

Abstract
Proteins are renowned for their specificity of function. There is, however, accumulating evidence that many proteins, from enzymes to antibodies, are functionally promiscuous. Promiscuity is of considerable physiological importance. In the immune system, cross-reactive or multispecific antibodies are implicated in autoimmune and allergy conditions. In most cases, however, the mechanism behind promiscuity and the relationship between specific and promiscuous activities are unknown. Are the two contradictory? Or can a protein exhibit several unrelated activities each of which is highly specific? To address these questions, we studied a multispecific IgE antibody (SPE7) elicited against a 2,4-dinitrophenyl hapten (DNP). SPE7 is able to distinguish between closely related derivatives such as NP (nitrophenol) and DNP, yet it can also bind a number of unrelated ligands. We find that, like DNP, the cross-reactants are themselves bound specifically—close derivatives of these cross-reactants show very low or no binding to SPE7. It has been suggested that cross-reactivity is simply due to “hydrophobic stickiness”, nonspecific interactions between hydrophobic ligands and binding sites. However, partitioning experiments reveal that affinity for SPE7 is unrelated to ligand hydrophobicity. These data, combined with crystal structures of SPE7 in complex with four different ligands, demonstrate that each cross-reactant is bound specifically, forming different hydrogen bonds dependant upon its particular chemistry and the availability of complementary antibody residues. SPE7 is highly homologous to the germline antinitrophenol (NP) antibody B1–8. By comparing the sequences and binding patterns of SPE7 and B1–8, we address the relationship between affinity maturation, specificity, and cross-reactivity.
Keywords: Catalytic promiscuity, multispecificity, moonlighting, autoimmunity, hydrophobicity
Promiscuity is not a new concept but its mechanism has never been explored in depth. Many enzymes have been shown to catalyse reactions and utilize substrates in addition to their supposed original function and not necessarily related to it (O’Brien and Herschlag 1999; James and Tawfik 2001; Copley 2003). Drug action is based almost solely on promiscuity—a drug commonly binds an active site that evolved to bind another molecule altogether. Likewise, many antibodies elicited against a particular antigen have also been shown to bind other, structurally unrelated antigens (Cameron and Erlanger 1977; Mariuzza and Poljak 1993; Webster et al. 1994). Such phenomenon have been referred to alternately as cross-reactivity, multispecificity, molecular mimicry, and moonlighting—although the terms properly differ in meaning (James and Tawfik 2003). Promiscuity, or multispecificity is thought to play a key role in enabling proteins to rapidly evolve new functions (O’Brien and Herschlag 1999; James and Tawfik 2003). While protein specificity has been widely studied, the mechanisms behind promiscuity, and the relationships between specificity and promiscuity are largely unexplored. How multiple functions are performed by a single protein sequence is mostly unclear. Are specificity and promiscuity contradictory; or can a protein exhibit a promiscuous activity that is itself highly specific?
A plausible possibility is that the very stereochemistry of protein binding sites predispose them to perform more than one function. There appear to be common active-site features such as rugged matrixes of apolar and polar residues that can promote binding or catalysis of more than one substrate and reaction (O’Brien and Herschlag 1999; DeLano et al. 2000; James and Tawfik 2001). Antibodies, for instance, generally have a high frequency of aromatic residues such as tryptophan or tyrosine in their binding sites. A long-held view has claimed that this imparts antibodies a property of “hydrophobic stickiness,” allowing several different antigens to bind through nonspecific hydrophobic interaction (Padlan 1994). Indeed, certain antibodies were shown to bind a range of ligands with affinities directly related to ligand hydrophobicity (Barbas 3rd et al. 1997). However, this view of nonspecific binding is at odds with the exquisite specificity with which proteins, such as enzymes and antibodies, are known to interact with their substrates. In the case of antibodies, it is imperative that all antigens are differentiated specifically to mount an effective and targeted immune response. How then do binding sites accomplish being both specific and cross-reactive?
The above question can be resolved if the cross-reactants are related or contain the same binding epitope as the original antigen. This type of cross-reactivity, where the target and cross-reactant are related and interact with the binding site in a similar manner has been termed molecular or antigen mimicry. Molecular mimicry constitutes by far the most common type of cross-reactivity described in the literature (Oldstone 1998). However, we know that proteins, and antibodies in particular, can also bind completely unrelated ligands (James et al. 2003). Theoretical models propose that the number of complementary binding ligands for any given binding site is a function of the ligand library size and the affinity cutoff with which the library is selected. Thus, alternative ligands can be found for any protein provided that a large enough chemical diversity is explored (Inman 1978; Perelson and Oster 1979). On the few occasions where proteins have been subject to systematic screens, these predictions have largely been borne out. A comprehensive antibody cross-reactant search was performed by Varga and colleagues, who screened an anti-DNP (2,4-dinitrophenol) IgE antibody (SPE7) against a library of over 2000 compounds and identified a number of unrelated compounds which competed against the immunizing hapten for binding (DNP; Varga et al. 1991). We have been investigating SPE7 to establish a mechanistic paradigm for promiscuity at the molecular level. Recently, we demonstrated that SPE7 can bind entirely unrelated antigens (from haptens to proteins) by switching between radically different preexisting structural isomers. One isomer possesses a deep and narrow binding site for small aromatic ligands, including the immunizing hapten. A protein antigen makes use of another, unrelated antibody isomer with a wide, shallow binding site (Foote 2003; James et al. 2003). However, there is also cross-reactivity within each structural isomer. Although the small haptenic cross-reactants identified by Varga are structurally unrelated, they all appear to bind the antibody isomer that possesses a deep and narrow binding site. This study focuses on the mechanism by which a single binding site binds a number of unrelated ligands. We describe the cloning, sequencing, and biochemical characterization of the SPE7 Fv fragment. We examined SPE7’s binding to a series of small molecule ligands—the immunizing hapten (DNP) and its derivatives, and several promiscuous ligands and derivatives thereof. We found that SPE7 exhibits exquisite specificity for both the immunizing hapten and the promiscuous ligands, and that ligand hydrophobicity is not the driving force behind cross-reactivity. We find that cross-reactants do not need to mimic the target antigen to bind, but bind the antibody by making different specific contacts with its combining site. The affinity and specificity of binding of the various ligands (cross-reactants and immunizing hapten alike) is controlled by the number and nature of specific hydrogen-bonding interactions between ligand and antibody. Finally, comparison of the sequence and binding pattern of SPE7 with its putative germline antibody (B1–8) indicate that there is no obvious link between affinity maturation and cross-reactivity.
