

Abstract
The mRNA of Escherichia coli yedU gene is induced 31-fold upon heat shock. The 31-kD YedU protein, also calls Hsp31, is highly conserved in several human pathogens and has chaperone activity. We solved the crystal structure of YedU at 2.2 Å resolution. YedU monomer has an α/β/α sandwich domain and a small α/β domain. YedU is a dimer in solution, and its crystal structure indicates that a significant amount of surface area is buried upon dimerization. There is an extended hydrophobic patch that crosses the dimer interface on the surface of the protein. This hydrophobic patch is likely the substrate-binding site responsible for the chaperone activity. The structure also reveals a potential protease-like catalytic triad composed of Cys184, His185, and Asp213, although no enzymatic activity could be identified. YedU coordinates a metal ion using His85, His122, and Glu90. This 2-His-1-carboxylate motif is present in carboxypeptidase A (a zinc enzyme), and a number of dioxygenases and hydroxylases that utilize iron as a cofactor, suggesting another potential function for YedU.
Keywords: YedU, Hsp31, X-ray crystallography, chaperone, catalytic triad, metal binding protein
Many bacterial genes encoding chaperones, proteases, transcription regulators, and other proteins are activated upon heat shock. Chaperones and proteases can increase a bacterium’s ability to survive under stress conditions by helping proteins fold and by removing damaged polypeptides. The mRNA of the Escherichia coli yedU gene is induced 31-fold upon heat shock (Richmond et al. 1999). Expression of the YedU protein, the 282-residue product of the yedU gene, is enhanced in an hns-deletion strain (Yoshida et al. 1993), a genetic background known to derepress stress gene expression (Hengge-Aronis 1996). Primary sequence analysis shows that YedU is highly conserved in several microbial genomes (Fig. 1 ▶).
Figure 1.

Sequence alignment of YedU with its homologs. Sfle, Shigella flexneri (gi:24113342, percentage identity 98%); Pflu, Pseudomonas fluorescens (gi:23059816, 61%); Saur, Staphylococcus aureus (gi:15923541, 56%); Paer, Pseudomonas aeruginosa (gi:11347751, 55%); Vcho, Vibrio cholerae (gi:9658312, 61%). The secondary structures of YedU are shown on top of the alignment. H, helix; S, strand.
It was recently reported that YedU has chaperone activity (Sastry et al. 2002; Malki et al. 2003). At 45°C, it can suppress the aggregation of folding intermediates of alcohol dehydrogenase (ADH) and promote its reactivation when the temperature is dropped to 23°C. Furthermore, 1-anilino-8-naphthalenesulfonate (bis-ANS) binding experiments suggest that hydrophobic domains of YedU are more exposed at the elevated temperature. Although an ATPase activity of YedU has not been detected, ATP can suppress its chaperone activity at 45°C in vitro.
Remote homology (less than 15% identity) is detected between YedU and the PfpI intracellular cysteine protease family using PSI-BLAST (Altschul et al. 1997). YedU is almost twice as long as the members of the PfpI protease family, due to an N-terminal extension and several large insertions. PH1704 from Pyrococcus horikoshii is the first member of the PfpI protease family whose structure has been determined (Du et al. 2000; PDB accession code 1G2I). The active form of PH1704 is a hexamer, and its catalytic triad is composed of Cys100, His101, and a Glu474 contributed by an adjacent subunit. Cys100 and His101 are conserved in YedU, but Glu474 is not. The strongest E. coli homolog of PfpI is not YedU but YhbO (gi:606010, 47% identity with PfpI), which is annotated as a “hypothetical protein.” This suggests that YhbO protein is a PfpI protease family member and most likely has a conserved function. YedU may have structural similarity, but may differ in function.
We solved the crystal structure of YedU at 2.2 Å resolution by multiple isomorphous replacement (MIR). An extended hydrophobic patch was found on the surface of the protein, crossing the dimer interface, and may serve as a substrate-binding site for its chaperone activity. The structure also revealed a catalytic triad similar to that seen in protease PH1704, but with access to the Cys residue restricted by a narrow channel. Additionally, a zinc(II) ion is coordinated by a 2-His-1-carboxylate motif, reminiscent of carboxypeptidase A (Christianson et al. 1989) and a number of other metalloenzymes (Que 2000). The presence of these potential active sites raises the possibility that YedU may have multiple functions.
