

Abstract
Heteronuclear NMR methods have been used to probe the conformation of four complexes of Escherichia coli dihydrofolate reductase (DHFR) in solution. 1HN, 15N, and 13Cα resonance assignments have been made for the ternary complex with folate and oxidized NADP+ cofactor and the ternary complex with folate and a reduced cofactor analog, 5,6-dihydroNADPH. The backbone chemical shifts have been compared with those of the binary complex of DHFR with the substrate analog folate and the binary complex with NADPH (the holoenzyme). Analysis of 1HN and 15N chemical shifts has led to the identification of marker resonances that report on the active site conformation of the enzyme. Other backbone amide resonances report on the presence of ligands in the pterin binding pocket and in the adenosine and nicotinamide–ribose binding sites of the NADPH cofactor. The chemical shift data indicate that the enzyme populates two dominant structural states in solution, with the active site loops in either the closed or occluded conformations defined by X-ray crystallography; there is no evidence that the open conformation observed in some X-ray structures of E. coli DHFR are populated in solution.
Keywords: Dihydrofolate reductase, Met 20 loop, conformational change, enzyme mechanism
Dihydrofolate reductase (5,6,7,8-tetrahydrofolate:NADP+ oxidoreductase, EC 1.5.1.3; DHFR) uses NADPH to reduce 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate and is one of the best-characterized enzymes to date. Because of the central role that DHFR plays in maintenance of the cellular pools of tetrahydrofolate and its derivatives, which are essential for purine and thymidylate biosynthesis, the enzyme is an important target for several anticancer and antibacterial drugs (Blakley 1969; Hitchings Jr. 1989). DHFR from Escherichia coli is well suited for investigating physical and chemical events crucial for catalysis because of the wealth of structural, kinetic, mutagenesis, dynamic, and computational studies that have been performed. Many of these studies point to the importance of surface exposed loops in the catalytic function and mechanism of DHFR (Li et al. 1992; Falzone et al. 1994b; Gekko et al. 1994; Cameron and Benkovic 1997; Sawaya and Kraut 1997; Miller and Benkovic 1998a,b; Radkiewicz and Brooks III 2000; Miller et al. 2001; Osborne et al. 2001). These surface loops exhibit ligand-dependent conformational changes (Sawaya and Kraut 1997), a feature that is common to many enzymes (Gerstein et al. 1994) and is proposed to play a functional role during catalysis.
More than 60 X-ray structures of substrate, cofactor, and inhibitor complexes of E. coli DHFR have been reported and the structural features have been reviewed in detail by Sawaya and Kraut (1997). DHFR retains an essentially rigid scaffold comprised of a central eight-stranded β-sheet and four α-helices (Matthews et al. 1977). However, the Ala 9–Asn 23 loop (designated the Met 20 loop, or loop 1) adopts three distinct conformations in the solid state (Sawaya and Kraut 1997), which are stabilized by hydrogen bonding interactions with two other surface exposed loops, the FG loop (residues 117–131) and GH loop (residues 142–149). Sawaya and Kraut (1997) have argued that only two of these conformations, the closed and occluded configurations (Fig. 1 ▶), prevail during the catalytic cycle and suggest that the third “open” conformation is stabilized by crystal packing contacts involving Met 20 loop residues. Based on a series of isomorphous X-ray crystal structures, they proposed that the Met 20 loop conformational changes and its interactions with the FG and GH loops are critical for modulating ligand specificity and hence DHFR function.
Figure 1.

The Met 20 loop conformations and hydrogen bonding patterns in the closed and occluded structures of E. coli DHFR. The figure shows a superposition of the α-carbon traces, for residues in the central regions of the Met 20, FG, and GH loops, of the E:folate:NADP+ (closed, shown in red) and E:folate (occluded, shown in green) complexes (Protein Data Bank accession numbers 1rx2 and 1rx7, respectively; Sawaya and Kraut 1997). The different hydrogen bonding interactions between the Met 20 loop and the FG and GH loops in the closed and occluded conformations are indicated. The location of bound folate (blue) and NADP+ (yellow) in the E:folate:NADP+ complex are shown.