Results and Discussion
Cloning sequencing and generation of SPE7 Fab and Fv fragments
We cloned the cDNA for antibody SPE7’s Fab fragment from hybridoma SPE7.49 (generously provided by Prof. Zelig Eshhar; Eshhar et al. 1980); for cloning primers see Table 1 and Materials and Methods. Following sequencing (Fig. 1 ▶), The Fab and Fv fragments of SPE7 were expressed in Escherichia coli and purified to homogeneity by Ni-NTA chromatography followed by gel filtration. While the Fab fragment expressed poorly, the Fv fragment gave a reasonable yield. We followed binding to SPE7 by measuring the quenching in intrinsic antibody fluorescence that occurs upon hapten complexation. Quenching was observed with both intact SPE7 (data not shown) and Fv (Fig. 2 ▶). Intact SPE7 contains nonbinding-site tryptophans that contribute to the fluorescence. The fraction of quenched amplitude relative to the total fluorescence of the free protein was therefore much greater with the Fv. Nevertheless, the affinities measured for both intact IgE and the Fv fragment were essentially identical. All following references to experiments with SPE7 refer to SPE7 Fv unless otherwise stated.
Table 1.
Sequences of the SPE7 primers
| Primer | Primer sequence (5′–3′)a |
| VλB | GATGCTGTTGTGACTCAGGAATC |
| VλF | ACCTACGACAGTCAGTTTGG |
| Mλ1Fo | GGAACAGTCAGCACGGGA |
| IgE-C2-Fo | CGCAGCGATGAATGGAGTAGCTCC |
| IgE-B1-Bc | GAGGTSMARCTGCAGSAGTCWGG |
| IgE-B9-Bc | GAGGTSCARCTGCAGGAGTCW |
| H_Fo_Eco | GACGCCTAAGGAATTCTTACTAGTGATGGTGATG |
| H_Bc_Avo | CGGCAGTCCAGCGTACCTAGGCTAATAAGCATGC |
a S = C/G, M = A/C, R = A/G, W = A/T.
Figure 1.

Nucleotide and amino acid sequence of the Fab fragment of antibody SPE7 (variable and 1st constant domains). Sequences were derived from cDNA produced from hybridoma SPE7.49 (see Materials and Methods).
Figure 2.

Quenching of SPE7’s fluorescence upon binding of DNP-Ser and alizarin-red. The ligands were added to 0.3 μM SPE7 Fv in 1-μL aliquots of varying concentration. The fluorescence measured at 341 nm (in arbitrary units) was fit to ligand concentration using a standard quadratic expression (see Materials and Methods) to yield dissociation constants (Kd) of 20 ± 2 nM for DNP-Ser and 40 ± 5 nM for alizarin-red.
Characterization of the binding pattern of SPE7
We determined binding constants for a range of small molecules that promiscuously bind SPE7 (including those identified by Varga et al. 1991) with affinities between 20 nM to 200 μM. Given the wide range of affinities tested, different methods had to be applied depending on the affinity constant of the ligand. Fluorescence quenching was used for ligands with Kd < 10 μM. Competitive ELISA was applied in two different modes depending on the affinity constant of the ligand (Table 2). For low-affinity ligands (Kd > 10 μM), we adapted the traditional competitive ELISA method (Voller 1978) using immobilized DNP–Asn:BSA. As the affinity of DNP–Asn for SPE7 (0.7 μM) is significantly less than DNP–Lys (Table 2), it provides a much better reference for low-affinity ligands. For ligands with higher affinity, we made use of DNP–Biotin to assay DNP binding and competitive inhibition in solution (after incubation, complexes were captured onto streptavidin-coated plates; for details of the assay, see Materials and Methods). The affinities measured for a series of ligands with submicromolar affinities show good agreement between the different methods. Representative results of these experiments are shown in Figures 2 ▶ and 3 ▶, and the resulting affinities are summarized in Table 2. As many of the cross-reactants contain a ketone group (e.g., alizarin or acenaphthenequinone), the involvement of a Schiff-base covalent bond with an antibody lysine residue was ruled out by borohydride reduction experiments (see Materials and Methods).
Table 2.
Structures and affinities of various SPE7 ligands

The immunizing hapten (indicated in bold), DNP derivatives, and several unrelated haptens are shown in the left column. In the right column, is shown alizarin and a series of related derivatives. Binding affinities were determined by fluorescence quenching,a or by competitive ELISA with either DNP-Biotinb or DNP-Asn-BSAc (for details, see Materials and Methods). Those compounds which gave less than 50% inhibition in competition ELISAs at a concentration of 200 μM are indicated as having an affinity >200 μM, and are effectively considered to be nonbinding..
Figure 3.
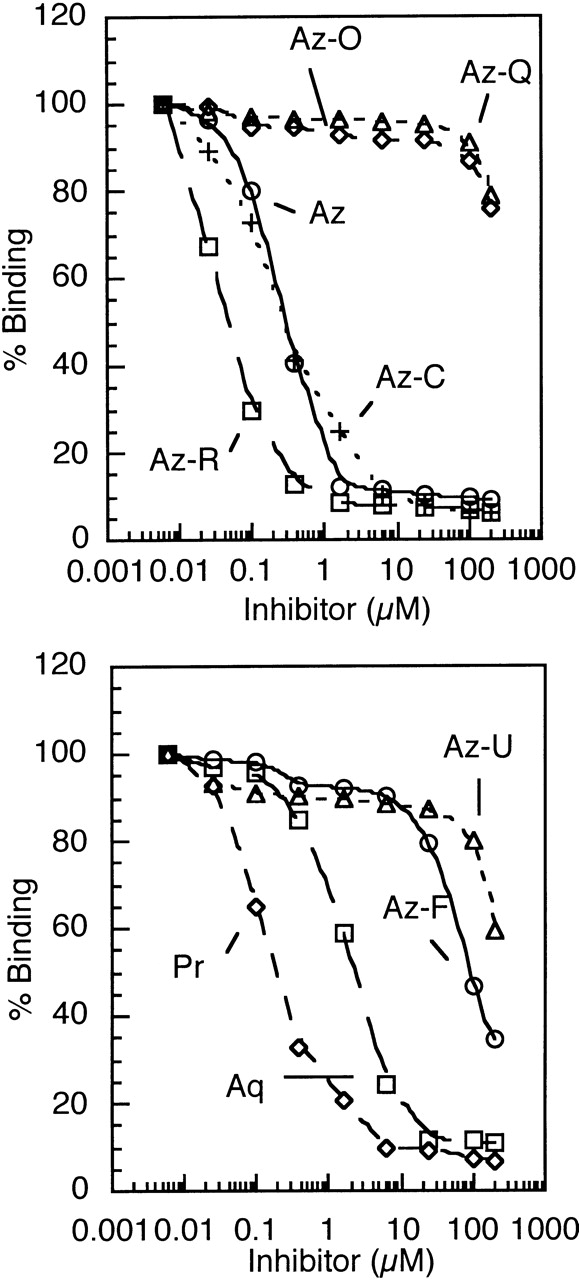
Inhibition of SPE7 Fv binding to immobilized DNP-Asn by various cross-reactants as measured by competitive ELISA. Noted are the abbreviated names of the various cross-reactants, the full names and structures of which are given in Table 2. Residual SPE7 binding to immobilized DNP-Asn was determined by measuring capture of SPE7 using anti-myc-HRP secondary antibody (see Materials and Methods). Binding, as a percentage of the binding observed in the absence of any inhibitor, was plotted against inhibitor concentration. IC50s were determined as the concentration of ligand needed for 50% inhibition and are given in Table 2.
