

Abstract
To understand the molecular basis of glycosyltransferases’ (GTFs) catalytic mechanism, extensive structural information is required. Here, fold recognition methods were employed to assign 3D protein shapes (folds) to the currently known GTF sequences, available in public databases such as GenBank and Swissprot. First, GTF sequences were retrieved and classified into clusters, based on sequence similarity only. Intracluster sequence similarity was chosen sufficiently high to ensure that the same fold is found within a given cluster. Then, a representative sequence from each cluster was selected to compose a subset of GTF sequences. The members of this reduced set were processed by three different fold recognition methods: 3D-PSSM, FUGUE, and GeneFold. Finally, the results from different fold recognition methods were analyzed and compared to sequence-similarity search methods (i.e., BLAST and PSI-BLAST). It was established that the folds of about 70% of all currently known GTF sequences can be confidently assigned by fold recognition methods, a value which is higher than the fold identification rate based on sequence comparison alone (48% for BLAST and 64% for PSI-BLAST). The identified folds were submitted to 3D clustering, and we found that most of the GTF sequences adopt the typical GTF A or GTF B folds. Our results indicate a lack of evidence that new GTF folds (i.e., folds other than GTF A and B) exist. Based on cases where fold identification was not possible, we suggest several sequences as the most promising targets for a structural genomics initiative focused on the GTF protein family.
Keywords: Glycosyltransferase, fold recognition, sequence-similarity searching, protein structure prediction, structural genomics
The glycosylation reaction is of great biological importance to both prokaryotes and eukaryotes, and is catalyzed by enzymes forming a large protein family, the glycosyltransferases (GTFs). These enzymes transfer a sugar moiety from an activated glyconucleotide to an acceptor, which may be a growing oligosaccharide, a lipid, or a protein. In addition to its central role in all synthetic processes involving carbohydrates, GTFs are important drug targets in the fight against cancer, as well as against bacterial, viral, and fungal infections (Breton and Imberty 1999; Unligil and Rini 2000; Davies 2001). GTFs also provide an important technological tool, as they have opened new perspectives in the chemoenzymatic synthesis of oligosaccharides (Sears and Wong 1996; Davies 2001). Despite the many applications of these enzymes, the precise molecular events in the catalytic mechanism of GTFs have remained elusive. This lack of information is due to difficulties in expressing the enzymes, which are frequently membrane-bound, and in characterizing enzymatic mechanisms with complex substrates. However, it is now well established that the substrate specificity and stereospecificity of the glycosylation reaction depend on the enzymes’ three-dimensional (3D) architecture, especially in the vicinity of the binding site (Tvaroska et al. 2000,Tvaroska et al. 2002; Andre et al. 2001, 2002).
At the sequence level, there are now a large number of open reading frames (ORFs) that correspond to GTFs. A database classifying GTF sequences into families based on sequence similarity and substrate/product stereochemistry is available and currently contains 56 potential families (see http://afmb.cnrs-mrs.fr/CAZY/; Campbell et al. 1997). At the 3D-structural level, currently only 13 GTF protein structures are available, which can be grouped into two folds: GTF A and GTF B. These two basic folds are shown in Figure 1 ▶. The GTF A fold belongs to the α/β family, consisting of parallel β-strands, flanked on both sides by α-helices, and has been described as containing an N-terminal glyconucleotide donor-binding pocket and a C-terminal acceptor-binding domain (Unligil and Rini 2000). The GTF B fold is also a member of the α/β family. In contrast to the GTF A type of fold, the GTF B fold comprises two Rossmann-fold-like domains separated by a deep cleft. The glyconucleotide donor-binding pocket is located at the bottom of the cleft, where it interacts solely with the C-terminal domain, and the N-terminal domain is predicted to be responsible for acceptor binding (Unligil and Rini 2000). Undoubtedly, these structures provide a wealth of information about substrate binding, specificity, and possible catalytic mechanisms for most of the known GTFs (Gastinel et al. 2001; Persson et al. 2001; Tarbouriech et al. 2001).
Figure 1.

The two most abundant folds found in the GTF protein family: GTF A and GTF B folds. (A) The typical GTF A structure of the spore coat polysaccharide biosynthesis protein Spsa from Bacillus subtilis (PDB code 1qg8A; Charnock and Davies 1999). (B) The typical GTF B type fold adopted by the Escherichia coli protein MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis (PDB code 1f0kA; Ha et al. 2000).
For filling in the information gap between protein sequences and structures, knowledge-based methods (i.e., comparative modeling) have been found to be useful (Blundell et al. 1987; Baker and Sali 2001). The central step in knowledge-based protein structure prediction is the search for structurally similar templates for a given query sequence. Once an appropriate structural template is identified, information about the 3D shape for the query sequence could be suggested. This is particularly helpful in identifying interaction partners for the query protein and in providing insights into its potential function (Domingues et al. 2000). Different methods are used to deduce protein homologies. At the sequence level, homologies can be recognized by pairwise searches in which the query sequence is scanned against sequences stored in a database, using software programs such as BLAST (Altschul et al. 1990) and FASTA (Pearson and Lipman 1988). Marked improvements in detecting higher numbers of remote homologies (i.e., by matching more and more dissimilar sequences) have been obtained using PSI-BLAST (Altschul et al. 1997) and Hidden Markov Models (HMMs; Eddy 1996; Sonnhammer et al. 1997).
In addition to such sequence-based approaches for structural template identification, several fold-recognition techniques have been developed which incorporate structural information at a variety of levels. These fold-recognition methods are broadly classified into two categories based on the nature of the algorithm used. Profile-based methods operate by gathering both sequence and structural information (Rice and Eisenberg 1997; Kelley et al. 2000; Shi et al. 2001). Threading methods are based on mean force fields derived from databases of known structures (Godzik et al. 1992; Jones et al. 1992; Sippl 1995; Bryant 1996). These methods were developed to push fold recognition beyond the level of sequence-based similarity searches. The overall good performances of these techniques have been widely addressed in a series of Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiments (Levitt 1997; Murzin 1999; Sippl et al. 2001). In addition to providing the remotely homologous template to be used for comparative modeling in case-by-case studies, fold recognition has also been used in automatic prediction experiments. There, fold recognition was found to enhance genome annotation by suggesting 3D fold information for a number of genome sequences (Pawlowski et al. 2001).
In the field of comparative modeling of GTFs, fold recognition approaches have been used in case-by-case studies for identifying structural templates for the bovine α-1,3-galactosyltransferase (Rao and Tvaroska 2001), porcine α3-galactosyltransferase (Imberty et al. 1999), and human α3-fucosyltransferase (de Vries et al. 2001). Studies have also adressed the occurence of specific and conserved peptidic motifs to identify remote homologs in the GTF family (Breton and Imberty 1999; Breton et al. 2002). However, to our knowledge, automatic fold assignment was not carried out on sequences from this protein family.
As stated earlier, the number of available GTF 3D structures is still quite limited, although the number of GTF sequences delivered by genome sequencing projects keeps increasing. Since remote homology is frequently found within the GTF family (i.e., different GTFs sharing the same fold at low sequence identity; Unligil and Rini 2000), fold recognition methods will be an important tool to direct and accelerate the mapping of GTF sequence landscape to protein structural space. To address the relationship between GTF sequences and structures, it is very natural and interesting to ask, “How many GTFs can find an adapted structural template among currently known protein structures?” And, “In addition to the GTF A and GTF B folds, should one expect some other GTF folds?” The present study attempted to answer these questions.
