

Abstract
We have used site-specific mutagenesis to study the contribution of Glu 74 and the active site residues Gln 38, Glu 41, Glu 54, Arg 65, and His 85 to the catalytic activity and thermal stability of ribonuclease Sa. The activity of Gln38Ala is lowered by one order of magnitude, which confirms the involvement of this residue in substrate binding. In contrast, Glu41Lys had no effect on the ribonuclease Sa activity. This is surprising, because the hydrogen bond between the guanosine N1 atom and the side chain of Glu 41 is thought to be important for the guanine specificity in related ribonucleases. The activities of Glu54Gln and Arg65Ala are both lowered about 1000-fold, and His85Gln is totally inactive, confirming the importance of these residues to the catalytic function of ribonuclease Sa. In Glu74Lys, kcat is reduced sixfold despite the fact that Glu 74 is over 15 Å from the active site. The pH dependence of kcat/KM is very similar for Glu74Lys and wild-type RNase Sa, suggesting that this is not due to a change in the pK values of the groups involved in catalysis. Compared to wild-type RNase Sa, the stabilities of Gln38Ala and Glu74Lys are increased, the stabilities of Glu41Lys, Glu54Gln, and Arg65Ala are decreased and the stability of His85Gln is unchanged. Thus, the active site residues in the ribonuclease Sa make different contributions to the stability.
Keywords: Ribonuclease Sa, active-site mutants, catalytic activity, specificity, thermal stability
In enzymes, the number of amino acids that participate in catalysis is generally only a small fraction of the total number of amino acids that the enzyme contains. However, this differs considerably among enzymes. Catalase is a large enzyme (≈2000 amino acids) with a small substrate (H2O2), while ribonuclease Sa (RNase Sa) is a small enzyme (96 amino acids) with a large substrate (RNA). The amino acids most likely to participate in catalysis at the active site of an enzyme are: His, Asp, Arg, Glu, and Lys (Bartlett et al. 2002). It has long been thought that residues that participate in catalysis are probably optimized for their participation in catalysis but not for their contribution to enzyme stability (Williams 1972; Richards 1974; Warshel 1978). This idea is supported by experimental studies (Meiering et al. 1992; Shoichet et al. 1995; Beadle and Shoichet 2002) and theoretical studies (Warshel 1998; Elcock 2001). It is also consistent with the idea that enzymes and their active sites often need to be “flexible” (Endrizzi et al. 2000; Hammes-Schiffer 2002). As proteins become larger, we would expect them to become more stable because they bury more hydrophobic side chains and more peptide groups, and both contribute to the stability (Pace 2001). However, protein stability does not correlate with increasing size. As proteins become larger, the content of charged amino acids increases to a greater extent than the content of polar and hydrophobic amino acids (Kajander et al. 2000). This may allow proteins to destabilize themselves by burying charged groups to counterbalance the expected enhanced stability resulting from the burial of more peptide groups and nonpolar side chains (Kajander et al. 2000). It may be that larger enzymes are better able to tolerate amino acids at the active site that destabilize than smaller enzymes.
RNase Sa, a guanosine specific ribonuclease, is the smallest microbial ribonuclease, and one of the smallest enzymes. The structure of the enzyme and its complexes with 2′-GMP, 3′-GMP and guanosine 2′,3′-cyclophosphorothioate were determined in crystals at atomic resolution (Sevcik et al. 1991, 1993, 1996, 2002), and the solution structure has been determined with NMR (Laurents et al. 2001). The stability of RNase Sa has been studied as a function of temperature, pH, and salt concentration (Pace et al. 1998). In addition, many mutants have been studied to gain a better understanding of the forces stabilizing the native, biologically active conformation (Grimsley et al. 1999; Pace et al. 2000, 2001; Shaw et al. 2001). The pKs of the ionizable residues have been determined (Huyghues-Despointes et al. 2003; Laurents et al. 2003). The catalytic properties of the enzyme were studied and compared to those of other guanosine-specific ribonucleases (Yakovlev et al. 1992).