The studied cross-reactant ligands are not structurally related to the immunizing hapten (DNP) and have very different chemistries, demonstrating that antibody cross-reactivity does not have to be with closely related molecules. To assess how specific this cross-reactivity is, we measured binding of a large number of derivatives related either to the original hapten (DNP–Lys; Table 2) or to one of the cross-reactants. Closely related derivatives of each ligand have little or no affinity, demonstrating that each cross-reactant interacts specifically. For example, SPE7 binds NIP with ~2000-fold lower affinity than DNP–Lys (Table 2), although NIP is much more closely related to DNP than any of the other cross-reactants. We also tested a range of alizarin derivatives, including some that differ only in the number or position of hydroxyl groups (Table 2, right panel). While SPE7 binds alizarin (Az) with an affinity of 0.2 μM, it shows almost no affinity for anthraquinone (Az-Q; which is identical except that it lacks two hydroxyl groups). Similarly, the presence of a hydroxyl group on the benzene rings (e.g., anthraflavic acid) dramatically reduces affinity (Table 2). Notably, our expanded screen for crossreactants has identified an alizarin derivative (Az-R; Kd = 40 nM) with similar affinity to the immunizing hapten DNP–Lys.
Cross-reactant binding is not driven by hydrophobic stickiness
Stacking interactions of aromatic ligands with binding site tryptophans or tyrosines often play an important role in antibody binding. Indeed, structural analysis of SPE7 reveals that the bound haptens stack against a tryptophan side chain at L93 (James et al. 2003; see also Fig. 5 ▶). It has been argued that cross-reactivity is the result of nonspecific hydrophobic interaction—a phenomenon generally referred to as hydrophobic stickiness (Padlan 1994). We measured the hydrophobicity of several cross-reactants, including a range of alizarin derivatives, by partition between n-octanol and water (see Materials and Methods). As can be seen in Figure 4 ▶, there is, in fact, no correlation between hydrophobicity and affinity. On the contrary, the highest affinity ligands are actually the most hydrophilic. Furthermore, substitutions around the alizarin aromatic ring system show that affinity does not increase with hydrophobicity. Anthraquinone (Az-Q), which is considerably more hydrophobic than alizarin, exhibits very low affinity to SPE7. Alizarin-red (Az-R) has a fivefold higher affinity than alizarin, despite it being 10,000 times more soluble in water (ΔlogP ≈ 4).
Figure 5.
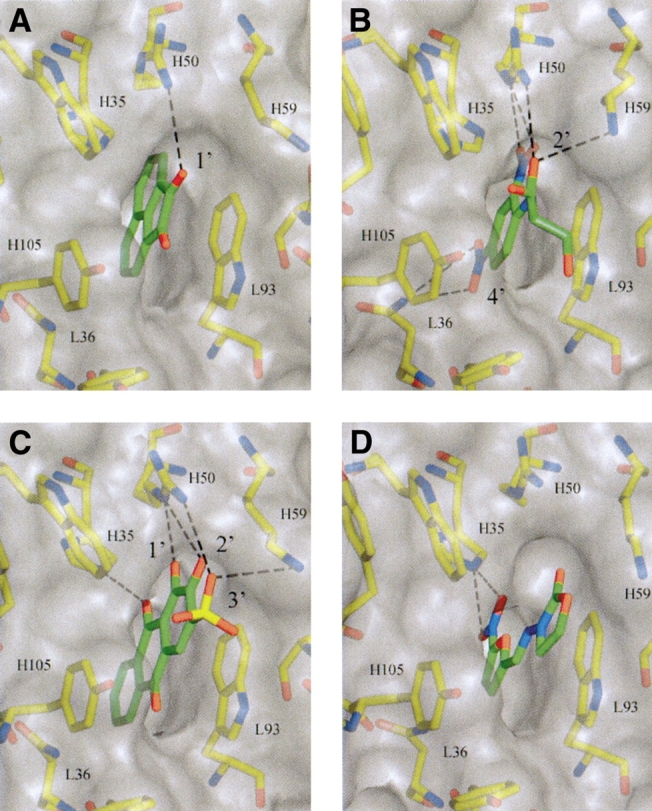
Details of the binding site of SPE-7 in complex with (A) Acenaphthenequinone, (B) DNP-Ser, (C) Alizarin-red, and (D) Furazolidone. The binding site is presented as a semitransparent surface together with interacting residues. Putative hydrogen bond contacts between SPE7 and the haptens are shown. The figure was generated with the program AESOP.
Figure 4.
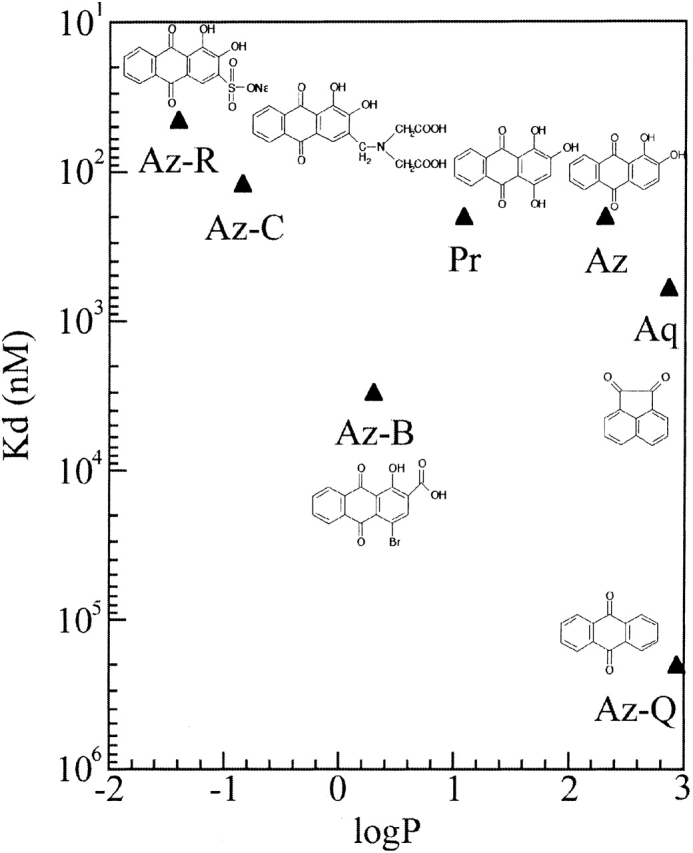
Relationship between ligand hydrophobicity and affinity for SPE7. Affinities were taken from Table 2. Hydrophobicity was determined by measuring ligand partition between an aqueous buffer and a hydrophobic organic solvent (n-octanol; see Materials and Methods). The partition coefficient (P) is the ratio of ligand concentration in n-octanol versus water. A positive logP value indicates that a ligand is more soluble in n-octanol than in water, and, therefore, more hydrophobic, while a negative logP indicates greater solubility in water and lower hydrophobicity.
