] [Google Scholar]
- 35.Gurtu VE, Verma S, Grossmann AH, Liskay RM, Skarnes WC, Baker SM. Maternal effect for DNA mismatch repair in the mouse. Genetics. 2002;160:271–277. doi: 10.1093/genetics/160.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas DC, Umar A, Kunkel TA. Measurement of heteroduplex repair in human cell extracts. Methods: Companion Methods Enzymol. 1995;7:187–197. [Google Scholar]
- 37.Chen C, Merrill BJ, Lau PJ, Holm C, Kolodner RD. Saccharomyces cerevisiae pol30 (proliferating cell nuclear antigen) mutations impair replication fidelity and mismatch repair. Mol Cell Biol. 1999;19:7801–7815. doi: 10.1128/mcb.19.11.7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X, Platt JL, Cascalho M. Dimerization of MLH1 and PMS2 limits nuclear localization of MutLa. Mol Cell Biol. 2003;23:3320–3328. doi: 10.1128/MCB.23.9.3320-3328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 40.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLa in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 41.Drotschmann K, Clark AB, Tran HT, Resnick MA, Gordenin DA, Kunkel TA. Mutator phenotypes of yeast strains heterozygous for mutations in the MSH2 gene. Proc Natl Acad Sci USA. 1999;96:2970–2975. doi: 10.1073/pnas.96.6.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stearns T, Botstein D. Unlinked noncomplementation: isolation of new conditional-lethal mutations in each of the tubulin genes of Saccharomvces cereyisiae. Genetics. 1988;119:249–260. doi: 10.1093/genetics/119.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]