The identification of the residues that define the catalytic properties of RNase Sa has not been studied directly using site-directed mutants. A comparison of the amino acid sequences of the microbial ribonucleases and their crystal structures suggests that the key residues of RNase Sa involved in catalysis are Glu 54, Arg 65, and His 85 (Hill et al. 1983; Sevcik et al. 1993). The structures also suggest that the specificity of RNase Sa for the guanine base is due to a network of hydrogen bonds involving the backbone groups of Gln 38, Asn 39, and Arg 40, and the side chain carboxyl group of Glu 41 (Fig. 1 ▶). Residue Glu 74 does not belong to the enzyme active site, but its replacement with Lys may be responsible for the previously observed decrease in activity of the 5K RNase Sa basic variant (Shaw et al. 2001). The goal of the research described here was to gain a better understanding of the contribution of these residues to the catalytic activity and conformational stability of RNase Sa. We report steady-state kinetic studies and thermal denaturation studies of wild-type RNase Sa and the following mutants: Gln38Ala, Glu41Lys, Glu54Gln, Arg65Ala, Glu74Lys, and His 85Gln.
Figure 1.

The active site of ribonuclease Sa in the complex with exo guanosine 2′,3′-cyclophosphorothioate. Possible hydrogen bonds are represented by dotted lines. The PDB identifier for RNase Sa structure is 1RSN (Sevcik et al. 1993).
Results and Discussion
The role of Gln 38 in substrate binding
Based on the crystal structure, the backbone amide group of Gln 38 forms a hydrogen bond to the N7 atom of the guanine base of nucleotides bound at the active site of RNase Sa, but the side chain of Gln 38 forms no intermolecular hydrogen bonds with the substrate or intramolecular hydrogen bonds with other groups in the protein. The kinetic parameters characterizing the cleavage of poly(I) by RNase Sa and its mutants are shown in Table 1. The activity of the Gln38Ala mutant of RNase Sa is 13 times lower than that of the native enzyme. The increase in KM suggests that the substrate does not bind as well to the mutant enzyme, and this is accompanied by an eightfold decrease in kcat. This suggests that the catalytic groups of the enzyme are not as well positioned for catalysis in the Gln38Ala mutant as they are in the native enzyme. This could be due to a small change in the conformation of the enzyme or to a small change in the positioning of the substrate.
Table 1.
Kinetic parameters characterizing the cleavage of poly(I) and GpU by RNase Sa and six mutants at 25°C and pH 6.5
| RNase | Substrate | kcat (sec−1) | KM × 104 (M) | kcat/KM × 10−4 (M−1 sec−1) |
| Wild type | poly(I) | 178 ± 10 | 1.51 ± 0.15 | 117.9 |
| GpU | 1.9 ± 0.1 | 5.5 ± 0.3 | 0.35 | |
| Gln38Ala | poly(I) | 23 ± 2 | 2.5 ± 0.2 | 9.2 |
| Glu41Lys | poly(I) | 173 ± 11 | 1.6 ± 0.2 | 108 |
| GpU | 1.8 ± 0.1 | 10.0 ± 0.5 | 0.18 | |
| Glu54Gln | poly(I) | 0.50 ± 0.05 | 3.1 ± 0.3 | 0.16 |
| Arg65Ala | poly(I) | 0.21 ± 0.03 | 2.0 ± 0.2 | 0.11 |
| Glu74Lys | poly(I) | 34 ± 4 | 1.7 ± 0.2 | 20 |
| GpU | 0.50 ± 0.05 | 5.7 ± 0.3 | 0.09 | |
| His85Glna | poly(I) | — | — | <0.007 |
a No activity was observed at enzyme concentrations below 1 • 10−6 M.
The role of Glu 41 in substrate binding
Glu 41 in RNase Sa is a conserved residue in all guanine-specific RNases where it forms a hydrogen bond with the N1 atom of the guanine base of substrates (Hill et al. 1983; Sevcik et al. 1993). The replacement of equivalent residues in barnase (Glu60) and RNase T1 (Glu46) by Gln substantially reduced the enzyme activity (Steyaert et al. 1991; Bastyns et al. 1994; Chitester and Walz 2002). Consequently, it seemed likely that the Glu41Lys mutant of RNase Sa would have an even lower activity than mutants in which the Glu is replaced by Gln. However, the activity of the Glu41Lys mutant is almost the same as native RNase Sa (Table 1). Because the carboxylate of Glu 41 is a hydrogen bond acceptor and the amino group of a Lys can only act as a hydrogen bond donor, the Lys cannot be simply replacing the Glu in its interactions with substrate. This may suggest that Glu 41 does not participate in substrate binding in solution as it does in the crystal (Sevcik et al. 1993), or that the Lys is able to bind to the polyanionic substrate in a different way to compensate for the loss of the interactions with Glu. To evaluate the contribution of interaction of the charged side chain of Lys41 of the Glu41Lys mutant with the poly(I) phosphate groups into the enzymatic reaction rate we compared activities of Glu41Lys and the wild-type RNase Sa with dinucleoside phosphate GpU as substrate. With this substrate the Glu41Lys activity was half of that for the wild-type RNase Sa (Table 1). This shows that the interaction of Lys41 with poly(I) cannot be responsible for the high activity of Glu41Lys on a polymeric substrate. Therefore, Glu41 of RNase Sa is not involved in substrate binding. In this respect RNase Sa differs from other guanyl RNases. It should be noted that hydrogen bonding between protein and O6 atom of guanyl base is enough to realize guanyl specificity.