Results and Discussion
Monomer structure
The YedU structure was determined at 2.2 Å resolution by multiple isomorphous replacement with anomalous scattering (MIRAS) using Hg and KI derivatives. The first three residues and the last residue were disordered in the structure. A model containing 278 residues, a zinc(II) ion, and 120 ordered water molecules was refined to a final R-factor and Rfree of 20.30% and 24.30%, respectively.
After the structure of the protein was modeled, a difference map demonstrated a strong density between the side chains of His85, Glu90, His122, and their symmetry equivalents on a crystallographic twofold axis. An X-ray fluorescence experiment indicated that zinc was present in the native crystals. We added a zinc(II) ion to the model, and the occupancy was estimated to be about 40%. A crystal soaked in ZnCl2 yielded a structure with zinc(II) ion at full occupancy. The low occupancy of zinc(II) ion in the native crystal was probably due to the absence of zinc(II) ion in buffers during protein purification and crystallization. Because no zinc(II) ions were provided during purification and crystallization, this suggests that the zinc(II) ion may be the biologically relevant metal ion, and that it survives protein purification. The metal binding site will be described in a separate section.
The overall secondary structure is a mixture of helices and sheets (Fig. 2A ▶). The globular structure (~50 Å × 45 Å × 30 Å) can be subdivided into two domains, DOM1 (residues 48–56, 71–210, and 230–282) and DOM2 (1–47, 57–70, and 211–229). DOM1 is an α/β/α sandwich fold similar to protease PH1704 (Fig. 2B ▶). The central β-sheet consists of seven strands (S7, S6, S3, S8, S9, S13, and S12), with S12 as the only antiparallel strand. The central β-sheet is flanked by helices H2, H3, H4, and H11 on one side and by helices H5, H6, H7, H8, and H10 on the other. DOM2 is smaller than DOM1 and is composed of an antiparallel β-sheet (S1↑-S2↓-S5↓-S4↑) and two helices, H1 and H9.
Figure 2.

Overall structure of YedU and structural comparison with protease PH1704. (A) Stereoview of the YedU monomer. H, helix; S, strand; orange and green code for helix and strand, respectively. The upper part of the structure is a sandwich domain (DOM1) composed of a central seven-strand β-sheet flanked by helices. Below the sandwich domain, there is a smaller domain (DOM2) composed of an antiparallel β-sheet (S1↑-S2↓-S5↓-S4↑) and two helices (H1 and H9). Helices H3 and H4 at the right side of the sandwich domain are different from the corresponding region of (B), the structure of PH1704 (Du et al. 2000; PDB acc. code 1G2I), a member of the PfpI cysteine protease family. (C) Stereoview of the YedU homodimer looking down the dimer twofold axis; subunits A and A′ are in green and red, respectively. In subunit A, the β-turn L2 and loops L5 and L9 on the interface are labeled. The potential catalytic triad residues are shown as ball-and-stick in magenta, and the residues coordinating zinc(II) ion are shown as ball-and-stick in cyan. In subunit A′, atom γS of Cys184 and the zinc(II) ion are shown as spheres in magenta and cyan, respectively. (D) The molecular surface of YedU homodimer looking down the molecular twofold axis. Hydrophobic residues (Pro, Ala, Val, Leu, Ile, Met, Phe, Tyr, and Trp) are yellow; positively charged residues (His, Lys, and Arg) are blue; negatively charged residues (Asp and Glu) are red; all other residues are white. Arrows point to the β-turn L2.
Dimerization
Size exclusion chromatography indicates that YedU is a dimer in solution. There are two crystal contacts that are candidates for the solution dimer interface, each at a crystallographic twofold axis. Both contacts bury a significant amount of surface area. The zinc binding site is present in one contact, which buries about 2020 Å2 of solvent-accessible surface area (1010 Å2 from each monomer). The side chains of His85, Glu90, His122, and their symmetry mates coordinate two zinc(II) ions at the center of the contact. Together with the zinc coordinating residues, the van der Waals interactions between residues Pro41, Pro43, Tyr82, Ala86, Pro117, Gln121, Ser124, Asn128, and their symmetry mates form the core of this crystal contact with 1340 Å2 of buried surface area. Side chains of additional residues forming the peripheral part of this contact do not pack well and some are indeed disordered in the electron density map, but nevertheless contribute to the total buried surface area calculated. These disordered side chains include those of Lys42, Arg45, Lys47, Glu92, Lys130, and Glu144. Most residues involved in this contact, including the zinc coordinating residues, are not conserved among all homologs.