Cross-reactant binding is mediated by specific hydrogen bonds
Analysis of cross-reactant derivatives, and of alizarin in particular, reveals that SPE7 is able to differentiate between very closely related molecules (Table 2). The hydroxy groups at the 1′ and 2′ positions of the anthraquinone ring system seem to determine specificity. Removal of these groups negates binding (as in Az-Q), as does replacing them with different groups—for example, in 4-bromo-1-hydroxy-anthraquinone-2-carboxylic acid (Az-B) where the 2′ hydroxyl is replaced by a carboxylic acid and affinity reduced by 15-fold. Groups, including hydroxyls, at other positions, for example, at the 3′ or 4′ positions, seem to have little effect on affinity. Alizarin complexone (Az-C) with an additional substituent at the 3′ position and purpurin with an hydroxy group at the 4′ position, bind with similar affinity to Alizarin. The positioning of the hydroxyl groups at one of the benzene groups of the anthraqinone system is also crucial—1′,5′ and 2′,6′ dihydroxy anthraquinones (anthrafurin and anthraflavic acid, respectively), where the hydroxy groups are positioned on the two opposite benzene rings, exhibit very low affinity (Kd > 200 μM). Two derivatives were found to exhibit higher affinity than alizarin—these are alizarin complexone (Az-C, Kd = 0.12 μM) and alizarin red (Az-R, Kd = 0.045 μM; Table 2). Both derivatives posses a negatively charged group at the 3′ position.
This binding pattern analysis suggests that binding of both the immunizing hapten and cross-reactants is dependent on different but specific stereochemistry. These findings are in good agreement with the recently published crystal structures and presteady-state kinetics of SPE7 Fv bound to DNP–Ser, alizarin-red, and furazolidone (James et al. 2003). In addition to these, we include here the structure of SPE7 in complex with the cross-reactant acenaphthenequinone (Fig. 5 ▶). Details of the structure determination and refinement are given in Table 3. Comparison of the four hapten-bound structures reveals that they all bind to a very similar combining-site configuration with only slight alterations in the position of a few antibody side chains. This is in contrast to the binding of a protein cross-reactant, which makes use of another, radically different combining-site configuration of SPE7 (James et al. 2003). The structures show that while the haptens all stack against a tryptophan of the L3 loop (L93), they make different sets of hydrogen bonds dependent upon their different chemistries (Fig. 5 ▶; Table 4). For instance, the hydrogen bond contacts made by DNP–Ser with L36 and L93 are absent in alizarin-red, while furazolidone contacts only H35 and acenaphthenequinone only H50. The importance of the 1′ and 2′ position in the alizarin derivatives is clear; the hydroxyl groups act as hydrogen bond acceptors for the side chain of arginine H50 (from CDR loop H2). The finding that negatively charged groups at the 3′ position, as in Az-C and Az-R, result in higher affinities also correlates with the structures. These groups are positioned to form charged hydrogen bonds with the amino group of lysine H59. Hydrogen bonding and electrostatic interaction effectively improve the affinity of the hydrophobic anthraquinone ring moiety from an affinity of less than 100 μM (Az-Q) to 0.045 μM (Az-R; Table 2)—a ~2000-fold improvement in affinity.
Table 3.
Structure determination parameters of the acenaphthenequinone: SPE7 complex
| Spacegroup | P212121 |
| Cell | a = 78.73, b = 78.64, c = 167.56 |
| Copies per a.u. | 4 |
| Buffer | 22% PEG 8K, 0.1 M Sodium Cacodylate, 0.2 M Sodium Acetate pH 5.6 |
| Resolution | 2.4 Å |
| Unique reflections | 30,262 |
| Rmerge | 0.111 (0.465) |
| Redundancy | 4.0 |
| Completeness (%) | 98.1 (98.4) |
| Average I/σI | 7.5 (2.2) |
| *Final Rcryst | 0.269 |
| *Final Rfree | 0.271 |
| Bond r.m.s.d. | 0.007 |
| Angle r.m.s.d. | 1.3° |
Numbers in parentheses refer to the highest resolution shell (2.55–2.4 Å).
Table 4.
Summary of putative interactions between SPE7 and various binding ligands
| Antibody | |||||
| Residue | Atom | DNP-Ser | Furazolidone | Aq | Az-R |
| L36 | Nδ2 | O41 | |||
| O42 | |||||
| H50 | Nɛ | O62 | O1 | ||
| H50 | Nη2 | O61 | O5 | O2 | O3 |
| O1 | O6 | ||||
| H59 | N2 | O1 | O4 | ||
| L93 | Nɛ1 | Oδ | O1 | O2 | |
| H35 | Nɛ2 | O62 | O2 | O | |
| O3 | |||||
Hydrogen bonds are indicated in plain text; residues and atoms involved in planar stacking or charged interactions are indicated in italics. The atom annotations in this table refer to the atoms as listed in the respective PDB files.
Further evidence that it is specific hydrogen bond contacts that mediate cross-reactant binding rather than changes in the context of hydrophobic interaction is provided by the structure of the acenaphthenequinone (Aq):SPE7 complex presented here (Table 3). As can be seen in Figure 5a ▶, Aq is bound in a similar manner to DNP–Ser (the immunizing hapten) and Az-R—the quinone ring system stacks against Trp93L. The 1′ oxygen of Aq’s carbonyl is positioned to hydrogen bond with the side chain of Arg50H, which has its NH1 group within 3.2 Å of the 1′ oxygen. The structures also show that Aq has a considerably lower affinity for SPE7 than Az-R, not because of steric hindrance or poor complementarity to the hydrophobic pocket but because it does not have the same hydrogen bond forming potential as Az-R. While the 1′ hydroxy of Aq is positioned to hydrogen bond in a similar manner to the 2′ hydroxy from Az-R, it lacks the 2′ hydroxy, the negatively charged sulphate group, and keto group of the anthraquinone system (Fig. 5 ▶; Table 4). Thus, although Aq is considerably more hydrophobic than Az-R, it does not have suitably placed groups with which additional specific contacts can be made. The same is also true within the alizarin derivatives, where the basic anthraquinone ring, although considerably more hydrophobic, has a 2000-fold lower affinity than its hydroxylated derivatives. However, it is not just the number of hydrogen bond interactions that affect ligand affinity. Ligand:binding-site shape complementarity, opposing charge repulsions, differing solvation energies for different ligands, and also differences in the strength of the hydrogen bonds themselves will all affect affinity. Furazolidone, like Aq, forms only one hydrogen bond. The two ligands bind SPE7 with similar affinities (1.2 and 0.6 μM, respectively), although different antibody residues are involved in forming the complex as well as different ligand moieties.
This study of antibody SPE7 suggests that hydrophobic stickiness alone can provide only very low affinity binding (Kd > 200 μM; Table 2). The physiological relevance of such low affinity binding is questionable. To become activated and begin clonal expansion, B-cells have to bind antigens above a threshold affinity. Even the primary response, mediated by low-affinity IgM antibodies, requires micromolar affinities (<10 μM). Thus, hydrophobic stacking, unless coupled to additional, specific interactions, will not allow antigens to stimulate B-cell activation and thereby initiate clonal selection and affinity maturation. In the case of SPE7, it seems that these additional interactions involve specific hydrogen bonds to the 1′ and 2′ hydroxyls (in the anthraquinone system) and with antibody residues Arg50H and Lys59H. Both the number and the precise position of these groups have a profound effect on the affinity.