The functional role of Glu 54, Arg 65, and His 85 at the active site of RNase Sa
Based on homology between the primary structures of RNase Sa and other guanine-specific RNases, Glu 54, Arg 65, and His 85 are thought to play key catalytic roles at the active site of RNase Sa. In the microbial ribonuclease family, the catalytic mechanism of RNase T1 has been studied in most detail: first by the Walz group (Osterman and Walz 1978) and the Saenger group (Heinemann and Saenger 1982; Pace et al. 1991; Koellner et al. 1992), and more recently, using protein engineering, by the Steyaert group (Steyaert 1997) and the Walz group (Chitester and Walz 2002). The results suggest that the transesterification reaction catalyzed by the microbial ribonucleases consists of a nucleophilic displacement at the phosphorus atom of the 5′ leaving group by the 2′ entering oxygen atom. The reaction proceeds through the formation of an intermediate pentacovalent phosphorus atom. In the case of RNase Sa, the role of the side chain carboxyl of Glu 54 is to accept a proton from the ribose 2′OH group, and the role of the protonated imidazole ring of His 85 is to donate a proton to the leaving 5′O group. The role of the positively charged side chain of Arg 65 is to promote formation of a negatively charged, pentacovalent intermediate state of the phosphate group.
The substitution of Glu 54, Arg 65, or His 85 results in dramatic decreases in the RNase Sa activity (Table 1). This confirms their important contribution to the catalytic function. His85Gln has no measurable catalytic activity. Similar substitutions of the functionally equivalent His 102 in barnase with Asp (Paddon and Hartley 1987) or Ala (Meiering et al. 1992) also resulted in complete inactivation of the enzyme. In the Glu54Gln mutant, the affinity for the substrate was only halved, but there was a 356-fold decrease in kcat. In the Arg65Ala mutant, the affinity for substrate decreased slightly, but kcat was lowered by approximately three orders of magnitude. This is consistent with the idea that the role of this residue is to promote the formation of the transition state of the reaction by favorable coulombic interactions with the negatively charged pentacovalent phosphate (Meiering et al. 1992; Yakovlev et al. 1998).
The effect of the Glu74Lys substitution on the RNase Sa activity
Glu 74 is located over 15 Å from the active site of RNase Sa. Nevertheless, replacement of this residue with lysine results in a sixfold decrease in the enzyme activity for poly(I) as substrate and in a fourfold decrease for GpU, mainly due to lowering kcat (Table 1). Because this substitution replaced a negative with a positive charge, we thought this change might alter the pK values of groups involved in catalysis. To test this, we measured the pH dependence of the kcat/KM ratio for the cleavage of poly(I) by this mutant and by the wild-type enzyme (Fig. 2 ▶). This allowed us to estimate the ionization constants of the ionizable groups of the catalytic residues (Table 2). The changes are small and not in the direction expected for replacing a negative charge with a positive charge. This suggests that the decrease in the activity of the mutant is not due to changes in the pK values of the catalytic groups. Instead, it more likely reflects a change in the orientation of the enzyme catalytic groups near the site of the bond to be cleaved in the substrate.
Figure 2.

pH dependence of log(kcat/KM) for the cleavage of poly(I) by RNase Sa (filled circles) and its Glu74Lys mutant (open circles). Curves were calculated using the data in Table 2 and equation 1.
Table 2.
Kinetic parameters and ionization constants of ionogenic groups of RNase Sa and its Glu74Lys mutant, which define the pH dependence of kcat/KM for poly(I) hydrolysis (Fig. 2 ▶)
| Parameter | Wild type | Glu74Lys |
| kcat/KM 10−5 (M−1 sec−1) | 17.0 ± 1.5 | 2.7 ± 0.2 |
| pKA0 | 4.39 ± 0.23 | 4.46 ± 0.16 |
| pKB0 | 5.68 ± 0.09 | 5.66 ± 0.06 |
| pKC0 | 7.02 ± 0.08 | 7.22 ± 0.06 |
| pKD0 | 8.35 ± 0.24 | 8.78 ± 0.26 |
See Materials and Methods for details of the analysis.