The second contact occurs on another crystallographic twofold axis and buries about 2080 Å2 of solvent-accessible surface area (Fig. 2C ▶). Most of the residues involved in this contact are located on three secondary structural elements, β-turn L2 between strands S1 and S2, loop L5 between strands S3 and S4, and loop L9 between strand S6 and helix H3. The β-turn L2 inserts into the other subunit by loop “handshaking,” making extensive van der Waals contacts with the other subunit. L5 plus Tyr59 of S4 are located at the center of the contact and interact with L2′, L5′, and L9′ of the other subunit. Seven residues (Leu99–Tyr105) of L9 are buried or partially buried; they make contacts with L2′, L5′, and strand S5′ of the other subunit. There are three pairs of salt bridges between Glu14 (of L2) and Arg58′ (L5′), Asp15 (L2) and Lys123′ (H4′), Glu57 (L5) and Lys102′ (L9′), and their symmetry-related equivalents. Leu67 of strand S5 is also buried in the interface. Other residues that are partially buried include Phe19 and Ile158. Most of these residues making the contact are conserved or conservatively substituted throughout family members. Although the buried surface areas are comparable in two crystal contacts, the former contact with the zinc(II) ion is less extensive because the side chains that form the peripheral part of it are indeed disordered. Thus, the latter more conserved and better packed contact without the zinc binding site most likely occurs in the solution dimer.
A chaperone active site
YedU is able to suppress the aggregation of folding intermediates and promote their reactivation (Sastry et al. 2002; Malki et al. 2003). Examination of the structure revealed two concave hydrophobic patches separated by a hydrophobic ridge on the molecular twofold axis (Fig. 2D ▶). The ridge is comprised of solvent-exposed hydrophobic residues (Val10, Ile12, Phe18, and Phe19) from strands S1 and S2, and their connecting L2 β-turn. The concave hydrophobic patches are comprised of solvent-exposed residues Tyr105, Trp106, and Met108 from loop L9 and Met116, Pro117, and Phe118 from the following helix H3. These residues are conserved or conservatively substituted throughout family members.
These solvent-exposed hydrophobic patches of YedU are reminiscent of those found in other molecular chaperones, which provide hydrophobic binding sites for their protein substrates. The interior surfaces of the apical domain of the trans GroEL cavity (Xu et al. 1997) and small heat shock protein oligomers (Kim et al. 1998; van Montfort et al. 2001) present hydrophobic surfaces. DnaK, a member of the Hsp70 family, has a β-sandwich domain with an extended hydrophobic pocket (Zhu et al. 1996). Hsp33, a redox-regulated molecular chaperone, has two regions that may serve as substrate binding sites: a large saddle-shaped hydrophobic surface and a pronounced groove with a mixture of polar and nonpolar residues (Vijayalakshmi et al. 2001). Molecular chaperones use various strategies to regulate the exposure of their hydrophobic binding sites. The exposure of GroEL apical domain hydrophobic patches is regulated by ATP hydrolysis and co-chaperonin GroES binding (Xu et al. 1997). The small heat shock proteins (sHSPs) associate into large assemblies at ground state and expose their hydrophobic binding sites upon a temperature-induced dissociation of the large assembles into smaller ones (van Montfort et al. 2001).
YedU displays chaperone activity at elevated temperatures, and bis-ANS binding to YedU is enhanced at high temperatures (Sastry et al. 2002). This suggests that local unfolding may occur at the chaperone active site to enhance interaction with protein folding intermediates. We propose that at elevated temperatures, the β-turn L2 between strands S1 and S2 of YedU may become more dynamic. In a dynamic state, movement of the β-turns away from the molecular twofold axis will expose packed hydrophobic residues Ile12, Ala13, and Phe19 and flatten the separating ridge, thus joining two concave hydrophobic patches to form one larger hydrophobic groove that crosses the molecular twofold axis. The L2 β-turn structure is stabilized by a salt bridge between Glu14 and Arg58′ from the other subunit. At elevated temperatures, a salt bridge between Glu14 and Arg127′ may help to stabilize an alternative conformation. The dimensions of the large hydrophobic groove will be ~35 Å long and 8–12 Å wide, large enough to accommodate a nine-residue extended polypeptide. It is very likely that this hydrophobic groove on the surface of YedU serves as a substrate-binding site for its reported chaperone activity at elevated temperatures in vitro (Sastry et al. 2002).