The anti-NP antibody B1–B8 is SPE7’s putative germline precursor
An IgBLAST search (Altschul et al. 1990) indicated that SPE7 is most closely related to antibody B1–8 (Fig. 6 ▶). B1–8 is a primary response IgM raised against 4-hydroxy-3-nitrophenyl acetate (NP; Table 2) (Bothwell et al. 1981). Like B1–8, the heavy chain V, D, and J regions of SPE7 are constructed from the V(186-2), DFL16.2, and JH2 genes, respectively. In joining the V and D regions an additional residue (Met99) has been incorporated into the reading frame. In SPE7, the first D-region residue has been substituted from an Aspartate to a Tryptophan. The heavy chain is combined with a light chain constructed from the Vλ1 V gene and j1v1 J gene. λ-light chains are unusual as they are only used if the κ-genes fail to recombine; however, they are common in NP/DNP binding antibodies (Taketani et al. 1995). A number of different anti-NP antibodies have been sequenced, and most have been found to derive from a small subset of V, D, and J genes (Taketani et al. 1995). Despite these general similarities, the sequence identity between B1–8 and SPE7 is unusually high. SPE7 uses identical germline V(D) and J genes in both chains, including the Vλ1 light chain gene. In fact, SPE7 differs from B1–8 within CDR regions only at one position within CDR H2 (60:Ser → Gly), four positions in CDR H3 (99:Tyr → Met), (100:Asp → Trp), (104:Ser → Thr), (105:Ser → Tyr), and one position in CDR L3 (98:Trp → Leu). SPE7 is an affinity matured antibody, and was isolated following repeated challenges with DNP antigen (Eshhar et al. 1980). The differences observed between it and B1–8 are likely to be the result of somatic hypermutation (e.g., the mutation in H2). It therefore seems that SPE7 originated from a germline antibody with a sequence highly similar, if not identical, to B1–8.
Figure 6.

Heavy- and light-chain amino acid sequence of SPE7 Fv aligned with antibody B1–8. Residues with absolute identity are highlighted and in bold. CDRs are indicated as boxed sections.
Cross-reactivity and affinity maturation
A comparison of the binding patterns of SPE7 and its putative germline equivalent B1–8, indicates that SPE7 has a vestigial ability to bind the same NP derivatives as B1–8, albeit with lower affinity. B1–8 binds NP with an affinity of about 2 μM, whereas SPE7 binds it with 50-fold lower affinity (Table 2). Conversely, B1–8 has very poor affinity for DNP, less than 100 μM. Several mutants of B1–8 have been explored (Bruggemann et al. 1986), one of which (B1–8.V4) has a single point mutation in CDR H2 (50:Arg → Gly). This single mutation results in more than a 100-fold reduction in affinity for NIP (the immunizing hapten) but increases affinity for DNP to around 10 μM (Bruggemann et al. 1986). Interestingly, we found that neither the V4 mutant nor wild-type B1–8 bind the SPE7 cross-reactants (e.g., furazolidone, acenaphthenequinone, or alizarin; measured by competitive ELISA). It seems that the observed pattern of multispecific binding is unique to SPE7, and has arisen during the course of affinity maturation. In every other way, SPE7 behaves like a matured antibody; it binds DNP with higher affinity than its putative precursor B1–8 and distinguishes between DNP and NP with exquisite specificity. The fact that discrimination of the target antigen (DNP) is improved but multispecific binding to unrelated ligands is still possible may appear contradictory, yet we suggest that it is not. After all, cross-reactive ligands do not have to share the same binding site determinants as the primary ligand. This is consistent with the biochemical evidence that cross-reactants engage SPE7 through a unique pattern of hydrogen bond interactions, dependent upon their specific chemistry. It is, of course, highly likely that changes during affinity maturation will affect binding of cross-reactants that bear structural resemblance to the primary antigen. In the case of SPE7, the increased specificity toward DNP is manifested in lower affinity (relative to its progenitor, B1–8) toward derivatives such as NP or NIP. However, the modified binding site that binds DNP with higher affinity and specificity, has the capacity to bind new and completely unrelated ligands. Thus, when it comes to promiscuous or multispecific binding, maturation would be expected to alter the pattern rather than the number of promiscuously-bound ligands as the stereochemical landscape of the binding site is changed. We, therefore, suspect that B1–8 has its own unique pattern of multispecificity, distinct from SPE7.
Whether B1–8, a germline antibody, exhibits a broader spectrum of promiscuous binding relative to the matured variant SPE7 remains to be examined. To our knowledge, no study has been performed to compare the multispecificity of a mature antibody with its germline precursor. It has been suggested that certain properties of germline antibodies may predispose them toward multispecific binding. For example, long flexible loops and a high occurrence of unrestrictive residues such as glycine might promote conformational diversity, and hence, the ability to bind several unrelated ligands, or, a concentration of aromatic residues in the binding site might lead to cross-reactivity through hydrophobic stickiness (Padlan 1994). However, none of these theories have been demonstrated experimentally. Certainly, there is nothing in the sequence comparison of B1–8 and SPE7 to support them: the loop lengths have not changed upon maturation and the number of aromatic residues has decreased, if only by one residue (Fig. 6 ▶).
Immunological implications
The results presented above suggest that, contrary to common assumption, multispecificity cannot be explained in terms of general hydrophobic stickiness. Binding to very different ligands occurs through specific interactions with SPE7’s binding site. SPE7, therefore, combines two beneficial features in terms of the antibody immune response. First, by binding to a number of different antigens it increases the size of the immune repertoire and thus the likelihood of intercepting an invading antigen. If a significant number of antibodies display similar characteristics in vivo, then the primary immune repertoire may not be limited to the number of circulating B-cells. Second, by ensuring specificity for each antigen, the potential for cross-reaction to an antigen mimic is limited. Potentially, this characteristic could significantly reduce the probability that antibodies raised against a bacterial protein will cross-react with a self-protein that resembles the antigen and cause autoimmunity. The fact that autoimmunity can be mediated by antigen mimicry (Cohen 2001) highlights that such protection is not complete. There has been considerable success in determining a causal relationship between specific infections and autoimmune conditions where antigens are related by molecular mimicry (Oldstone 1998). However, our results suggest that cross-reactivity to unrelated antigens may be an even greater and yet underappreciated risk. There are many cases where a link has been established between an infection and an autoimmune disease but the mechanism of cross-reactivity is not known (Fairweather et al. 1998; Bar Meir et al. 2000). Linking unrelated antigens to a single autoimmune antibody is not trivial even when a likely pathogen is known, and may require detailed systematic analysis. However, establishing these relationships at the molecular level may open the door to effective prophylactic treatment.