Thermal stability of RNase Sa mutants
The stability of the proteins was studied using differential scanning calorimetry and typical results are shown in Figure 3 ▶. The studies were done at pH 5.5 and at pH 7 where most previous studies of the wild-type RNase Sa stability were done (Pace et al. 1998). The maximum activity of RNase Sa is near pH 6.5 (Fig. 2 ▶). The reversibility of the thermal denaturation is good, generally between 90%–95%. The ratio R = ΔHcal/Δ Heff is between 0.89 and 1.04 for all measurements, suggesting a close approximation to a two-state unfolding process. The results are summarized in Table 3.
Figure 3.

Temperature dependence of the partial molar heat capacity of RNase Sa (1) and six mutants: Gln38Ala (2), Glu54Gln (3), Arg65Ala (4), His85Gln (5), Glu41Lys (6), and Glu74Lys (7) at pH 7.
Table 3.
Thermal denaturation parameters of RNase Sa and six mutants
| RNase | Td (°C) | ΔTda (°C) | ΔΔGb (kcal/mole) | ΔHcal (kcal/mole) | ΔHeff (kcal/mole) | Rc |
| pH 7.0 | ||||||
| Wild type | 49.0 | — | — | 92 | 103 | 0.89 |
| Gln38Ala | 52.5 | 3.5 | 1.0 | 109 | 110 | 0.99 |
| Glu41Lys | 46.5 | −2.5 | −0.7 | 90 | 100 | 0.90 |
| Glu54Gln | 43.1 | −5.9 | −1.7 | 81 | 87 | 0.93 |
| Arg65Ala | 45.6 | −3.4 | −1.0 | 81 | 90 | 0.90 |
| Glu74Lys | 52.1 | 3.1 | 0.9 | 101 | 113 | 0.89 |
| His85Gln | 49.1 | 0.1 | 0 | 90 | 89 | 1.01 |
| pH 5.5 | ||||||
| Wild type | 55.6 | — | — | 97 | 101 | 0.96 |
| Gln38Ala | 56.9 | 1.3 | 0.4 | 98 | 103 | 0.95 |
| Glu41Lys | 50.5 | −5.1 | −1.5 | 97 | 101 | 0.96 |
| Glu54Gln | 47.7 | −7.9 | −2.3 | 89 | 92 | 0.97 |
| Arg65Ala | 51.1 | −4.5 | −1.3 | 81 | 78 | 1.04 |
| Glu74Lys | 56.0 | 0.4 | 0.1 | 103 | 99 | 1.04 |
| His85Gln | 55.5 | −0.1 | 0 | 94 | 105 | 0.90 |
a ΔTd = Td (mutant) − Td (wild type).
b ΔΔG (= ΔTd × ΔSd) is the difference in the free energies of unfolding of wild-type and mutant RNase Sa at Td, and ΔSd (= ΔHcal/Td) is the entropy of unfolding of wild-type RNase Sa. A negative sign indicates a decrease in stability.
cR = ΔHcal/ΔHeff.
The Gln38Ala mutant is slightly more stable than RNase Sa (Table 3). This was expected, because the Gln side chain is exposed to solvent and not hydrogen bonded. In contrast, the amide group of Asn39 forms three good hydrogen bonds, and the Asn39Ala mutant is over 2 kcal/mole less stable than wild-type RNase Sa (Hebert et al. 1998). The backbone atoms of both of these residues form hydrogen bonds to a guanosine residue in nucleotides bound to RNase Sa.
The Glu41Lys and Glu74Lys mutants were studied previously (Pace et al. 2000). The Glu41Lys mutant is about 1 kcal/mole less stable and the Glu74Lys mutant 1 kcal/mole more stable than RNase Sa at pH 7 (Table 3; Pace et al. 2000). The side chains of both Glu 41 and Glu 74 are exposed to solvent and form no hydrogen bonds. In our previous study, we showed that an increase in stability was expected for both mutants based on more favorable Coulombic interactions of the positive charge of the amino group with the excess negative charges on the folded state of RNase Sa. The observed decrease in stability for Glu41Lys led us to the surprising conclusion that Coulombic interactions between the charged groups are more favorable in the denatured state ensemble than in the native state for this mutant (Pace et al. 2000).