Although no data have been reported to show that ATP directly binds to YedU in solution, there are in vitro biochemical studies suggesting that ATP suppresses the chaperone activity of YedU at elevated temperatures (Sastry et al. 2002). The proposed substrate-binding site found on the surface of YedU is composed of residues from both subunits of the homodimer. The structural elements making the dimer interface are predominantly loops (Fig. 2C ▶), which are relatively easily adapted to an alternative conformation upon ligand binding. If ATP does bind directly to YedU in solution, the binding of ATP may rearrange the dimer interface and disrupt the continuity of the hydrophobic substrate-binding site, resulting in a suppression of the chaperone activity of YedU. Alternatively, ATP binding could stabilize the native state and prevent local unfolding at elevated temperatures by mass action, thus inhibiting chaperone activity. Difference maps from an ATP-soaked crystal do not reveal a binding site for ATP. The binding site may be blocked in this crystal form.
The potential catalytic triad
A potential catalytic triad is formed by Cys184, His185, and Asp213 (Fig. 3A,B ▶). The triad shares the same handedness with that in protease PH1704 and papain-type cysteine proteases; that is, the Cys184 interacts with δN of His185 and Asp213 interacts with ɛN of His185. An additional acidic residue Glu76 makes a hydrogen bond to Cys184, analogous to Glu15 in protease PH1704. Asp 213 of the catalytic triad also forms a hydrogen bond with an additional histidine His154. The imidazole ring of His154 and NH moieties of His154 and Gly153 may form the “oxyanion hole” that could stabilize the substrate during transition state. The nucleophile Cys100 of protease PH1704 has 12 Å2 of solvent-accessible surface area, and bulky compounds such as gelatin have been shown as a substrate of this protease. In YedU, solvent-accessible surface area of the potential nucleophile Cys184 is 6 Å2, and its accessibility is restricted by a narrow channel which is about 6 Å wide and 11 Å deep (Fig. 3C ▶). The channel is predominately hydrophobic, composed of residues Tyr28, Phe208, Ile219, Tyr221, Ile246, and Phe263, with a negatively charged opening at the protein surface. The potential catalytic triad and channel residues are conserved or conservatively substituted throughout family members. There are two pockets near Cys184 (Fig. 3D ▶). Pocket 1 is lined by residues His154, Asp213, Thr216, Tyr221, and Met222. The opening of pocket 2 to the channel is hydrophilic because of residues His73 and Asp104, whereas the interior of pocket 2 is hydrophobic and composed of residues Pro20, Ser24, Leu25, Tyr28, Thr29, Phe68, Trp106, and Tyr221. Additionally, a narrow passageway connects the channel to a neighboring cavity which is about 7 Å deep and 8 Å wide. The cavity is predominately hydrophobic with a wide and amphipathic opening (Fig. 3E ▶). The narrow connecting passageway is located at the bottom of the cavity, and its width at the narrowest point is less than 4 Å between His73 and Pro262. The neighboring cavity and pockets 1 and 2 may serve as the substrate-binding pockets and define the substrate specificity.
Figure 3.
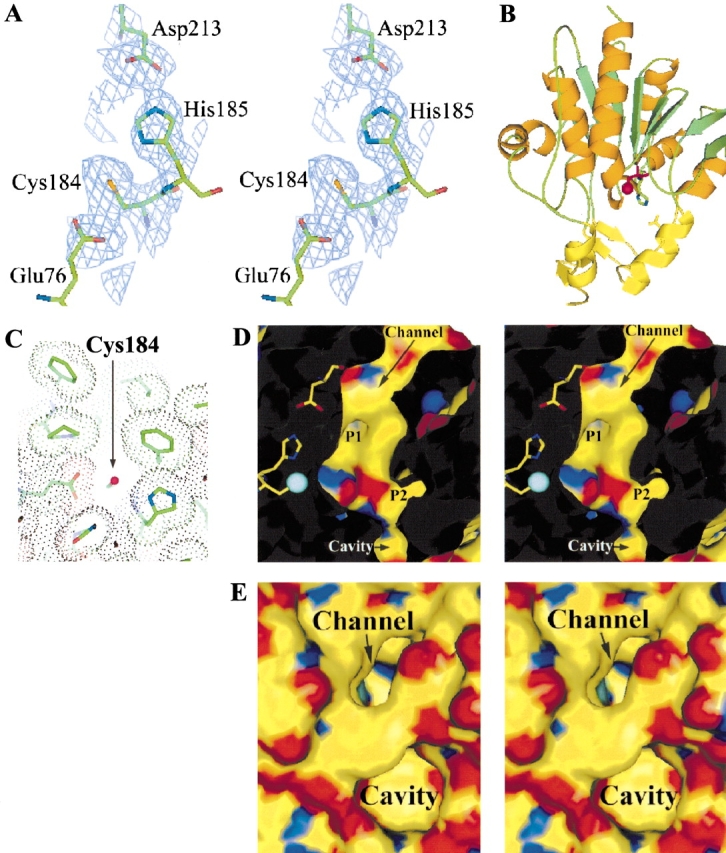
Cys184, His185, and Asp213 form a potential catalytic triad. (A) A stereogram of the triad with the corresponding solvent-flattened experimental map contouring at the 1σ contour level. (B) Asp213 is contributed to the catalytic triad by DOM2, which is shown in yellow. (C) The nucleophile Cys184 (shown as a magenta sphere) is accessible through a narrow channel. A van der Waals dot surface is shown for the channel-forming residues. (D) Two potential side chain binding pockets P1 and P2. Atom γS of Cys 184 is shown in cyan. (E) A cavity is located near the deep channel. Atom γS of Cys 184 is shown in cyan, which can be seen at the bottom of the channel.