Results and Discussion
Sequence clustering
We retrieved 7500 GTF sequences from public databases, and reduced this set to 5188 unique sequences. Using sequence-clustering techniques, we identified 262 clusters in this original set. The details of the algorithms we used to perform the clustering are outlined in the Materials and Methods section. However, it is important to point out here that we performed this clustering with the constraint that the same fold should be found among the sequences in each cluster. It should also be mentioned that a weak sequence similarity may still exist between sequences from different clusters, because a very low cutoff E-value (1e-10) was chosen when assigning cluster membership. Consequently, a representative sequence was selected in every one of the identified 262 clusters.
The sizes of the clusters vary markedly. The largest cluster consists of 1043 sequences, containing almost 20% of all of the GTF sequences. The five largest clusters consist of 2866 sequences, amounting to 55% of all of the GTFs addressed in this paper. There are 132 singlet clusters.
Fold recognition
The 262 representative GTF sequences from the reduced data set were further processed by three different fold-recognition and two sequence-based searching methods. The results are summarized in Table 1. The fold recognition methods 3D-PSSM (Kelley et al. 2000), FUGUE (Shi et al. 2001), and GeneFold (Jaroszewski et al. 1998) provided confident fold assignments for 102, 138, and 74 representative sequences out of the 262 initial ones, respectively. When considering all GTFs represented by these seed sequences, we established that the three fold-recognition methods were able to confidently assign structure to 3695 (71.2%), 3774 (72.7%), and 3635 (70.1%) GTF sequences out of the initial 5188, respectively. The deduced success rates of the different fold-recognition methods were further compared to those obtained with sequence-based similarity searching methods (i.e., BLAST and PSI-BLAST). These were used to assign folds to the 262 GTF sequences by searching against a sequence database representing all known protein 3D structures (i.e., PDB sequence database; Berman et al. 2000). We found that the performance of every one of the three fold-recognition methods was significantly better than that of BLAST searching alone. Taking the results of 3D-PSSM as an example, one can confidently identify the folds adopted by the member sequences of 72 more clusters (about 1203 GTF sequences) compared to simple BLAST searching. In addition, fold recognition was also found to outperform PSI-BLAST-based searching. For example, 3D-PSSM was able to identify folds in 35 more clusters (about 373 GTF sequences) than PSI-BLAST did. In the present study, FUGUE confidently assigned folds to more cluster members than any other fold-recognition or sequence-based similarity searching method.
Table 1.
The results of fold recognition and sequence-based similarity searching methods applied to the glycosyltransferase protein family
| Method | Standard for confident hit | No. of clustersa out of 262 | No. of sequencesb out of 5188 |
| 3D-PSSM | E-val3D-PSSM < 0.05 | 102 | 3695 |
| FUGUE | ZFUGUE > 6.0 | 138 | 3774 |
| GeneFold | ST > 400 | 74 | 3635 |
| BLAST | E-value < 0.001 | 30 | 2492 |
| PSI-BLAST | E-value < 0.001 | 67 | 3322 |
a The number of clusters with confident hits.
b The total number of sequences represented by those clusters where a confident hit was identified.
Complementary to the performance evaluation of the different methods by the number of folds identified confidently in the subset of the 262 representative GTF sequences, we carried out a pairwise assessment. For this, two characteristic numbers were considered: (1) the number of clusters where two given methods found a significant fold hit, and (2) the number of clusters where two given methods generated an identical significant fold hit. The evaluation of similarity of 3D protein shapes was carried out with the help of the combinatorial extension of the optimal path (CE) method (Shindyalov and Bourne 1998), as indicated in Materials and Methods. A z-score (ZCE ) generated by the CE structural alignment method was used as a quantitative measure of similarity between two folds. If ZCE was larger than 4.2, the two folds were regarded as identical. A summary of the pairwise performance evaluation of the different methods is shown in Table 2. In most of the clusters where confident hits were found by BLAST and PSI-BLAST, fold-recognition methods assigned folds with high certainty. For example, in the 30 clusters where folds were identified by BLAST, 3D-PSSM and FUGUE assigned an identical significant top fold hit in 23 and 28 clusters, respectively. Furthermore, when considering the 67 clusters identified by PSI-BLAST, 3D-PSSM and FUGUE were able to identify an identical significant top fold hit in 52 and 62 clusters, respectively. In addition, largely consensual results were also found between 3D-PSSM and FUGUE. For those 102 folds identified by 3D-PSSM, FUGUE was able to provide an identical hit for 87 clusters. However, a poor performance for GeneFold was observed, as the results obtained with this method were quite different compared to the sequence-based searching and the other two fold-recognition methods. Indeed, GeneFold was able to identify the same top fold hit in only 32 and 28 clusters in comparison to the 67 and 102 folds assigned by PSI-BLAST and 3D-PSSM, respectively. The comparable performances of 3D-PSSM and FUGUE are certainly due to the similar fold-recognition algorithms used, as both fall in the category of the 1D/3D profile-based fold-recognition methods. More precisely, 3D-PSSM and FUGUE share the following three basic characteristics: (1) PSI-BLAST searching is included in both methods in order to make extensive use of sequence information; (2) both methods use structure-based profiles; and (3) the standards for identification of confident hits are well defined.
Table 2.
Comparison of the consensus among different methods
| 3D-PSSM | FUGUE | GeneFold | BLAST | PSI-BLAST | |
| 3D-PSSM | — | 92 (87) | 51 (32) | 24 (23) | 54 (52) |
| FUGUE | — | 51 (38) | 29 (28) | 64 (62) | |
| GeneFold | — | 18 (15) | 37 (28) | ||
| BLAST | — | 30 (30) | |||
| PSI-BLAST | — |
The value outside the parentheses denotes the total number of clusters where both methods were able to assign a confident hit. The value inside the parentheses denotes the number of clusters where the same significant top hit is identified by both methods. Comparable numbers outside and inside the parentheses indicate equal performance of both of the methods.
Among the 262 representative GTF sequences, there were cases where 3D-PSSM and FUGUE suggested several significant hits. It was interesting to perform a more detailed analysis in such situations, as we expected to obtain further insights into the performance of the fold-recognition methods. When facing solutions with several confident hits, we focussed our attention on the regions aligned between the query and the hit sequences. We realized that folds for several domains situated on the query sequence could be confidently identified in this way, provided that the regions of query-hit alignments overlap by less than 20%. In situations where query-hit alignments occurred in nearly the same regions (overlap larger than 80%), the similarity between these confident hits was assessed on the structural level by using the CE algorithm.
A fully automated analysis was performed for query sequences with several significant hits identified by 3D-PSSM or FUGUE by considering all of these hits as well as their alignments. Of the 102 representative sequences confidently identified by 3D-PSSM, 48 sequences gave several confident hits, but after analysis we established that folds for two domains could be confidently assigned in the case of only four sequences. In the remaining 44 cases, we applied CE for structural similarity assessment, and found that 41 sequences share structurally related folds (ZCE > 4.2). In only three cases, structurally unrelated folds (ZCE ≤ 4.2 for at least one pair of hits) are assigned to an identical region in the query sequence. Similarly, of the 138 sequences with confident top hits identified by FUGUE, several significant hits were assigned to 77 GTFs. In the case of only 15 sequences, folds for two unrelated domains were confidently assigned to the same query. In the remaining 62 cases, we applied CE for structural similarity assessment and found that 54 domains share structurally related folds. In only eight cases, structurally unrelated folds are assigned to an identical region in the query sequence. Compared with 3D-PSSM, FUGUE confidently identified more sequences with two domains, at the expense of an increased uncertainty in identifying structurally unrelated folds assigned to an identical region in the query sequence. Independently of the fold recognition methods being applied, we established that among the hits where two domains were confidently identified, in most of the cases the top-scoring fold belongs to the GTF A or GTF B class. Based on this observation, we took into account only the top hits in our further analysis.