The Glu54Ala mutant is about 2 kcal/mole less stable than RNase Sa. This is expected, because Glu 54 is largely buried at the active site and has favorable Coulombic interactions with the neighboring positive charges of Arg 65, Arg 69, and His 53. Decreases in stability are also observed when the equivalent Glu residues are replaced with Ala in barnase (Meiering et al. 1992) and RNase T1 (Steyaert and Wyns 1993).
The Arg65Ala mutant is 1 kcal/mole less stable than RNase Sa. Arg 65 is largely buried at the active site. The mutant will lose a favorable Coulombic interaction with Glu 54, but the repulsive interactions with the other positive charges at the active site will be reduced so the decrease in stability is lower than for the Glu54Ala mutant.
We previously studied the effect of net charge and salt concentration on the pK of His 85 in RNase Sa (Huyghues-Despointes et al. 2003). The side chain of His 85 is partially buried at the active site. It is not hydrogen bonded, but there are four charged residues (Glu 54, Arg 65, Arg 69, Asp 84) within 10 Å. The His 85 pK = 6.35 in 0.1 M NaCl, and depends only slightly on salt concentration. This is very similar to the pK expected based on model peptides, and this suggests that electrostatic interactions with the neighboring charges largely cancel. The fact that the stability of His85Gln is almost identical to that of RNase Sa at both pH 5.5 and 7.0 provides further support for this idea.
Conclusion
Our results clearly show that Glu 54, Arg 65, and His 85 make important contributions to the catalytic activity of wild type RNase Sa. It is interesting that the activity of Glu74Lys is substantially lower than the wild type because the mutation site is over 15 Å from the active site. It is suprising that the activity of Glu41Lys is almost identical to the wild type because the carboxyl of Glu 41 is clearly involved in binding the guanine base in crystal structures of RNase Sa with nucleotides bound. Glu 41, Glu 54, and Arg 65 make important favorable contributions to the stability of RNase Sa. Gln 38 and Glu 74 contribute unfavorably to the stability, but neither of their side chains are directly involved in binding or catalysis. Thus, unlike most of the enzymes that have been studied, for RNase Sa two of the residues actively involved in catalysis and one involved in substrate specificity make important favorable contributions to the stability. Perhaps because RNase Sa is one of the smallest enzymes, it cannot tolerate unfavorable contributions to the stability as well as a larger enzyme might.
Materials and methods
Oligonucleotides for the construction of E41K and E74K mutants of RNase Sa were synthesized by the Gene Technologies Laboratory at Texas A&M University. Oligonucleotides for the construction of Q38A, E54Q, R65A, and H85Q mutants were synthesized by the Integrated DNA Technologies. Poly(I), GpU and PIPES were obtained from Sigma.
The E41K and E74K variants of RNase Sa were constructed, expressed, and purified according to Hebert et al. (1998). The Q38A, E54Q, R65A, and H85Q mutants were constructed using the QuickChange site-directed mutagenesis kit from Stratagene. All mutant genes were sequenced at the Gene Technologies Laboratory at Texas A&M University to confirm the introduction of the mutation. The Q38A, E54Q, R65A, and H85Q mutants were expressed and purified according to Shaw et al. (2001). Protein purity was confirmed using SDS-polyacrylamide gel electrophoresis.
Steady-state kinetic studies of ribonucleases catalyzing hydrolysis of poly(I) and GpU were performed at 25°C and pH 6.5. The buffer used was 0.05 M Tris, 0.1 M potassium chloride, and 0.05 M sodium acetate. Concentrations of RNase Sa and its mutants were determined spectrophotometrically using the same molar extinction coefficient ɛ280 = 12,300 M−1 cm−1 (Hebert et al. 1997). The poly(I) and GpU concentrations were measured using ɛ248 = 10,000 M−1 cm−1 at pH 7.8 (Chamberlin and Patterson 1965), and ɛ280 = 10,600 M−1 cm−1 at pH 7.0 (Zabinski and Walz 1976), respectively. Initial reaction rates were determined by recording changes in absorption at 248 nm for poly(I) and 280 nm for GpU, using the difference molar extinction coefficients of 1330 M−1 cm−1 (Yakovlev et al. 1992) and 850 M−1 cm−1 (Zabinski and Walz 1976), respectively. To obtain kinetic parameters, initial rates were measured for seven to eight substrate concentrations. The rate of substrate hydrolysis changed hyperbolically as the substrate concentration increased. Values of kcat and KM were determined from Lineweaver-Burk plots using a weighted least-square procedure.