YedU was examined for general protease activity by using azocasein as a substrate, the same assay used for PfpI protease (Blumentals et al. 1990). No general endopeptidase activity was observed (data not shown), consistent with the results reported by Malki and coworkers (2003). This result is consistent with the limited accessibility of Cys184 seen in the crystal structure. The dimension of the channel suggests that the end of a peptide may be able to access the active nucleophile Cys184. However, there was no detectable exopeptidase activity after 10-min incubation of Dps peptide or Pep302 peptide with YedU at ~1:1 molar ratio, as described in the Materials and Methods section. Peptides Dps (sequence STALKC) and Pep302 (sequence GQGQQYGGY) were used as substrates because of their immediate availability. It is possible that these peptide sequences are inconsistent with the substrate specificity of YedU if it is indeed an exopeptidase.
The zinc(II) ion and 2-His-1-carboxylate motif
Although the overall fold of YedU DOM1 is highly similar to the α/β/α sandwich fold of protease PH1704, the Val115-Arg127 region of YedU DOM1 is significantly different from the corresponding region of protease PH1704. The Val115-Arg127 region of YedU folds as two helices (H3 and H4), whereas two strands are present in the corresponding region of protease PH1704. Interestingly, the protein fold seems to be rearranged to facilitate the coordination of a zinc(II) ion or another metal ion. The zinc(II) ion observed in the crystal is coordinated by the side chains of His85, Glu90, and His122 contributed by a short loop between helices H3 and H4 (Fig. 4 ▶). In crystals, the zinc(II) ion and its symmetry mate are brought together due to crystal packing. In solution, the zinc(II) ion is exposed to the solvent (Figs. 2C ▶, 4 ▶). It is possible that a water molecular can participate in coordination of the zinc(II) ion in solution and serve a catalytic role. Additionally, the adjacent ɛO of Gln121 may be linked to the potential catalytic water molecule and serve as the general base. Using X-ray fluorescence, we found that the metal ion present in the crystals is zinc(II). No metal ions were added during purification and crystallization, indicating that zinc(II) ion could be the biologically relevant metal ion. It is also possible that iron is the biologically relevant metal ion and that it was lost during purification and replaced by the ubiquitous and more stable zinc(II) ion. Further exploration of this potential catalytic site must embrace both metals as potentially relevant cofactors. A catalytic zinc(II) ion coordinated by the side chains of two histidines and a glutamic acid with similar geometry was found in carboxypeptidase A (Christianson et al. 1989). In addition, a common 2-His-1-carboxylate motif coordinating an iron(II) ion was found in four enzyme families that catalyze different reactions (Que 2000), including extradiol cleaving catachol dioxygenases, Rieske dioxygenases, pter-independent hydroxylases, and isopenicillin N synthetase, suggesting other potential functions for YedU.
Figure 4.

A zinc(II) ion is coordinated by side chains of His85, Glu90, and His122. The zinc(II) ion is shown as a sphere in cyan.
Summary
The presence of a catalytic triad and a metal binding site, in combination with the reported chaperone activity, raises the possibility that YedU is a moonlighting protein, that is, a protein that has multiple functions (Jeffery 1999). If YedU is indeed a multifunctional protein in vivo, it may participate in bacteria responses not only to elevated temperatures, but also to other stress conditions such as oxidative stress. Studies of YedU will deepen our understanding of stress responses and the underlying mechanisms.
As we were finishing this manuscript, a report appeared describing the structure of YedU in a different crystal form (Quigley et al. 2003). Our results are consistent with theirs, including the preferred dimer interface, solvent-exposed hydrophobic surface as potential substrate-binding site for chaperone activity, and the catalytic triad. In addition, we identify the metal binding site, which was not reported in the their structure. The absence of the zinc(II) ion in the previous report is likely due to the presence of EDTA in their crystallization experiments.