Three types of contradictory results can be seen in Table 2:
In some cases, sequence-based searching could identify a significant hit, whereas fold recognition could not. Such a finding was observed previously in genome annotation using fold recognition. In the annotation of the Mycobacterium genitalium ORFs reported by Kelley et al. (2000), not all of the assignments made by PSI-BLAST were confidently confirmed by 3D-PSSM (Kelley et al. 2000). A strong sequence signal, perhaps a motif highly conserved in close homologs, may be attenuated upon the inclusion of a large amount of diverse sequence and structural information in the fold-recognition procedure. Therefore, techniques such as PSI-BLAST must still be used for an initial screening, complemented by fold-recognition techniques to extend the range of detectable homologies.
It was also observed that both 3D-PSSM and FUGUE could outperform each other, depending on the particular sequence to be analyzed. The reason for such differences is that 3D-PSSM and FUGUE capture different aspects of similarity between distant protein homologs. Such behaviors were widely addressed in the series of CASP experiments (Fischer et al. 2000). It was established that the different fold-recognition methods are often complementary.
For a given cluster, a confident fold hit can be assigned by different methods, but these top hits differ significantly, even if they are mapped on the same region in the query sequence. In such cases, generally, additional information (obtained, i.e., by trying other fold-recognition methods or human expert judgment) must be used to decide which is the most appropriate fold hit.
The above results clearly demonstrate that structures can be successfully assigned to about 70% of the GTF sequences by the current fold-recognition methods, with a prediction rate higher than those obtained with sequence-based searching methods (i.e., BLAST and PSI-BLAST). Additionally, the results from 3D-PSSM and FUGUE are to a large extent in agreement, implying that the hits from these two methods are reliable. Finally, we have shown that a joint, jury-like prediction scheme combining the results of different fold-recognition methods enhances the confidence of fold assignment, and contributes to an increased detection rate of remote homologs (Lundström et al. 2001).
Structural clustering
To analyze and extract valuable information from the fold-recognition processing of the 262 representative sequences, we performed a structural clustering of all of the identified folds by again using CE structural alignment (Shindyalov and Bourne 1998). Consequently, a dissimilarity matrix was constructed, and the dimension was reduced by multidimensional scaling (MDS; Schiffman et al. 1981). We obtained in this way 2D maps of GTF structural space by using either 3D-PSSM or FUGUE. These maps are shown in Figure 2A and B ▶, respectively. In Figure 2A ▶, two main clusters can be found, with eight entries falling in the GTF A family and seven entries grouped in the GTF B class. The values of RMSDCE and ZCE between the two central, representative folds in the GTF A (1g8oA) and GTF B (1f6dA) clusters are 5.4 Å and 2.6 Å for 80 aligned residues, respectively. The numbers of GTFs sequences falling into the two basic clusters GTF A and GTF B are 2600 and 1057, respectively. Therefore, 3D-PSSM confidently identified nearly 70% (3657 of 5188) of GTFs sequences to adopt either the GTF A or the GTF B fold. However, one can see in Figure 2A ▶ the presence of five other points, representative of folds corresponding to 38 GTFs sequences. These points are significantly distant from the points forming the two main clusters GTF A and GTF B, and belong to the potential “new” GTF folds identified by 3D-PSSM.
Figure 2.
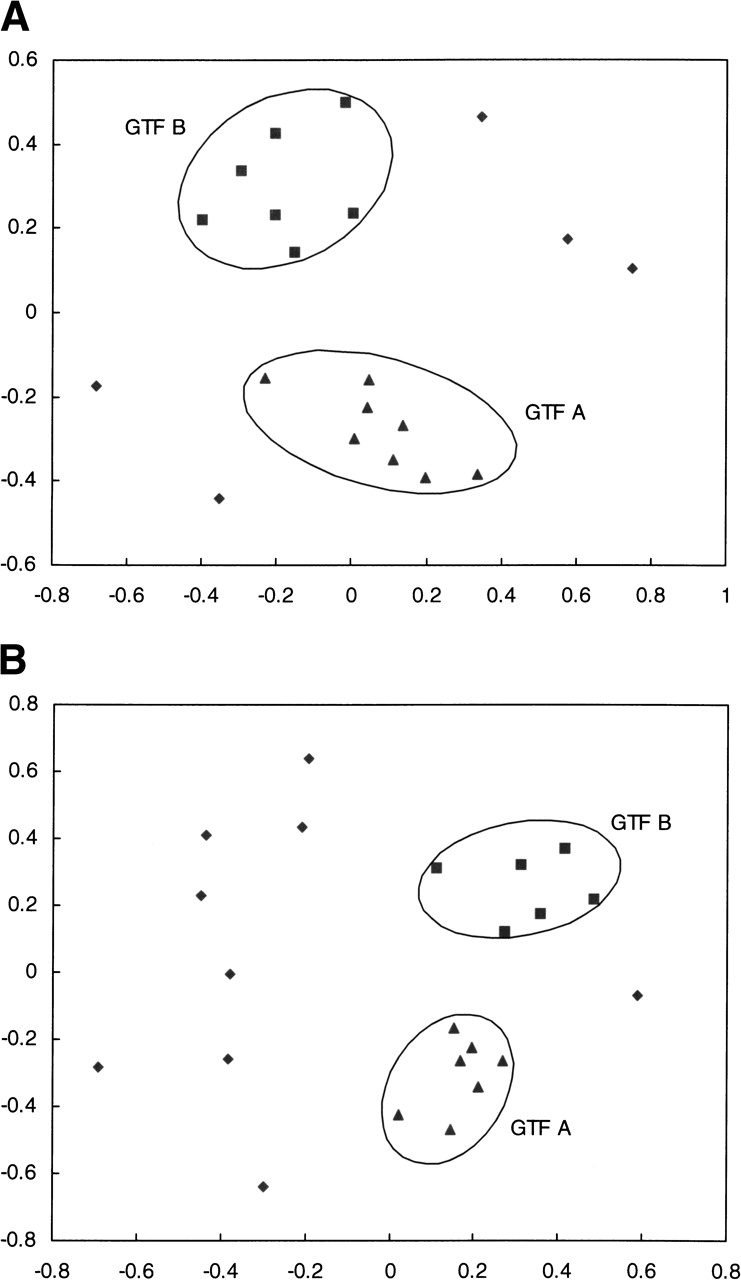
A 2D projection of the structural similarities from an all-against-all comparison of the hits generated from fold recognition by the MDS technique. For any two hits, the closeness in the 2D plot approximately represents the pair’s structural similarity. Based on (A) the results from 3D-PSSM; (B) the results from FUGUE. The hits are labeled according to the groups (▴, GTF-A; ▪, GTF-B; ♦, potential new GTF folds).
Data generated by FUGUE were processed by analogy to the analysis carried out on 3D-PSSM results. A graph summary of the analysis is shown in Figure 2B ▶. Again two main clusters of points can be identified, which are representative of the GTF A and GTF B fold types, with seven and six structural entries, respectively. A total of 2587 GTFs sequences are affiliated with the seven structures falling in cluster GTF A, and 1113 sequences are related to the six structures in cluster GTF B. The values of RMSDCE and ZCE for the comparison between the central hits (1ll3A and 1iirA) in GTF A and GTF B clusters are 5.1 Å and 3.5 for 136 aligned residues, respectively. The remaining nine entries may be regarded as potential “new” GTF folds. Only 74 GTF sequences are related to these marginal structures.