For RNase Sa and its Glu74Lys mutant, the pH dependence of catalytic parameters of poly(I) hydrolysis was determined in the pH interval 4.0–8.5 using the same buffer titrated to the desired pH with acetic acid. Initial reaction rates were determined using the same value of the difference molar extinction coefficient 1330 M−1 cm−1 because poly(I) is not titrated in the pH interval studied. Analysis of the pH dependences of kcat/KM was carried out according to Osterman and Walz (1978) using the equation
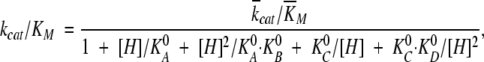 |
(1) |
where 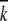 cat and
cat and 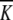 M are the pH independent values of the turnover number and the Michaelis constant, respectively; KA0, KB0, KC0, and KD0 are macroscopic acid dissociation constants characterizing pertinent groups on the free enzyme or substrate (Osterman and Walz 1978).
M are the pH independent values of the turnover number and the Michaelis constant, respectively; KA0, KB0, KC0, and KD0 are macroscopic acid dissociation constants characterizing pertinent groups on the free enzyme or substrate (Osterman and Walz 1978).
Microcalorimetric measurements were carried out on a DASM-4 microcalorimeter (NPO Biopribor) in 0.48-mL cells at a heating rate of 1 K/min on 0.4–1.2 mg/mL protein solutions. Curves were corrected for the instrumental baseline obtained by heating the solvent used for the protein solutions. The reversibility of denaturation was checked routinely by sample reheating after cooling in the calorimetric cell. The partial molar heat capacity of the protein (Cp), denaturation temperature (Td), calorimetric denaturation enthalpy (ΔHcal), and effective or van’t Hoff denaturation enthalpy (ΔHeff) were determined as described elsewhere (Makarov et al. 1991). To analyze functions of excess heat capacity, the SCAL2 software package developed at the Institute of Protein Research was used. The accuracy of the calorimetric and effective enthalpies was ±6%, that of Td ± 0.1°C. The buffers used were 0.01 M sodium acetate, 0.05 M potassium chloride for pH 5.5, and 0.03 M PIPES for pH 7.0.
Acknowledgments
This work was supported by NIH FIRCA grant TW01058, RFBR grants 02-04-48259 and 02-04-49110, the Physicochemical Biology program of the Russian Academy of Sciences, NATO grant LST.CLG.979534, INTAS-RFBR grant 97-245, NIH grant GM 37039, Welch Foundation grant BE-1060, and the Tom and Jean McMullin Professorship. We thank Larry Dangott and the Protein Chemistry Laboratory for help with this research.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03176803.
References
- Bartlett, G.J., Porter, C.T., Borkakoti, N., and Thornton, J.M. 2002. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 324 105–121. [DOI] [PubMed] [Google Scholar]
- Bastyns, K., Froeyer, M., Diaz, J.F., Volckaert, G., and Engelborghs, Y. 1994. The role of Glu-60 in the specificity of the recombinant ribonuclease from Bacillus amyloliquefaciens (barnase) towards dinucleotides, poly(A) and RNA. Biochem. J. 300 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle, B.M. and Shoichet, B.K. 2002. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 321 285–296. [DOI] [PubMed] [Google Scholar]
- Chamberlin, M.J. and Patterson, D.L. 1965. Physical and chemical characterization of the ordered complexes formed between polyinosonic acid, polycytidylic acid and their deoxyribo-analogues. J. Mol. Biol. 12 410–428. [DOI] [PubMed] [Google Scholar]
- Chitester, B.J. and Walz, F.G. 2002. Kinetic studies of guanine recognition and a phosphate group subsite on ribonuclease T1 using substitution mutants at Glu46 and Lys41. Arch. Biochem. Biophys. 406 73–77. [DOI] [PubMed] [Google Scholar]
- Elcock, A.H. 2001. Prediction of functionally important residues based solely on the computed energetics of protein structure. J. Mol. Biol. 312 885–896. [DOI] [PubMed] [Google Scholar]