Materials and methods
Recombinant expression and protein purification
Briefly, the open reading frame (ORF) of yedU from E. coli K-12 strain MG1655 was transferred into pET-30Xa/LIC vector (Novagen). After the construct’s identity was confirmed by DNA sequencing, BL21(DE3)pLysS cells (Novagen) bearing the expression vector were grown at 37°C and induced with isopropyl-β-D-thiogalactopyranoside (IPTG). The recombinant protein was first purified on a nickel-nitrilotriacetic acid (Ni2+-NTA) affinity column (QIAGEN). To remove the fusion tags, the recombinant protein was incubated overnight with Factor Xa protease (Novagen). The cleaved protein was loaded onto a MonoQ HR16/10 column and eluted with a 0–500 mM NaCl gradient over 20 column volumes (Amersham Pharmacia Biotech). The YedU-containing fractions from the MonoQ column were collected and further purified on a Superdex75 26/60 sizing column (Amersham Pharmacia Biotech) in 10 mM NaCl, 20 mM Tris, pH 8.2. The column was calibrated with gamma globulin (molecular weight, 158,000), bovine serum albumin (66,000), ovalbumin (44,000), and myoglobin (17,000). The final product of a 282-residue YedU protein was confirmed by mass spectrometry (Fisons).
Protease and exopeptidase assays
Protease activity was measured by the hydrolysis of azocasein (Blumentals et al. 1990). Assay mixtures containing 950 μL of 0.1% azocasein in 50 mM sodium phosphate buffer at pH 7.3 were mixed with 50 μL of 200 μM YedU in 5 mM DTT, 10 mM NaCl, and 20 mM Tris, pH 8.2. The final enzyme to substrate molar ratio was about 1:5. After 30 min, the reaction was terminated by the addition of 500 μL of 15% trichloroacetic acid (TCA) and incubation for 5 min on ice. The precipitate was removed by centrifugation, and the supernatant absorbance was measured at 440 nm. Substrate without YedU was used as a negative control, and trypsin (Sigma, with azocasein at a 1:50 molar ratio) was used as a positive control. The exopeptidase activity was measured by peptide digestion of Dps peptide (sequence STALKC) incubated with YedU at a molar ratio of 1:1 for 10 min in 1mM 2-mercaptoethanol (BME), 60mM NaCl, 10mM Tris, pH 7.0. The reaction was loaded onto a C18 reversed-phase column (Vydac). Digestion of the peptide substrate was analyzed by mass spectrometry (Fisons). As a negative control, reactions containing peptide only were also loaded on the C18 column and analyzed by mass spectrometry. The exopeptidase assay was repeated with the Pep302 peptide (sequence GQGQQYGGY). Both protease and exopeptidase assays were repeated in the presence of 2 mM ATP and 5 mM MgCl2. To test the potential metalloprotease activity, experiments were performed in the presence of 200 μM ZnCl2 or 1 mM ZnCl2. All experiments were performed first at room temperature and repeated at 37°C and 45°C.
Crystallization and heavy atom derivative preparation
YedU was concentrated to 15.0 mg/mL in a solution containing 5 mM DTT, 10 mM NaCl and 20 mM Tris, pH 8.2. Crystals were grown in hanging drops containing 2% PEG 600, 5mM DTT, 1.8 M ammonium sulfate, 0.1 M HEPES, pH 7.5 and frozen with 30% glycerol in liquid nitrogen. To prepare derivatives, the native crystals were soaked overnight in 1 mM p-chloromercuribenzoate (PCMB) or 5 min in 160 mM KI, respectively. A crystal was soaked 48 h in 1 mM ZnCl2 to recover full occupancy of the zinc(II) ion. To investigate nucleotide binding, crystals were soaked overnight in 1 mM adenosine triphosphate (ATP).