Concluding remarks
The issue of promiscuity is fundamental to our understanding of the function and evolution of proteins. For instance, multifunctionality appears to be behind the ability of a single protein to “moonlight” in several different roles (for reviews, see Jeffery 1999; Copley 2003). These roles can be structural or regulatory, as in Neurospora crassa mitrochondrial tyrosyl-tRNA synthetase, which has an additional role in the folding of active self-splicing Group 1 introns (Caprara et al. 1996). Alternatively, these roles can be catalytic as in Drosophila alcohol dehydrogenase whose flexible active site confers the ability to bind several different substrates (Benach et al. 1999). A picture is therefore emerging of a spectrum of enzymatic side activities, from catalysis of a range of substrates involving the same key-step chemistry as the enzyme’s primary activity, to catalysis of unrelated reactions via fundamentally different enzymatic mechanisms (James and Tawfik 2001; Copley 2003; James and Tawfik 2003). A similar spectrum is observed with binding—from cross-reactivity to multispecificity, namely, from binding of ligands related to the original ligand, to ligands of completely different structures (James et al. 2003). There seem to exist, however, many different terms that are used interchangeably to describe widely different degrees of promiscuity. Our findings highlight the importance of distinguishing between multiple ligand recognition (multispecificity, or promiscuity) and cross-reactivity by molecular mimicry. While cross-reactivity implies binding of ligands that are similar or overlap the antibody’s original antigen (Arevalo et al. 1994; Trinh et al. 1997), multispecificity refers to the binding of distinctly different ligands, sometimes using a different set of combining-site residues (Kramer et al. 1997), or even different conformations of the same antibody (James et al. 2003). This study has focused on the mechanism by which a single isomer with one binding pocket is capable of binding a number of structurally different ligands in a specific manner. We show that in the case of SPE7, the stereochemical heterogeneity of both the antibody binding site and the various ligands are exploited to generate specific contacts, with little need for structural rearrangement. How proteins exhibit specificity and promiscuity at the same time is an interesting question. In this work we have demonstrated that these properties need not be mutually exclusive. We have shown that the specific complexation of unrelated binding partners occurs through the use of alternative binding site residues, and that this behavior is intrinsic to the fabric of protein binding sites. Thus, promiscuous activities need not be nonspecific as such, but rather each protein may have its own unique pattern of promiscuous activities, and each activity, standing alone, is highly specific.
Materials and methods
Cloning sequencing and generation of SPE7 Fv and Fab fragments
The original SPE7 hybridoma clone expresses both κ and λ light chains (Eshhar et al. 1980), and the resulting IgE is available from Sigma. We cloned SPE7 Fab and Fv fragments from a subcloned hybridoma, SPE7.49 that produces antibodies with λ-light chains only (generously provided by Prof. Zelig Eshhar). cDNA from the SPE7.49 hybridoma, was prepared using the Reverse Transcriptase System (Promega). As the sequence of SPE7 was unknown, light-chain primers were synthesized based on the variable domain sequence of a cloned IgE anti-DNP antibody MPOC315 (Baldwin and Schultz 1989). Once these primers, VλF and VλB, were found to amplify the Fv domain from the prepared SPE7.49 cDNA, a Fab forward (3′) primer was designed (Table 1) based on a constant λ-light chain IgE sequence from the Kabat database (sequence 36λ1; Kabat 1991). To amplify the Fab portion of the heavy chain, a forward primer was designed based on the sequence of mouse IgE C2 region (second constant domain; Shinkai et al. 1988) and a degenerate back primer based on consensus antibody sequences, IgE-B1-Bc (Table 1). The final back primer used in cloning (IgE-B9-Bc) was a refined version of IgE-B1-Bc. The remaining degenerate positions in IgE-B9-Bc are located in the third position of the codon and are synonymous substitutions.
The PCR-amplified light- and heavy-chain DNA fragments were digested with NotI and PstI and PstI and NcoI, respectively, and cloned into a pUC119 poly-myc-his vector (generously provided by G. Winter). The resulting vector (pUC119–SPE7HλFab) contains both chains, each with an RBS and pelB leader and with a C-terminal myc and 6His tag on the heavy chain (the pelB leader mediates export of the antibody into the periplasm of the cell). The Fab fragment was found to express poorly (<0.1 mg per liter culture). An Fv expression construct (pUC119–SPE7HλFv) was created from the above Fab construct by digesting the gene encoding the light chain at a natural AvrII site, which occurs at the end of the light variable domain, and EcoRI, which flanks the polylinker cassette. The heavy chain was then amplified using a 5′ primer that introduces an AvrII site and appends a stop codon for the upstream light-chain variable domain and a 3′ primer that preserves the tags and introduces an EcoRI site (primers H_Fo_Eco and H_Bc_Avo; Table 1). The heavy chain was then recloned at the AvrII and EcoRI sites of pUC119–SPE7HλFab.
SPE7 Fv was expressed in E. coli TG1 cells freshly transformed with pUC119–SPE7HλFv. Cells were grown in LB medium to O.D600 0.8 at 30°C, transferred to 27°C, and induced with 1 mM IPTG. Induced cells were for grown for 6 h at 27°C and spun down at an RCF (relative centrifugal force) of 8300 for 20 min. All following steps were performed on ice or at 4°C. Cell pellets were resuspended in 1/40th volume of periplasm lysis buffer (20% sucrose, 2 mM EDTA, 50 mM Tris, pH 8.0) and incubated for 20 min. The periplasm extract was spun for 20 min at RCF 40,000 in a Sorval SS-34 and the supernatant dialyzed overnight against PBS. The dialyzed solution was incubated for 1 h with Qiagen Ni-NTA resin (2.5 mL per 250 mL periplasm extract) in binding buffer (50 mM phosphate pH 7.4, 350 mM NaCl2). The resin was spun down at RCF 1400 and washed twice with 250 mL of Wash Buffer (50 mM phosphate pH 7.4, 350 mM NaCl and 10 mM Imidizole). The washed resin was transferred to a column and Fv eluted in 2 mL of elution buffer (50 mM sodium phosphate pH 7.4, 350 mM NaCl2 and 200 mM Imidizole). The volume was reduced to ~1 mL and loaded onto a HiLoad S200 Superdex gel filtration column, preequilibrated in 5 mM HEPES pH 7 plus 10 mM NaCl2. Fractions corresponding to the Fv were identified by ELISA on DNP-BSA (see below) and SDS-PAGE, collected, concentrated, and stored at −20°C.
ELISA
DNP–biotin
DNP-hexyl-NH2 was synthesized by reacting 10 mmole N-Boc-diaminohexane with 10 mmole 2,4-dinitrofluorobenzene in acetonitrile. The product was purified by silica gel chromatography and treated with trifluoroacetic acid (TFA) to give DNP-hexyl-NH2 as TFA saltl; 0.22 mmole of DNP-hexyl-NH2 was added to 0.22 mmole of Biotin-amidocaproate-N-hydroxysuccinimide ester (Sigma) in 2 mL of acetonitrile, followed by 0.5 mmole of Et3N. The resulting solution was made up to 5 mL and stirred o/n. The product (DNP-biotin) was purified by HPLC on a reverse-phase RP C-18 column using a water/acetonitrile gradient plus 0.1% TFA.