“New” GTF folds
Our fold recognition studies demonstrated that unexpected folds could be assigned confidently to some of the GTF sequences, referred to as “new GTF folds.” 3D-PSSM identified five such folds, and nine where found by FUGUE. It should be emphasized here that these “new” GTF folds share common folds with already known protein structures, and they are “new” only in that they differ from the GTF A and GTF B shapes. Taking into account the three clusters identified as “new” folds by both 3D-PSSM and FUGUE, the number of unique clusters with potential “new” folds is 11. Results from both methods are compared in detail in Table 3.
Table 3.
Detailed results for those clusters with identified “new” GTF folds from 3D-PSSM or FUGUE
| 3D-PSSM | FUGUE | ||||||||||
| No. of cluster | NCBI-gia | Size of cluster | Hit | E-val3D-PSSM | Alignment lengthd | Class | Hit | ZFUGUE | Alignment length | Class | ZCEe |
| 1 | 2984014 | 3 | 1qg8Ab | 8.57e-03 | 221/882 | GTFA | 1czfA | 13.73 | 319/882 | New | 2.6 |
| 2 | 9963867 | 6 | N.C.H.c | — | — | — | 1bkcA | 6.00 | 238/238 | New | — |
| 3 | 13881785 | 2 | 1f89A | 6.85e-10 | 225239500/868 | New | 1fo6A | 23.10 | 233269519/868 | New | 6.7 |
| 4 | 15292321 | 20 | N.C.H. | — | — | — | 1knyA | 7.68 | 241/689 | New | — |
| 5 | 17133624 | 2 | 1f6dA | 3.51e-03 | 329/714 | GTFB | 1im8A | 8.33 | 219/714 | New | 2.6 |
| 6 | 1698601 | 7 | 1b04A | 3.19e-02 | 280/740 | New | N.C.H. | — | — | — | — |
| 7 | 2384786 | 9 | N.C.H. | — | — | — | 1gcqC | 10.96 | 62/868 | New | — |
| 8 | 3172046 | 12 | 1h54A | 1.19e-04 | 29640812/822 | New | 1h54A | 13.79 | 1727822/822 | New | 8.6 |
| 9 | 15230362 | 1 | 1klo_ | 2.03e-03 | 137/736 | New | 1c3jA | 8.35 | 297/736 | GTFB | 2.0 |
| 10 | 18699592 | 4 | N.C.H. | — | — | — | 1qmeA | 38.05 | 261/366 | New | — |
| 11 | 15159314 | 16 | 1fchA | 2.65e-08 | 232/686 | New | 1ft1A | 15.16 | 281/686 | New | 3.9 |
a GenPept accession number for representative sequence.
b Each hit is described by five characters, including the PDB code together with the chain name. In the case when the fifth character is “_”, the protein has only one chain.
c N.C.H., no confident hit available.
d Alignment length and aligned region on the query sequence. Figures in regular font style indicate the alignment length versus overall length for the query sequence. When figures are in bold font, the left (left superscript) and right (right superscript) bounds of the aligned region flank the figure, indicating the alignment length.
e Z-score of CE structural alignment. Pairs of hits with ZCE > 4.2 are regarded as identical.
In the case of the nine clusters for which 3D-PSSM or FUGUE gave nonconsistent solutions, we can only speculate that the proposed fold would be the real one. In contrast, we can predict with sufficient confidence that “new” GTF folds could be found in the two clusters where 3D-PSSM and FUGUE share the same top significant hit. These two clusters represent some 14 GTF sequences. A first cluster finds a template structure in fold 1f89A, adopted by the yeast α+β protein Ylr351C. This cluster contains only two sequences from the genome of Mycobacterium tuberculosis. The first sequence has a dolichol-phosphate-mannosyl transferase activity (Dpm1); the substrate specificity of the second sequence has not been identified. In this first cluster, we found that close to the common top hit generated by both 3D-PSSM and FUGUE were present hits with sufficiently high scores to be considered significant. It turned out that these were representative of the GTF A fold and are mapped in a region of the sequence different from the one generating the top hit (cf. Table 3). We therefore found a situation where a typical GTF fold is located on the same sequence together with a “new” fold of unknown function. More experimental work is necessary in this case to determine whether GTF function is retained when the GTF A domain is deleted. Only when this is verified can one conclude that a “new” GTF fold could be related to the sequences in this first cluster.
The second cluster contains 12 bacterial protein sequences, most of them exhibiting a cellobiose/cellodextrin phosphorylase activity. We found that a relevant template fold is 1h54A (maltose phosphorylase; MP), a dimeric enzyme that catalyzes the conversion of maltose and inorganic phosphate into β-D-glucose-1-phosphate. Every monomer consists of an N-terminal complex β-sandwich domain, a helical linker, an (α/α)6 barrel catalytic domain, and a C-terminal β-sheet domain. In contrast to the first cluster, we found no indications that any part of this protein sequence will fit GTF A or GTF B templates. The top hit provided by 3D-PSSM or FUGUE maps in the same region of the sequence, and the alignment length matches on almost all of the template (cf. Table 3). Furthermore, it was established that the (α/α)6 barrel has an unexpectedly strong structural and functional analogy with the catalytic domain of glucoamylase from Aspergillus awamori. The only conserved glutamate of MP (Glu487) superposes onto the catalytic residue Glu179 of glucoamylase and likely represents the general acid catalyst. When we scrutinized the 3D-PSSM model generated for the representative protein sequence gi3172046 for this second cluster, we found that a homologous residue (Asp) maps close to MP Glu487 and glucoamylase Glu179. All of the described observations provide us good confidence to assign this type of fold to GTF sequences with cellobiose/cellodextrin phosphorylase activity.
Structural genomics target selection
As a key research topic in the postgenomic era, structural genomics aims to use high-throughput structure determination and computational analysis to provide a 3D structure for every known protein (Brenner 2000; Sali 2001). Currently exhaustive structural determination for all known proteins appears to be prohibitively expensive, and therefore the selection of a structurally nonredundant set of targets is of primary importance. The principal requirement for target selection is to define a relatively small set of proteins with new, currently unknown folds in an initial large collection of sequences (Portugaly and Linial 2000; Frishman 2002). The selection of such targets is a challenging task, because it is extremely difficult to predict whether a given sequence will point to a novel protein fold or not. However, there are encouraging indications that the total number of stable protein folds is limited (Chothia 1992; Portugaly and Linial 2000; McGuffin and Jones 2002).
We speculate that there are two possible situations when considering those GTFs with unassigned folds in our study. On one hand, some GTFs could share a common fold with proteins of known structure, but could not be detected by current fold-recognition methods. We expect that when applying different state-of-the-art fold-recognition methods on such GTF sequences, variable results would be obtained, with top hits situated somewhere in the lower limits of certainty. On the other hand, some GTFs could adopt novel, unknown protein folds. We expect that for such sequences, various state-of-the-art fold-recognition methods might provide consistently nonconfident hits (McGuffin and Jones 2002), although a systematic analysis of possible correlations between low statistical scores from fold-recognition methods (i.e., 3D-PSSM and FUGUE) and the likelihood of finding novel folds is still not available. Due to the poor performance of GeneFold, only the results from 3D-PSSM and FUGUE were jointly utilized for such a target selection. As pointed out by the authors of 3D-PSSM (Kelley et al. 2000), those hits with E-val3D-PSSM larger than 1.0 should be regarded as hits of low confidence. When applying 3D-PSSM, we identified 70 clusters with such low-confidence hits (Fig. S-1a in the Supplemental Material). Similarly, FUGUE identified 59 clusters with very weak hits (i.e., ZFUGUE < 3.0; Fig. S-1b). However, low-confidence and uncertain predictions were jointly provided by the two methods for only 30 of the 262 clusters. In this set of 30 unknown folds, 19 clusters include only one sequence (singlets), whereas the remaining 11 clusters account for 261 GTF sequences. We therefore ended up with these 261 sequences as the most promising targets for structural genomics studies of the GTF family. We took into account an argument by Frishman (2002), requiring the targets for a structural genomics study to represent not only novel folds, but also as much as possible of the sequences in the initial data set, and this mainly for reasons of cost-effectiveness. Details regarding these 11 clusters representative of the 261 target sequences are listed in Table S-2. Precise choices of candidates for structural determination should be further guided by feasibility studies of the expression, purification, and crystallization behavior of the targets.