- Endrizzi, J.A., Beernink, P.T., Alber, T., and Schachman, H.K. 2000. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: Implications for allosteric regulation induced cell migration. Proc. Natl. Acad. Sci. 97 5077– 5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsley, G.R., Shaw, K.L., Fee, L.R., Alston, R.W., Huyghues-Despointes, B.M., Thurlkill, R.L., Scholtz, J.M., and Pace, C.N. 1999. Increasing protein stability by altering long-range coulombic interactions. Protein Sci. 8 1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer, S. 2002. Impact of enzyme motion on activity. Biochemistry 41 13335–13343. [DOI] [PubMed] [Google Scholar]
- Hebert, E.J., Grimsley, G.R., Hartley, R.W., Horn, G., Schell, D., Garcia, S., Both, V., Sevcik, J., and Pace, C.N. 1997. Purification of ribonucleases Sa, Sa2, and Sa3 after expression in Escherichia coli. Protein Expr. Purif. 11 162–168. [DOI] [PubMed] [Google Scholar]
- Hebert, E.J., Giletto, A., Sevcik, J., Urbanikova, L., Wilson, K.S., Dauter, Z., and Pace, C.N. 1998. Contribution of a conserved asparagine to the conformational stability of ribonucleases Sa, Ba, and T1. Biochemistry 37 16192–16200. [DOI] [PubMed] [Google Scholar]
- Heinemann, U. and Saenger, W. 1982. Specific protein–nucleic acid recognition in ribonuclease T1–2′-guanylic acid complex: An X-ray study. Nature 299 27–31. [DOI] [PubMed] [Google Scholar]
- Hill, C.P., Dodson, G.G., Heineman, U., Saenger, W., Mitsui, Y., Nacamura, K., Borisov, S., Tishenko, G., Polyakov, K., and Pavlovsky, S. 1983. The structural and sequence homology of a family of microbial ribonucleases. Trends Biochem. Sci. 8 364–369. [Google Scholar]
- Huyghues-Despointes, B.M., Thurlkill, R.L., Daily, M.D., Schell, D., Briggs, J.M., Antosiewicz, J.M., Pace, C.N., and Scholtz, J.M. 2003. pK values of histidine residues in ribonuclease Sa: Effect of salt and net charge. J. Mol. Biol. 325 1093–1105. [DOI] [PubMed] [Google Scholar]
- Kajander, T., Kahn, P.C., Passila, S.H., Cohen, D.C., Lehtio, L., Adolfsen, W., Warwicker, J., Schell, U., and Goldman, A. 2000. Buried charged surface in proteins. Structure Fold. Des. 8 1203–1214. [DOI] [PubMed] [Google Scholar]
- Koellner, G., Choe, H.W., Heinemann, U., Grunert, H.P., Zouni, A., Hahn, U., and Saenger, W. 1992. His92Ala mutation in ribonuclease T1 induces segmental flexibility. An X-ray study. J. Mol. Biol. 224 701–713. [DOI] [PubMed] [Google Scholar]
- Laurents, D., Perez-Canadillas, J.M., Santoro, J., Rico, M., Schell, D., Pace, C.N., and Bruix, M. 2001. Solution structure and dynamics of ribonuclease Sa. Proteins 44 200–211. [DOI] [PubMed] [Google Scholar]
- Laurents, D.V., Huyghues-Despointes, B.M., Bruix, M., Thurlkill, R.L., Schell, D., Newsom, S., Grimsley, G.R., Shaw, K.L., Trevino, S., Rico, M., et al. 2003. Charge–charge interactions are key determinants of the pK values of ionizable groups in ribonuclease Sa (pI = 3.5) and a basic variant (pI = 10.2). J. Mol. Biol. 325 1077–1092. [DOI] [PubMed] [Google Scholar]
- Makarov, A.A., Protasevich, I.I., Frank, E.G., Grishina, I.B., Bolotina, I.A., and Esipova, N.G. 1991. The number of cooperative regions in a pepsin molecule depends on the pH of the medium. Biochim. Biophys. Acta 1078 283–288. [DOI] [PubMed] [Google Scholar]
- Meiering, E.M., Serrano, L., and Fersht, A.R. 1992. Effect of active site residues in barnase on activity and stability. J. Mol. Biol. 225 585–589. [DOI] [PubMed] [Google Scholar]
- Osterman, H.O. and Walz, F.G. 1978. Subsites and catalytic mechanism of ribonuclease T1: Kinetic studies using GpA, GpC, GpG, and GpU as substrates. Biochemistry 17 4124–4130. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 2001. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry 40 310–313. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Heinemann, U., Hahn, U., and Saenger, W. 1991. Ribonuclease T1: Structure, function and stability. Angew. Chem. Int. Ed. Engl. 30 343–360. [Google Scholar]