DNP–Asn–BSA
DNP–Asn (1 mmole) was activated for conjugation by adding 2 mmole of 1,1′-Carbonyldiimidazole in DMF and incubating for 1 h. One to 50 μmoles of activated DNP–Asn were added to 20 mg/mL BSA in 0.1 M carbonate buffer pH 9.8. The solution was stirred at room temperature for 3 h and then extensively dialyzed against PBS. The hapten density (Hd; number of hapten molecules per BSA molecule) was determined by measuring the O.D. of the conjugates in a microtiter plate at 405 nm (ɛ405nm = 2350 O.D. units/M per 100 μL samples). DNP–BSA was prepared by reacting 10 μL of DNFB in 0.5 mL DMF with 20 mL of 3 μmole BSA in 0.1 M Carbonate buffer pH 9.8. NIP-BSA was generously provided by G. Winter.
Inhibitor solutions
As many of the inhibitors are relatively water insoluble, stock solutions were prepared in DMSO at 50 mM and stored at −20°C. DMSO stocks were freshly diluted in phosphate-buffered saline (PBS) to ≤1 mM with vortexing while ensuring that no precipitation occurred.
Competitive ELISA with DNP–biotin
This format of competitive ELISA was developed to enable the process of DNP binding and its competitive inhibition by various ligands to occur in solution. To this end, SPE7 (150 nM of Fv, or 20 nM of intact IgE; Sigma) was incubated for 2 h, with 10 nM DNP–biotin (see above) in the presence of different inhibitors. The antibody:DNP–biotin complex was then captured on streptavidin-coated plates (Roche; high capacity). After a 15-min incubation, the plates were rinsed and incubated with antimouse peroxidase (Jackson; #115-035-006; 1:2500 Dilution in PBS with 1% Tween20 [PBS/T]) for 1 h. Finally, plates were rinsed extensively in PBS/T and the peroxidase substrate added (BM blue; Roche). In reactions with the SPE7 Fv an anti-myc peroxidase antibody was used instead (Invitrogen; 1:1000 Dilution in PBS/T). Reactions were quenched with 0.5 M H2SO4 and read at 450–650 nm in a spectrophotometer plate reader. Ic50 values were calculated from a range of different inhibitor concentrations (500 μM–5 nM), and refer to the inhibitor concentration giving 50% of the signal observed in the absence of any ligand.
Competitive ELISA with DNP–Asn–BSA
This format of ELISA was used to determine affinity measurements of ligands with a Kd of 10 μM or lower. Nunc maxisorp plates were coated for 2 h with 2 μg/mL DNP–Asn–BSA (Hd = 13) at room temperature and then blocked with 1 mg/mL BSA for 20 min. A solution containing SPE7 preincubated with inhibitors (for 1 h) was added and allowed to incubate for 30 min. Plates were then rinsed, and any captured antibody was detected as described above. The same protocol was applied for ELISA with DNP–BSA and NIP–BSA, and with antibody B1–8 (ScFv of both wild-type and V4 mutant were generously provided by G. Winter) where the ELISA was performed at 2 μg/mL.
Affinity measurements by fluorescence quenching
Equilibrium binding constants were measured by fluorescence quenching using an F-4500 Hitachi Fluorescence Spectrophotometer. All experiments were performed at 20°C, with 1 mL of purified Fv at 0.3–1 μM in PBS. Ligands were added in 1-μL aliquots in steps of varying concentration and fluorescence measured at 341 nm following excitation at 280 nm. Fits were performed with Kaleidograph (Abelbeck software) using equation I:(Lindner et al. 1999) F = Fa + f′((− (L0 − A0 + Kd) ± (((L0 − A0 + Kd)2) + (4KdA0))1/2))/2; where F is the observed fluorescence, Fa is the molar antibody fluorescence, f′ is the molar change in fluorescence, (A0) is the total antibody concentration, (L0) is the total ligand concentration, and Kd is the dissociation constant.
Testing for Schiff-base formation
The following protocol was applied, to rule out the possibility that cross-reactivity is due to the formation of a Schiff base between a ketone group of the cross-reactants and a lysine residue in the SPE7 binding site; 0.2 μmoles of SPE7 IgE were incubated with 50 μmoles inhibitor in a final reaction volume of 100 μL for 1 h. Sodium cyanoborohydride (NaH3BCN) was then added to a final concentration of 1 mM to reduce any Schiff base that forms. Samples were diluted to 200 μL, dialyzed extensively against 0.1 M sodium acetate pH 5.5 and PBS, and the concentration of active SPE7 determined by ELISA on DNP–BSA as described above. No change in SPE7 binding titer was observed following this treatment, indicating that binding of ketonic cross-reactants such as alizarin or acenaphthenequinone is not mediated by Schiff-base formation.
Partitioning experiments
The hydrophobicities of various SPE7 ligands was determined using a partition assay between n-octanol and water essentially as described (Fastrez and Fersht 1973). Ligand solutions were diluted into 500 μL of PBS to a final concentration of 1 mM; 500 μL of n-octanol were added and the solutions vortexed gently for 3 h. The solvent mixtures were allowed to settle overnight, and 100 μL from each phase were added to 900 μL of 96% ethanol. The O.D. of the ethanolic solutions was measured at a suitable wavelength, to give the ligand concentration in the aqueous and organic phases. The partition coefficient (P) is equal to the O.D. observed in the octanol phase divided by the O.D. observed in the aqueous phase.
Crystal structure analysis
Determination of the structure of the Acenaphthtenequinone:SPE7 complex was performed with the Fv fragment of SPE7 essentially as described for the complexes of SPE7 with DNP, furazolidone, and alizarin-red (see Supporting Material Online for James et al. 2003). The coordinates have been deposited under the PDB accession code 1OAX.