Data collection and processing
YedU native crystals diffracted to 2.65 Å on a MacScience rotating anode X-ray generator with a DIP2030 imaging plate. The resolution was extended to 2.2 Å on The Gulf Coast Consortium Protein Crystallography PX1 beamline at The Center for Advanced Microstructures and Devices (CAMD, Baton Rouge, LA). The crystal form was in space group P6122 with cell dimensions a = b = 52.98 Å and c = 347.40 Å. Assuming one molecule per asymmetric unit, the calculated Matthews coefficient VM value was 2.26Å3/Da, and the solvent content was estimated to be 45.6% (Matthews 1968). Diffraction data were collected at 100 K from a native crystal, a Hg (PCMB) derivative, and a KI derivative (Table 1). The PCMB derivative was collected at 1.008 Å to enhance the Hg anomalous signal. The data were processed and scaled with DENZO and SCALEPACK (Otwinowski and Minor 1997). The crystal was tested for the presence of zinc by X-ray fluorescence. A scintillation counter was placed about 2 cm from the crystal perpendicular to the X-ray beam, and the energy of the X-rays was then scanned from 9640 eV to 9680 eV in steps of 1 eV. The resulting spectrum, although noisy, exhibited a clear jump over a 7-eV range centered at 9666 eV. The K edge for pure Zn is at 9659 eV; however, the edge is shifted to higher energy for Zn(II). A scan of the crystal’s mother liquor showed no sign of zinc.
Table 1.
Data collection and phasing statistics
| Data set | Native | PCMB (Hg) | KI |
| Space group | P6122 | P6122 | P6122 |
| Unit cell (Å) | |||
| a − b | 52.98 | 52.85 | 53.02 |
| c | 347.40 | 347.40 | 347.22 |
| Resolution range (Å) | 90–2.2 | 90–2.5 | 90–2.3 |
| (Outer shell) | (2.25–2.2) | (2.56–2.5) | (2.35–2.3) |
| Wavelength (Å) | 1.5418 | 1.008 | 1.5418 |
| Measurements | 290299 | 228337 | 202376 |
| Unique reflectionsa | 15151 (779) | 9885 (496) | 11811 (421) |
| Completenessa | 94.5 (77.5) | 90.1 (73.5) | 83.4 (48.3) |
| I/σ (I)a | 14.7 (3.9) | 12.7 (2.3) | 13.6 (2.5) |
| Rmergea,b | 0.075 (0.319) | 0.107 (0.664) | 0.105 (0.581) |
| Mean FOM (40–2.4 Å) | 0.40 | ||
| Mean FOM following solvent flattening | 0.61 | ||
| Refinement statistics | |||
| Resolution (Å) | 30.0–2.2 (2.25–2.2) | ||
| Number of reflections (working/test)a | 14142 (750)/714 (40) | ||
| Number of molecules | |||
| Protein residues | 278 | ||
| Water | 120 | ||
| Zinc(II) | 1 | ||
| R-factor/R-free (%) | 20.30/24.30 | ||
| Rmsd from ideality | |||
| Bond lengths (Å) | 0.006 | ||
| Bond angles (°) | 1.3 | ||
| Ramachandran analysis (%) | |||
| Most favored regions | 87.3 | ||
| Additional allowed regions | 11.4 | ||
| Generously allowed regions | 0.4 | ||
| Disallowed regions | 0.8 | ||
a Values for the outer resolution shell of data are given in parentheses.
bRmerge = ∑ |Ii − Im|/∑ Ii, where Ii is the intensity of the measured reflection and Im is the mean intensity of all symmetry-related reflections.
Phasing, structure determination, and refinement
A single mercury site was found using the program SOLVE (Terwilliger and Berendzen 1997). The mercury site was used to cross-phase the KI derivative. The overall figure of merit (FOM) was 0.40–2.4 Å. After solvent flattening with the program RESOLVE (Terwilliger 2000), the FOM was improved to 0.61. RESOLVE and TEXTAL (Holton et al. 2000) built two fragmented models of modest quality, each containing backbone atoms for less than 50% of the residues. XtalView (McRee 1999) was used to rebuild and connect the fragments manually for the 278-residue molecular model. Reflections of 30–2.2 Å resolution were used for final refinement in the program CNS (Brunger et al. 1998). After the protein atoms were well refined, waters were gradually added into the model. A zinc(II) ion was included in the model after it was confirmed by X-ray fluorescence. The final Rfree was 24.30% and detailed refinement statistics are summarized in Table 1. PROCHECK (Laskowski et al. 1993) indicates that the majority of the residues are in the most favorable regions, and two residues (Cys184 and Arg254) are in disallowed regions of the Ramachandran plot. The Cys184 and Arg254 residues were examined carefully, and their conformations were consistent with the experimental electron density map. Solvent-accessible surface areas were calculated using the program GETAREA (Fraczkiewicz and Braun 1998).