Conclusions
The glycosyltransferase protein family is of particular interest for testing and validation of fold-recognition techniques because diverse amino acid sequences are known to adopt only two typical protein folds ensuring sugar synthesis. Three fold-recognition approaches (3D-PSSM, FUGUE, and GeneFold) were employed here to identify the folds of some 5188 GTF sequences. Taking the results from 3D-PSSM and FUGUE into account, the overall performance of fold recognition presented in this study is summarized in Tables 4 and 5. The results obtained indicate that current fold-recognition methods can identify confidently a fold for nearly 70% of all known GTF sequences with a confidence of at least 95%, improving on remote homolog identification by the most sophisticated sequence-based method (PSI-BLAST; Table 5). In most of the remaining 30% of sequences, we found a “hidden” relationship to GTF A or GTF B folds; that is, the top hits from fold recognition still point to GTF A/B but without a significant statistical score. We found that the FUGUE method performs slightly better than 3D-PSSM, which is evidenced by the consistently greater numbers appearing in the lower triangular part of Table 4. Generally, the results from 3D-PSSM and FUGUE are to a large extent in agreement, certainly due to the similar fold-recognition algorithms on which they are based. The high degree of degeneracy of GTF amino acid sequences in protein structural space was confirmed by 3D clustering of the significant hits. We were not able to confidently detect other currently known folds that could support glycosyltransferase function. However, an interesting evolutionary relationship has been identified among three folds exhibiting glucoamylase, maltose phosphorylase, and glycosyltransferase activities. In order to direct structural genomics efforts for GTFs structural determination, appropriate targets were selected from those GTFs for which the different fold-recognition methods in use were consistently unable to identify a fold type. The research strategy reported here would also be useful to map sequence space on the set of known folds (shapes) for other protein families.
Table 4.
Cross-comparison of the overall performance of 3D-PSSM and FUGUE
| FUGUE 3D-PSSM | GTF A | GTF B | Othera | Not Identifiedb |
| GTF A | 54 (2566) | 0 (0) | 1 (3) | 4 (31) |
| GTF B | 1 (1) | 31 (1043) | 1 (2) | 5 (11) |
| Other | 0 (0) | 1 (1) | 3 (30) | 1 (7) |
| Not identified | 9 (20) | 33 (69) | 4 (39) | 114 (1365) |
Figures outside parentheses relate to clusters of GTF sequences. Figures in parentheses relate to sequences.
a The folds other than GTF A/B.
b The top hit from fold recognition is low-confidence, i.e. the top hit from 3D-PSSM with E-val3D-PSSM > 0.05 or the top hit from FUGUE with ZFUGUE < 6.0.
Table 5.
Summary of the rate of fold identification in the GTF family based on the results from 3D-PSSM and FUGUE
| GTF A/B folds | Confidenta | 85 (3609) | 32.4% (69.6%) |
| Non-confidentb | 52 (132) | 19.8% (2.5%) | |
| Other folds | Confidentc | 2 (14) | 0.8% (0.3%) |
| Non-confidentd | 9 (68) | 3.5% (1.3%) | |
| Not identifiede | — | 114 (1365) | 43.5% (26.3%) |
| Total | — | 262 (5188) | 100.0% (100.0%) |
Figures outside parentheses relate to clusters of GTF sequences (column 1, raw numbers; column 4, percentages). Figures in parentheses relate to sequences.
a Both 3D-PSSM and FUGUE generate the same significant hit as GTF A/B.
b The same confident hit as GTF A/B is not available from the results of 3D-PSSM and FUGUE, but at least the top hit from one method should be confidently assigned as GTF A/B, and the top hit from the other method could be confidently assigned as GTF A/B or not identified.
c Both 3D-PSSM and FUGUE have the same confident hit but other than GTF A/B.
d No same confident hit other than GTF A/B is found from the results of 3D-PSSM and FUGUE, but at least one method should generate a significant hit other than GTF A/B.
e Neither 3D-PSSM nor FUGUE can generate a confident hit.
Materials and methods
GTF sequence database
In this work we relied essentially on the CAZy database (Campbell et al. 1997) as a primary source of information. The CAZy database was compiled in two steps. First, glycosyltransferase sequences (i.e., NDP-sugar hexosyltransferase, EC 2.4.1.x) were collected from the Swissprot and EMBL/GenBank databanks and compiled into a preliminary sequence library. Second, representatives of each EC number were used as templates for BLAST similarity searches. Similar sequences were retrieved, even if they were still uncharacterized ORFs. The data set we constructed is composed of unique GTF sequences selected from among the sequences included in the CAZy database at the time this study was carried out. For this, we first downloaded the corresponding GenBank/GenPept or Swissprot accession numbers for all of the GTF sequences from the CAZy database (version as of 01/05/2002). Then, the sequences were retrieved from the NCBI GenBank/GenPept or Swissprot databases. For those GTFs with identical sequences, only one was kept for further processing. In this way, a nonredundant GTF sequence data set consisting of 5451 entries was compiled.
The length of the sequences varies from 12 to 4573 amino acids, as illustrated in Figure 3 ▶. More precisely, 18 and 72 sequences have lengths shorter than 50 and 100 aa, respectively, and 245 are longer than 1000 aa. Sequences with chain lengths shorter than 50 aa were excluded, and in the final data set there were only 54 sequences with chain length between 50 and 100 aa. Our purpose was to find the compromise between a data set that will be adapted to fold-recognition studies, and one with maximized information content, containing even some fragments of GTF amino acid sequences. The cutoff of 50 aa was deduced from two different sources. First, we noted that the fold-recognition server validation LiveBench experiment (Bujnicki et al. 2001) automatically excludes from further analysis sequences shorter than 100 aa. Second, we performed some preliminary experiments with the FUGUE and 3D-PSSM methods, by sending to them several fragments derived from the N terminus of the GTF B fold adopted by the UDP-glucosyltransferase of Amycolatopsis orientalis (PDB entry 1iir). Fragments were cut from the N terminus, spanning regions 1–30, 1–50, and 1–100, respectively. The FUGUE server recognized confidently all of the fragments and correctly identified structure 1iir as the template. In contrast, 3D-PSSM was not able to identify confident hits for fragments 1–30 and 1–50, and picked 1iir as a low-confidence template only for fragment 1–100. Based on these observations, we decided to apply the cutoff for chain length of 50 aa as a good compromise between the performance of current fold-recognition methods and the discovery spirit with which we tried to analyze sequence-structure relationships in the GTF protein family. On the other hand, sequences longer than 1000 aa often code for multidomain proteins, and cannot be processed by most of the current fold-recognition methods. In our study we also filtered out the sequences with lengths exceeding 1000 aa. Finally, 5188 GTF sequences (about 95% of the original 5451 GTFs) were kept for further study.
Figure 3.

The distribution of sequence lengths in the GTF protein family. In 5451 nonredundant GTF sequences, the length ranges from 12 to 4573 aa. About 69% of GTF sequences have an aa chain length in the range 200–550; only about 5% of GTFs have sequence longer than 1000 aa or shorter than 50 aa.