Acknowledgments
Financial support by the Wellcome Trust, the EU (through the ENDIRPRO network) and the Israel Science Foundation is gratefully acknowledged. We are grateful to G. Winter for supplying the B1–8 ScFv fragments, and to Z. Eshhar and T. Waks for the SPE7 hybridoma.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RBS, ribosome binding site
CDR, complementarity determining region
PBS, phosphate-buffered saline
PBS/T, PBS plus Tween-20
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03172703.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Arevalo, J.H., Hassig, C.A., Stura, E.A., Sims, M.J., Taussig, M.J., and Wilson, I.A. 1994. Structural analysis of antibody specificity. Detailed comparison of five Fab′-steroid complexes. J. Mol. Biol. 241 663–690. [DOI] [PubMed] [Google Scholar]
- Baldwin, E. and Schultz, P.G. 1989. Generation of a catalytic antibody by site-directed mutagenesis. Science 245 1104–1107. [DOI] [PubMed] [Google Scholar]
- Barbas 3rd, C.F., Heine, A., Zhong, G., Hoffmann, T., Gramatikova, S., Bjornestedt, R., List, B., Anderson, J., Stura, E.A., Wilson, I.A., et al. 1997. Immune versus natural selection: Antibody aldolases with enzymic rates but broader scope. Science 278 2085–2092. [DOI] [PubMed] [Google Scholar]
- Bar Meir, E., Amital, H., Levy, Y., Kneller, A., Bar-Dayan, Y., and Shoenfeld, Y. 2000. Mycoplasma–pneumoniae-induced thrombotic thrombocytopenic purpura. Acta Haematol. 103 112–115. [DOI] [PubMed] [Google Scholar]
- Benach, J., Atrian, S., Gonzalez-Duarte, R., and Ladenstein, R. 1999. The catalytic reaction and inhibition mechanism of Drosophila alcohol dehydrogenase: Observation of an enzyme-bound NAD-ketone adduct at 1.4 Å resolution by X-ray crystallography. J. Mol. Biol. 289 335–355. [DOI] [PubMed] [Google Scholar]
- Bothwell, A.L., Paskind, M., Reth, M., Imanishi-Kari, T., Rajewsky, K., and Baltimore, D. 1981. Heavy chain variable region contribution to the NPb family of antibodies: Somatic mutation evident in a γ 2a variable region. Cell 24 625–637. [DOI] [PubMed] [Google Scholar]
- Bruggemann, M., Muller, H.J., Burger, C., and Rajewsky, K. 1986. Idiotypic selection of an antibody mutant with changed hapten binding specificity, resulting from a point mutation in position 50 of the heavy chain. EMBO J. 5 1561–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, D.J. and Erlanger, B.F. 1977. Evidence for multispecificity of antibody molecules. Nature 268 763–765. [DOI] [PubMed] [Google Scholar]
- Caprara, M.G., Lehnert, V., Lambowitz, A.M., and Westhof, E. 1996. A tyrosyl-tRNA synthetase recognizes a conserved tRNA-like structural motif in the group I intron catalytic core. Cell 87 1135–1145. [DOI] [PubMed] [Google Scholar]
- Cohen, I.R. 2001. Antigenic mimicry, clonal selection and autoimmunity. J. Autoimmun. 16 337–340. [DOI] [PubMed] [Google Scholar]
- Copley, S.D. 2003. Enzymes with extra talents: Moonlighting functions and catalytic promiscuity. Curr. Opin. Chem. Biol. 7 1–8. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L., Ultsch, M.H., de Vos, A.M., and Wells, J.A. 2000. Convergent solutions to binding at a protein–protein interface. Science 287 1279–1283. [DOI] [PubMed] [Google Scholar]
- Eshhar, Z., Ofarim, M., and Waks, T. 1980. Generation of hybridomas secreting murine reaginic antibodies of anti-DNP specificity. J. Immunol. 124 775–780. [PubMed] [Google Scholar]
- Fairweather, D., Lawson, C.M., Chapman, A.J., Brown, C.M., Booth, T.W., Papadimitriou, J.M., and Shellam, G.R. 1998. Wild isolates of murine cytomegalovirus induce myocarditis and antibodies that cross-react with virus and cardiac myosin. Immunology 94 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fastrez, J. and Fersht, A.R. 1973. Mechanism of chymotrypsin. Structure, reactivity, and nonproductive binding relationships. Biochemistry 12 1067–1074. [DOI] [PubMed] [Google Scholar]
- Foote, J. 2003. Immunology. Isomeric antibodies. Science 299 1327–1328. [DOI] [PubMed] [Google Scholar]
- Inman, J. 1978. The antibody combining region: Speculation on the hypothesis of general multispecificity. Theoretical immunology (H. Bell et al., eds.), pp. 243–278. Marcel Dekker, Inc., New York.
- James, L.C. and Tawfik, D.S. 2001. Catalytic and binding poly-reactivities shared by two unrelated proteins: The potential role of promiscuity in enzyme evolution. Protein Sci. 10 2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. Conformational diversity and protein evolution—A 60-year-old hypothesis revisited. Trends Biochem. Sci. 28 361–368. [DOI] [PubMed] [Google Scholar]
- James, L.C., Roversi, P., and Tawfik, D.S. 2003. Antibody multispecificity mediated by conformational diversity. Science 299 1362–1367. [DOI] [PubMed] [Google Scholar]
- Jeffery, C.J. 1999. Moonlighting proteins. Trends Biochem. Sci. 24 8–11. [DOI] [PubMed] [Google Scholar]
- Kabat, E. 1991. Sequences of proteins of immunological interest, 5th ed. U.S. Department of Health and Human Services. NIH Publication No. 91-3242 Bethesda.
- Kramer, A., Keitel, T., Winkler, K., Stocklein, W., Hohne, W., and Schneider-Mergener, J. 1997. Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell 91 799–809. [DOI] [PubMed] [Google Scholar]
- Lindner, A.B., Eshhar, Z., and Tawfik, D.S. 1999. Conformational changes affect binding and catalysis by ester-hydrolysing antibodies. J. Mol. Biol. 285 421–430. [DOI] [PubMed] [Google Scholar]
- Mariuzza, R.A. and Poljak, R.J. 1993. The basics of binding: Mechanisms of antigen recognition and mimicry by antibodies. Curr. Opin. Immunol. 5 50–55. [DOI] [PubMed] [Google Scholar]
- O’Brien, P.J. and Herschlag, D. 1999. Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol. 6 R91–R105. [DOI] [PubMed] [Google Scholar]
- Oldstone, M.B. 1998. Molecular mimicry and immune-mediated diseases. FASEB J. 12 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padlan, E.A. 1994. Anatomy of the antibody molecule. Mol. Immunol. 31 169–217. [DOI] [PubMed] [Google Scholar]
- Perelson, A.S. and Oster, G.F. 1979. Theoretical studies of clonal selection: Minimal antibody repertoire size and reliability of self/non-self discrimination. J. Theor. Biol. 81 645–670. [DOI] [PubMed] [Google Scholar]
- Shinkai, Y., Nakauchi, H., Honjo, T., and Okumura, K. 1988. Mouse immunoglobulin allotypes: Multiple differences between the nucleic acid sequences of the IgEa and IgEb alleles. Immunogenetics 27 288–292. [DOI] [PubMed] [Google Scholar]
- Taketani, M., Naitoh, A., Motoyama, N., and Azuma, T. 1995. Role of conserved amino acid residues in the complementarity determining regions on hapten-antibody interaction of anti-(4-hydroxy-3-nitrophenyl) acetyl antibodies. Mol. Immunol. 32 983–990. [DOI] [PubMed] [Google Scholar]
- Trinh, C.H., Hemmington, S.D., Verhoeyen, M.E., and Phillips, S.E. 1997. Antibody fragment Fv4155 bound to two closely related steroid hormones: the structural basis of fine specificity. Structure 5 937–948. [DOI] [PubMed] [Google Scholar]
- Varga, J.M., Kalchschmid, G., Klein, G.F., and Fritsch, P. 1991. Mechanism of allergic cross-reactions—I. Multispecific binding of ligands to a mouse monoclonal anti-DNP IgE antibody. Mol. Immunol. 28 641–654. [DOI] [PubMed] [Google Scholar]
- Voller, A. 1978. The enzyme-linked immunosorbent assay (ELISA) (theory, technique and applications). Ric. Clin. Lab. 8 289–298. [PubMed] [Google Scholar]
- Webster, D.M., Henry, A.H., and Rees, A.R. 1994. Antibody-antigen interactions. Curr. Opin. Struct. Biol. 4 123–129. [DOI] [PubMed] [Google Scholar]