Acknowledgments
This work was supported by grants from The Robert A. Welch Foundation (H-1345) and the NIH (GM51332) to R.O.F. Diffraction data used in this study were collected on The Gulf Coast Protein Crystallography Consortium (GCPCC) PX1 beamline at The Center for Advanced Microstructures and Devices. This beamline is supported by NSF grant DBI-9871464 with co-funding from The National Institute for General Medical Sciences. This paper is dedicated to the memory of the late Dr. Benjamin Craft, who was instrumental in the development of the PX1 beamline. We thank Dr. Xiuzhen Fan for mass spectrometry, Drs. Edmund W. Czerwinski and John Caradonna for helpful discussions, and Dr. David A. Konkel for critically reading and editing the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03121403.
Data deposition. The coordinates and structure factors have been deposited in the Protein Data Bank (accession code 1ONS).
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumentals, I.I., Robinson, A.S., and Kelly, R.M. 1990. Characterization of sodium dodecyl sulfate-resistant proteolytic activity in the hyperthermophilic archaebacterium Pyrococcus furiosus. Appl. Env. Microbiol. 56 1992–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Christianson, D.W., Mangani, S., Shoham, G., and Lipscomb, W.N. 1989. Binding of D-phenylalanine and D-tyrosine to carboxypeptidase A. J. Biol. Chem. 264 12849–12853. [PubMed] [Google Scholar]
- Du, X., Choi, I.G., Kim, R., Wang, W., Jancarik, J., Yokota, H., and Kim, S.H. 2000. Crystal structure of an intracellular protease from Pyrococcus horikoshii at 2 Å resolution. Proc. Natl. Acad. Sci. 97 14079–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraczkiewicz, R. and Braun, W. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 19 319–333. [Google Scholar]
- Hengge-Aronis, R. 1996. Back to log phase: Sigma S as a global regulator in the osmotic control of gene expression in Escherichia coli. Mol. Microbiol. 21 887–893. [DOI] [PubMed] [Google Scholar]
- Holton, T.R., Ioerger, T.R., Christopher, J.A., and Sacchettini, J.C. 2000. Determining protein structure from electron-density maps using pattern matching. Acta Crystallogr. D 56 722–734. [DOI] [PubMed] [Google Scholar]
- Jeffery, C.J. 1999. Moonlighting proteins. Trends Biochem. Sci. 24 8–11. [DOI] [PubMed] [Google Scholar]
- Kim, K.K., Kim, R., and Kim, S.-H. 1998. Crystal structure of a small heat-shock protein. Nature 394 595–599. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Malki, A., Kern, R., Abdallah, J., and Richarme, G. 2003. Characterization of the Escherichia coli YedU protein as a molecular chaperone. Biochem. Biophys. Res. Commun. 301 430–436. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Que, L. 2000. One motif—Many different reactions. Nat. Struct. Biol. 7 182–184. [DOI] [PubMed] [Google Scholar]
- Quigley, P.M., Korotkov, K., Baneyx, F., and Hol, W.G. 2003. The 1.6 Å crystal structure of the class of chaperones represented by Escherichia coli Hsp31 reveals a putative catalytic triad. Proc. Natl. Acad. Sci. 100 3137–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond, C.S., Glasner, J.D., Mau, R., Jin, H., and Blattner, F.R. 1999. Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res. 27 3821–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry, M.S., Korotkov, K., Brodsky, Y., and Baneyx, F. 2002. Hsp31, the Escherichia coli yedU gene product, is a molecular chaperone whose activity is inhibited by ATP at high temperatures. J. Biol. Chem. 277 46026–46034. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D 56 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayalakshmi, J., Mukhergee, M.K., Graumann, J., Jakob, U., and Saper, M. 2001. The 2.2 Å crystal structure of Hsp33: A heat shock protein with redox-regulated chaperone activity. Structure 9 367–375. [DOI] [PubMed] [Google Scholar]
- van Montfort, R.L.M., Basha, E., Friedrich, K.L., Slingsby, C., and Vierling, E. 2001. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8 1025–1030. [DOI] [PubMed] [Google Scholar]
- Xu, Z., Horwich, A.L., and Sigler, P.B. 1997. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperone complex. Nature 388 741–750. [DOI] [PubMed] [Google Scholar]
- Yoshida, T., Ueguchi, C., Yamada, H., and Mizuno, T. 1993. Function of the Escherichia coli nucleoid protein, H-NS: Molecular analysis of a subset of proteins whose expression is enhanced in an hns deletion mutant. Mol. Gen. Genet. 237 113–122. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Zhao, X., Burkholder, W.F., Gragerov, A., Ogata, C.M., Gottesman, M.E., and Hendrickson, W.A. 1996. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]