- Pace, C.N., Hebert, E.J., Shaw, K.L., Schell, D., Both, V., Krajcikova, D., Sevcik, J., Wilson, K.S., Dauter, Z., Hartley, R.W., et al. 1998. Conformational stability and thermodynamics of folding of ribonucleases Sa, Sa2 and Sa3. J. Mol. Biol. 279 271–286. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Alston, R.W., Shaw, K.L. 2000. Charge–charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 9 1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N., Horn, G., Hebert, R.J., Bechert, J., Shaw, K., Urbanikova, L., Scholtz, J.M., and Sevcik, J. 2001. Tyrosine hydrogen bonds make a large contribution to protein stability. J. Mol. Biol. 312 393–404. [DOI] [PubMed] [Google Scholar]
- Paddon, C.J. and Hartley, R.W. 1987. Expression of Bacillus amyloliquefaciens extracellular ribonuclease (barnase) in Escherichia coli following an inactivation mutation. Gene 40 231–239. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1974. The interpretation of protein structures: Total volume, group volume distributions and packing density. J. Mol. Biol. 82 1–14. [DOI] [PubMed] [Google Scholar]
- Sevcik, J., Dodson, E., and Dodson, G.G. 1991. Determination and restrained least-squares refinement of the structures of ribonuclease Sa and its complex with 3′-guanylic acid at 1.8 Å resolution. Acta Crystallogr. B47 240–253. [PubMed] [Google Scholar]
- Sevcik, J., Zegers, I., Wyns, L., Dauter, Z., and Wilson, K.S. 1993. Complex of ribonuclease Sa with a cyclic nucleotide and a proposed model for the reaction intermediate. Eur. J. Biochem. 216 301–305. [DOI] [PubMed] [Google Scholar]
- Sevcik, J., Dauter, Z., Lamzin, V.S., and Wilson, K.S. 1996. Ribonuclease from Streptomyces aureofaciens at atomic resolution. Acta Crystallogr. D 52 327–344. [DOI] [PubMed] [Google Scholar]
- Sevcik, J., Lamzin, V.S., Dauter, Z., and Wilson, K.S. 2002. Atomic resolution data reveal flexibility in the structure of RNase Sa. Acta Crystallogr. D 58 1307–1313. [DOI] [PubMed] [Google Scholar]
- Shaw, K.L., Grimsley, G.R., Yakovlev, G.I., Makarov, A.A., and Pace, C.N. 2001. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 10 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet, B.K., Baase, W.A., Kuroki, R., and Matthews, B.W. 1995. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. 92 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyaert, J. 1997. A decade of protein engineering on ribonuclease T1—Atomic dissection of the enzyme–substrate interactions. Eur. J Biochem. 247 1–11. [DOI] [PubMed] [Google Scholar]
- Steyaert, J. and Wyns, L. 1993. Functional interactions among the His40, Glu58 and His92 catalysts of ribonuclease T1 as studied by double and triple mutants. J. Mol. Biol. 229 770–781. [DOI] [PubMed] [Google Scholar]
- Steyaert, J., Opsomer, C., Wyns, L., and Stassens, P. 1991. Quantitative analysis of the contribution of Glu46 and Asn98 to the guanosine specificity of ribonuclease T1. Biochemistry 30 494–499. [DOI] [PubMed] [Google Scholar]
- Warshel, A. 1978. Energetics of enzyme catalysis. Proc. Natl. Acad. Sci. USA 75 5250–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel, A. 1998. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 273 27035–27038. [DOI] [PubMed] [Google Scholar]
- Williams, R.J. 1972. The entactic state. Cold Spring Harb. Symp. Quant. Biol. 36 53–62. [DOI] [PubMed] [Google Scholar]
- Yakovlev, G.I., Moiseyev, G.P., Bezborodova, S.I., Both, V., and Sevcik, J. 1992. A comparative study on the catalytic properties of guanyl-specific ribonucleases. Eur. J. Biochem. 204 187–190. [DOI] [PubMed] [Google Scholar]
- Yakovlev, G.I., Struminskaya, N.K., Znamenskaya, L.V., Kipenskaya, L.V., Leschinskaya, I.B., and Hartley, R.W. 1998. Contribution of arginine-82 and arginine-86 to catalysis of RNases from Bacillus intermedius (binase). FEBS Lett. 428 57–58. [DOI] [PubMed] [Google Scholar]
- Zabinski, M. and Walz, F.G. 1976. Subsites and catalytic mechanism of ribonuclease T1: Kinetic studies using GpC and GpU as substrates. Arch. Biochem. Biophys. 175 558–564. [DOI] [PubMed] [Google Scholar]