Sequence clustering
Generally, fold recognition requires more computational resources than sequence-based similarity searching. It was expected that fold identification would take a prohibitively long time if carried out on all of the 5188 GTF sequences, especially when using the web-based fold recognition servers (i.e., 3D-PSSM and FUGUE). However, it is clearly not necessary to perform fold recognition on every GTF sequence, because many GTF sequences have very high sequence similarity, sharing therefore the same fold. For these reasons, a systematic and exhaustive clustering was performed on the initial set of 5188 GTF sequences, with the constraint that every entry within the same cluster should correspond to the same protein fold. This last requirement is believed to hold true if sequence similarity is significant (Chothia and Lesk 1986; Rost 1999). Subsequently, one representative sequence from each cluster was selected to compile a new reduced GTF subset, which was further sent to fold recognition.
The following algorithm was applied for clustering the GTF sequences: (1) A sequence is chosen at random as a seed for the current cluster. (2) A BLAST search is executed with this sequence as a query against all other GTFs. Sequences with E-value less than 1e-10 are assigned to the current cluster. (3) For those sequences newly assigned to the current cluster, a BLAST run is executed against the remaining GTF sequences to find possible new cluster members. In this step, we made an extensive use of similarity by transitivity in the sequence space (Yona et al. 2000). To prevent unrelated proteins from clustering together, a more strict standard was adopted at this step; that is, the qualified new member was required to have not only an E-value less than 1e-10, but additionally a similar sequence length [i.e., |L1−L2|/max(L1,L2) < 30%]. (4) The above step would be repeated until no sequence could be merged into the current cluster. (5) For each member in the newly built cluster, the Number of Directly Similar sequences within this cluster (NDS) was calculated by intracluster sequence-based cross-comparisons. The E-value for two directly similar sequences was again set to 1e-10. Then a representative sequence for the current cluster was selected, by choosing the one with a maximal value of NDS. (6) The same procedure is iterated for the remaining GTF sequences to build the other clusters. Eventually, 262 clusters were formed out of the original set of 5188 GTF sequences, and the 262 representative sequences were further processed by fold-recognition methods.
Fold recognition
3D-PSSM
3D-PSSM (Kelley et al. 2000) is a profile-based method relying on both multiple sequence alignments and multiple structural alignments. Central to the method is the so-called Three-Dimensional Position-Specific Scoring Matrix (3D-PSSM) that combines data from multiple-sequence profiles as implemented in PSI-BLAST with structure-based profiles, taking into account secondary structure and solvent accessibility. However, the truly innovative component of the approach resides in the use of structural alignments of remote homologs to generate sequence profiles that are accurately aligned yet more diverse than those generated through PSI-BLAST. The fold library for 3D-PSSM is based mainly on the SCOP database (Murzin et al. 1995) and included 7485 structures at the time that the present study was undertaken.
The 262 GTF sequences, forming the representative subset, were submitted automatically to the 3D-PSSM fold-recognition server (http://www.sbg.bio.ic.ac.uk/servers/3dpssm/) by running a Perl script. 3D-PSSM scans a submitted query sequence against its fold library, and potential homologs are suggested. Results were downloaded automatically for further analysis. According to the 3D-PSSM authors’ experience, all hits with E-val3D-PSSM less than 0.05 should be regarded as confident at the 95% certainty level.
FUGUE
FUGUE is a profile-based fold-recognition program, making extensive use of both multiple sequence and structural information (Shi et al. 2001). It is based on environment-specific substitution tables and structure-dependent gap penalties, where scores for amino acid matching and insertions/deletions are evaluated depending on the local environment of each amino acid residue in known structures (Shi et al. 2001). Given a query sequence, FUGUE scans its fold library, which is based on the HOMSTRAD database (Mizuguchi et al. 1998), calculates the sequence-structure compatibility scores, and produces a list of potential homologs and alignments. At the time the present study was performed, the FUGUE fold library contained 3914 templates.
By analogy to the protocol applied when using the 3D-PSSM fold-recognition server, the 262 sequences were sent to the FUGUE server (http://www-cryst.bioc.cam.ac.uk/~fugue/) automatically by running a dedicated Perl script. In addition, the results were automatically downloaded from the web site for further analysis. As pointed out by the authors Shi et al. (2001), hits with Z-scores larger than 6.0 should be considered confident at the 99% confidence level, and thus considered significant.
GeneFold
The third fold-recognition method we used is GeneFold (Godzik et al. 1992; Jaroszewski et al. 1998). Licensed by Tripos Inc., GeneFold is integrated into the SYBYL molecular modeling environment (SYBYL 6.8 2000). It uses both sequence and structural information to measure sequence-structure compatibility using three different scoring functions (Jaroszewski et al. 1998). The first scoring function evaluates sequence similarity only. The second scoring function evaluates a hybrid sequence/structure similarity score, where sequence, local conformational preferences, and burial terms are taken into account. The third, most elaborate scoring function derives a full hybrid score based on the compatibility of sequence, secondary structure, local conformational preferences, and burial terms between a query sequence and a structural template from the fold library. The results of sequence-structure matches using the above three functions are returned as a list of templates, ordered by decreasing scores, that are possible matches for the target sequence.
The original fold library distributed by Tripos Inc. consisted of 1824 entries representing all of the protein structures in the release of the PDB databank as of April 1998. In the past five years however, many protein structures with new folds have been deposited in PDB databank, and therefore the original GeneFold library was clearly outdated. For the purposes of our study, we updated the GeneFold library with all entries included in the 3D-PSSM fold library. At the time our study was preformed, 7485 protein structures were present in the 3D-PSSM fold library. However, as GeneFold supports a maximum size of 2500 structures per library, three new libraries were built up, with sizes of 2410, 2413, and 2414 structures, respectively. As can be seen, a total of 248 entries were not included in the libraries out of the 7485 initial ones, as GeneFold does not support structures with multiple conformations for the surface residues, with disordered chain terminals, or for which only the Cα coordinates are provided (Godzik et al. 1992).
The processing of the 262 sequences by GeneFold was executed on an SGI O2+ workstation by running a dedicated Perl script. For every one of the query sequences, GeneFold scanned the three libraries to find potential hits. Since GeneFold provides three different scores for a hit, we used a “jury” method to combine these three scores into a unique score (Lundström et al. 2001). Therefore, we did the following modifications:
-
A unique total score (ST) was introduced:
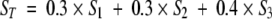
(1) where S1, S2, and S3 denote the three different scores, respectively. Then, all of the hits generated from these three fold libraries were ranked by order of decreasing ST.
In contrast to 3D-PSSM and FUGUE, statistical bounds were not derived for GeneFold scores which can guarantee confidence in the derived hits. However, we regarded a hit as highly confident if the total score was higher than 400. This value was derived from a validation study we carried out on a restrained set of protein sequence-structure pairs taken from the CASP4 experiment (Sippl et al. 2001).
BLAST and PSI-BLAST searching
In a manner similar to that used for our fold-recognition studies, the subset of the 262 representative GTFs sequences was processed by the sequence-based similarity searching methods BLAST and PSI-BLAST. For this we used the standalone version of the BLAST program (Altschul et al. 1990, 1997). The nonredundant (NR) and structural (PDB) sequence databases were downloaded from (ftp://ncbi.nlm.nih.gov/blast/) in their updates dated 14 May 2002. The NR sequence database consists of all nonredundant GenBank CDS translation, PDB, SwissProt, PIR, and PRF entries (Altschul et al. 1990, 1997). The PDB database contains all of the sequences derived from protein structures deposited in the PDB Databank (Berman et al. 2000). BLAST searching was executed by using all 262 sequences as queries against the PDB sequence database. After an adjustment to the size of the NR database, all of the hits with an E-value less than 0.001 were considered confident.
PSI-BLAST is a sensitive sequence-similarity search method, performed in an iterative manner. First, an initial BLAST search is carried out, and the hits are ranked according to their alignment scores. Second, a profile in the form of a score matrix model is calculated from a certain number of the sequences taken from the top of the hit list. Third, an additional search is executed as a profile-sequence comparison using the generated score-matrix model to find a new set of hits. This search loop is repeated until no more new hits can be found or the maximum number of iterations is reached. To assign a fold to every GTF sequence, PSI-BLAST searching was executed in two stages. First, all 262 sequences were run against the NR sequence database by PSI-BLAST for three iterations. Based on the score matrix model built in this first search, we further searched with PSI-BLAST against the PDB sequence database for one round to find the potential structurally similar hits. The E-values for including sequences in the score matrix model and assessing the significant similar hits were both set to 0.001.
Confidence levels
The confidence levels provided for BLAST, PSI-BLAST, and 3D-PSSM are based on expectation values (E-values). By definition, the E-value is a parameter that describes the number of hits one can “expect” to see just by chance when searching a database of a particular size. It decreases exponentially with the score that is assigned to a match between two sequences. The lower the E-value, or the closer it is to zero, the more “significant” the match is. Currently, the most extensively studied E-value statistic is the one associated with BLAST. On the other hand, FUGUE uses an alternative scoring based on Z-scores, evaluated as the number of standard deviations above the mean score obtained by chance. Limited information is provided by the authors of both FUGUE and 3D-PSSM on the precise method of calculating confidence levels in general. However, an initiative such as LiveBench (Bujnicki et al. 2001) can provide some basis for the rationale of our results. The LiveBench project is a continuous benchmarking program for a number of participating fold-recognition servers. Every week the results are collected and evaluated using automated model assessment programs. The LiveBench experiment thus provides a simple evaluation of the sensitivity and specificity of the available servers and provides a way to assess the confidence of the obtained predictions. In the current LiveBench program, the 95% confidence levels for the 3D-PSSM and FUGUE servers are situated at cutoffs for E-values < 0.119 and for Z-scores > 4.8, respectively (cf. http://www.cs.bgu.ac.il/~dfischer/CAFASP3/summaries/thresholds.html). In our study, in order to declare a 3D-PSSM hit confident, we used an E-value cutoff of 0.05, as recommended by the authors of that method. Similarly, in order to declare a FUGUE hit confident, we applied a Z-score cutoff of 6.0, deduced by the FUGUE authors. In both cases, we used more restrictive cutoffs than the ones obtained in a real application, such as the LiveBench experiment. Therefore, we expect our assignments to be at least 95% correct in a CASP-like experiment.
Structural alignment
In order to rationalize the results from the fold-recognition studies and to establish the structural relationships among the identified hits, it is important to reliably assess protein structural similarity. More precisely, evaluation of protein structural similarity was needed mainly in the following two situations: (1) For the same query sequence, it was necessary to compare among them the hits obtained by the different fold-recognition methods. (2) In order to classify all the hits identified, a structural clustering was carried out based on an all-against-all comparison of the generated hits.
Several structural alignment methods have been developed (Taylor and Orengo 1989; Holm and Sander 1993; Shindyalov and Bourne 1998; Lu 2000). In our work we used CE, a structural alignment method proposed by Shindyalov and Bourne (1998). This algorithm involves a combinatorial extension (CE) of an alignment path defined by aligned fragment pairs, in contrast to the conventional techniques based on dynamic programming and Monte Carlo optimization. Two main parameters (RMSDCE and ZCE) for characterizing a given structural superposition are returned along with the resulting sequence alignment. The parameter RMSDCE is the root mean square deviation (Å) based on Cα positions in the two structures at the optimal superposition. ZCE is the z-score from the CE statistical model. Although the value of RMSDCE is intuitive to reveal structural similarity between two structures, it is not sufficient. For example, a structure alignment with a lower RMSDCE can be more significant than one with a higher RMSDCE if the number of aligned residues is greater in the first alignment. In the present study, ZCE was used to measure the structural similarity of the hits derived by fold-recognition methods. As pointed out by Shindyalov and Bourne, a family level similarity can be found for structures with ZCE ≥ 4.5. In contrast, superfamily level similarity appears for structures with ZCE values between 4.0 and 4.5, whereas the similarity for those structures with ZCE ≤ 3.7 is usually very low. The source codes of the CE program were downloaded from http://cl.sdsc.edu/ce.html, and compiled for use in our local computer.
Structural clustering
Our study led us to the conclusion that an important structural degeneration is present among the otherwise diverse GTF amino acid sequences. For example, hits generated from some 102 of the 262 clusters map into only 20 different protein structures. As a matter of fact, this degeneration may be even higher, because some of these structures still share high similarity. To investigate the relationships between the hits produced by fold recognition, 3D clustering was undertaken by using the CE structural alignment method (Shindyalov and Bourne 1998). First, a structural dissimilarity function operating on two protein structures was defined as follows:
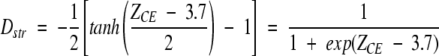 |
(2) |
where tanh is the hyperbolic tangent function, and the value of Dstr varies from 1.0 to 0.0 with the increase of the structure similarity (i.e., ZCE) between two hits. We used this type of sigmoid function to ensure smoothness properties for the dissimilarity function Dstr. First, we took the 20 hits generated by 3D-PSSM and we calculated Dstr between any pair of these, to obtain a 20 × 20 dissimilarity matrix. To provide a visual representation of the structural relationships among these 20 hits, we applied multidimensional scaling (MDS; Schiffman et al. 1981). In this way we reduced the dimension of the original 20 × 20 dissimilarity matrix to 20 × 2. Finally, structural similarity relationships were displayed as a 2D plot (see Fig. 2A ▶). The structural relationship for the 22 different hits generated by FUGUE were derived similarly, and are displayed in Figure 2B ▶.
Electronic supplemental material
The supplemental material contains two tables and one figure showing (1) the detailed fold-recognition results for the 262 representative GTFs using 3D-PSSM, FUGUE, and PSI-BLAST, (2) the potential targets for a GTF structural genomics initiative based on the results from 3D-PSSM and FUGUE, and (3) distribution of the statistical scores for the top hits of the 262 representative GTF sequences generated by the different fold-recognition methods. All of the tables, figures, and figure legends are included in the file supplement.pdf.
Acknowledgments
We thank Dr. Laure Jolly for a number of discussions concerning glycosyltransferases, and the referees whose constructive comments were very helpful in improving the quality of this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: See www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03131303.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre, I., Tvaroska, I., Rao, M., and Kozar, T. 2001. Designing modulators for glycosyltransferases based in crystal structure-determined atomic coordinates of reactive groups and molecular modeling of the active sites. Patent Appl. WO 2001085748.
- Andre, I., Tvaroska, I., and Carver, J. 2002. Design of inhibitors for glycosyltransferases based on the conformation of the sugar-phosphate linkage in sugar nucleotide for the glycosyltransferases. U.S. Patent 6415234.
- Baker, D. and Sali, A. 2001. Protein structure prediction and structural genomics. Science 294 93–96. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell, T.L., Sibanda, B.L., Sternberg, M.J., and Thornton, J.M. 1987. Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326 347–352. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E. 2000. Target selection for structural genomics. Nat. Struct. Biol. 7 (Suppl.) 967–969. [DOI] [PubMed] [Google Scholar]
- Breton, C. and Imberty, A. 1999. Structure/function studies of glycosyltransferases. Curr. Opin. Struct. Biol. 9 563–571. [DOI] [PubMed] [Google Scholar]
- Breton, C., Heissigerova, H., Jeanneau, C., Moravcova, J., and Imberty, A. 2002. Comparative aspects of glycosyltransferases. Biochem. Soc. Symp. 23–32. [DOI] [PubMed]
- Bryant, S.H. 1996. Evaluation of threading specificity and accuracy. Proteins 26 172–185. [DOI] [PubMed] [Google Scholar]
- Bujnicki, J.M., Elofssonm, A., Fischer, D. and Rychlewski, L. 2001. LiveBench-1: Continuous benchmarking of protein structure prediction servers. Protein Sci. 10 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, J.A., Davies, G.J., Bulone, V., and Henrissat, B. 1997. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 326 (Pt. 3) 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnock, S.J. and Davies, G.J. 1999. Structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 38 6380–6385. [DOI] [PubMed] [Google Scholar]
- Chothia, C. 1992. Proteins. One thousand families for the molecular biologist. Nature 357 543–544. [DOI] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, G.J. 2001. Sweet secrets of synthesis. Nat. Struct. Biol. 8 98100. [DOI] [PubMed] [Google Scholar]
- de Vries, T., Knegtel, R.M., Holmes, E.H., and Macher, B.A. 2001. Fucosyltransferases: Structure/function studies. Glycobiology 11 119R–128R. [DOI] [PubMed] [Google Scholar]
- Domingues, F.S., Koppensteiner, W.A., and Sippl, M.J. 2000. The role of protein structure in genomics. FEBS Lett. 476 98–102. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1996. Hidden Markov models. Curr. Opin. Struct. Biol. 6 361–365. [DOI] [PubMed] [Google Scholar]
- Fischer, D., Elofsson, A., and Rychlewski, L. 2000. The 2000 Olympic Games of protein structure prediction; fully automated programs are being evaluated vis-a-vis human teams in the protein structure prediction experiment CAFASP2. Protein Eng. 13 667–670. [DOI] [PubMed] [Google Scholar]
- Frishman, D. 2002. Knowledge-based selection of targets for structural genomics. Protein Eng. 15 169–183. [DOI] [PubMed] [Google Scholar]
- Gastinel, L.N., Bignon, C., Misra, A.K., Hindsgaul, O., Shaper, J.H., and Joziasse, D.H. 2001. Bovine α1,3-galactosyltransferase catalytic domain structure and its relationship with ABO histo-blood group and glycosphingolipid glycosyltransferases. EMBO J. 20 638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik, A., Kolinski, A., and Skolnick, J. 1992. Topology fingerprint approach to the inverse protein folding problem. J. Mol. Biol. 227 227–238. [DOI] [PubMed] [Google Scholar]
- Ha, S., Walker, D., Shi, Y., and Walker, S. 2000. The 1.9 Å crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci. 9 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233123–138. [DOI] [PubMed] [Google Scholar]
- Imberty, A., Monier, C., Bettler, E., Morera, S., Freemont, P., Sippl, M., Flockner, H., Ruger, W., and Breton, C. 1999. Fold recognition study of α3-galactosyltransferase and molecular modeling of the nucleotide sugar-binding domain. Glycobiology 9 713–722. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L., Rychlewski, L., Zhang, B., and Godzik, A. 1998. Fold prediction by a hierarchy of sequence, threading, and modeling methods. Protein Sci. 7 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Levitt, M. 1997. Competitive assessment of protein fold recognition and alignment accuracy. Proteins (Suppl.) 1 92–104. [DOI] [PubMed] [Google Scholar]
- Lu, G.G. 2000. TOP: A new method for protein structure comparisons and similarity searches. J. Appl. Crystallogr. 33 176–183. [Google Scholar]
- Lundström, J., Rychlewski, L., Bujnicki, J., and Elofsson, A. 2001. Pcons: A neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10 2354–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin, L.J. and Jones, D.T. 2002. Targeting novel folds for structural genomics. Proteins 48 44–52. [DOI] [PubMed] [Google Scholar]
- Mizuguchi, K., Deane, C.M., Blundell, T.L., and Overington, J.P. 1998. HOMSTRAD: A database of protein structure alignments for homologous families. Protein Sci. 7 2469–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G. 1999. Structure classification-based assessment of CASP3 predictions for the fold recognition targets. Proteins 37 88–103. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Pawlowski, K., Rychlewski, L., Zhang, B., and Godzik, A. 2001. Fold predictions for bacterial genomes. J. Struct. Biol. 134 219–231. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, K., Ly, H.D., Dieckelmann, M., Wakarchuk, W.W., Withers, S.G., and Strynadka, N.C. 2001. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8 166–175. [DOI] [PubMed] [Google Scholar]
- Portugaly, E. and Linial, M. 2000. Estimating the probability for a protein to have a new fold: A statistical computational model. Proc. Natl. Acad. Sci. 97 5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, M. and Tvaroska, I. 2001. Structure of bovine α-1,3-galactosyltransferase and its complexes with UDP and DPGal inferred from molecular modeling. Proteins 44 428–434. [DOI] [PubMed] [Google Scholar]
- Rice, D.W. and Eisenberg, D. 1997. A 3D-1D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence. J. Mol. Biol. 267 1026–1038. [DOI] [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12 85–94. [DOI] [PubMed] [Google Scholar]
- Sali, A. 2001. Target practice. Nat. Struct. Biol. 8 482–484. [DOI] [PubMed] [Google Scholar]
- Schiffman, S.S., Reynolds, M.L., and Young, F.W. 1981. Introduction to multidimensional scaling. Academic Press, New York.
- Sears, P. and Wong, C.H. 1996. Intervention of carbohydrate recognition by proteins and nucleic acids. Proc. Natl. Acad. Sci. 93 12086–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J., Blundell, T.L., and Mizuguchi, K. 2001. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310 243–257. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1995. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 5 229–235. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J., Lackner, P., Domingues, F.S., Prlic, A., Malik, R., Andreeva, A., and Wiederstein, M. 2001. Assessment of the CASP4 fold recognition category. Proteins (Suppl.) 5 55–67. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E.L., Eddy, S.R., and Durbin, R. 1997. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 28 405–420. [DOI] [PubMed] [Google Scholar]
- SYBYL 6.8. 2000. Software. Tripos Inc., St. Louis, MO.
- Tarbouriech, N., Charnock, S.J., and Davies, G.J. 2001. Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: A comparison with related NDP-sugar glycosyltransferases. J. Mol. Biol. 314 655–661. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. and Orengo, C.A. 1989. Protein structure alignment. J. Mol. Biol. 208 1–22. [DOI] [PubMed] [Google Scholar]
- Tvaroska, I., Andre, I., and Carver, J. 2000. Ab initio molecular orbital study of the catalytic mechanism of glycosyltransferases: Description of reaction pathways and determination of transition-state structures for inverting N-acetylglucosaminyltransferases. J. Am. Chem. Soc. 122 8762–8776. [Google Scholar]
- ———. 2002. Molecular modeling of catalytic mechanism of inverting and retaining glycosyltransferases. Abstr. Pap. Am. Chem. Soc. 223 U478–U479. [Google Scholar]
- Unligil, U.M. and Rini, J.M. 2000. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 10 510–517. [DOI] [PubMed] [Google Scholar]
- Yona, G., Linial, N., and Linial, M. 2000. ProtoMap: Automatic classification of protein sequences and hierarchy of protein families. Nucleic Acids Res. 28 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]