A number of NMR studies have implicated an important role for Met 20 loop motions in DHFR function. In the apo-enzyme, the Met 20 loop fluctuates between two conformations at a rate comparable with the rate of THF dissociation, which is the rate-limiting step in the catalytic cycle (Falzone et al. 1994b). Detailed 15N relaxation studies have shown that the psec–nsec time scale backbone dynamics in the Met 20 and FG loops differs significantly for the closed and occluded conformations (Osborne et al. 2001). The importance of these loops to DHFR function has been established by site directed mutagenesis. A 500-fold decrease in hydride transfer is observed when the central portion of the Met 20 loop is replaced with a glycine (Li et al. 1992). In addition, substitution or deletion of FG loop residues that exhibit significant changes in dynamics between the closed and occluded configurations dramatically alter the kinetics of DHFR, even though these residues are >19Å away from the active site (Cameron and Benkovic 1997; Miller and Benkovic 1998a). Likewise, mutations that perturb the hydrogen bonding interactions between the Met 20 loop and the FG or GH loops have a significant influence on ligand affinity and enzyme kinetics (Miller and Benkovic 1998b; Miller et al. 2001). Clearly, a large body of data suggests that the interplay of the Met 20 loop with the FG and GH loops, which varies between the closed and occluded configurations, is crucial to the catalytic function of DHFR.
Given the critical role of the Met 20 and neighboring loops in DHFR catalysis, it would be advantageous to have a simple diagnostic method to characterize the loop conformation for wild-type and mutant DHFR complexes in solution. With this goal in mind, we have undertaken complete backbone NMR assignments for a number of wild-type DHFR complexes to identify diagnostic resonances whose chemical shifts report on the conformational state of the enzyme. Importantly, we show that the pattern of chemical shifts can be used to determine the conformation of the Met 20 loop and report on the presence of ligands in the pterin binding pocket and in the adenosine and nicotinamide-ribose binding sites of the NADPH cofactor.
Results and Discussion
In this paper, backbone 1HN, 15N, and 13Cα chemical shifts were compared for four complexes of DHFR: the binary complex with the substrate analog folate (denoted E:folate), the binary complex with NADPH (E:NADPH), the ternary complex with folate and NADP+ (E:folate:NADP+), and the ternary complex with folate and a reduced cofactor analog, 5,6-dihydroNADPH (E:folate:DHNADPH). The assignment of backbone resonances in these complexes is described in the Materials and Methods section.
X-ray crystallographic studies of the E:NADPH and E:folate:NADP+ complexes (Protein Data Bank codes 1rx1 and 1rx2, respectively) show that the Met 20 loop adopts the closed conformation (in space groups where there are no crystal lattice contacts that influence the loop conformation), whereas the X-ray structure of the E:folate complex (1rx7) shows that it adopts an occluded loop conformation (Sawaya and Kraut 1997). No X-ray structure has been reported for the E:folate:DHNADPH complex, but we have shown previously through NMR experiments that it also contains an occluded Met 20 loop conformation (Osborne et al. 2001).
Assignments of the E:folate:DHNADPH and E:folate:NADP+ complexes
Resonance assignments for the E:folate:DHNADPH and E:folate:NADP+ complexes have not been previously reported and were made by triple-resonance methods using uniformly 13C/15N-labeled DHFR (see Materials and Methods section). Representative 1H–15N HSQC spectra of these two complexes are shown in Figure 2 ▶. A high percentage of backbone assignments were obtained for both complexes: All but two (G56, G97) of the expected 148 backbone 1HN/15N resonances were assigned for the E:folate:DHNADPH complex, whereas 139 backbone amide peaks (94%) were assigned in the E:folate:NADP+ complex (resonances of I14, E17, T46, W47, E48, G56, G97, H124, and D132 could not be identified). The 13Cα and 13Cβ resonances were assigned for 157 of the 159 residues (all except M16 and P55) in the E:folate:DHNADPH complex and ~97% of the residues in the E:folate:NADP+ complex (excluding residues 45–47, 55, 123, 132). For the E:folate:DHNADPH complex a high proportion of 13C and 1H resonances of nonaromatic side chains were assigned from analysis of the C(CO)NH–TOCSY experiments (Grzesiek et al. 1993) in combination with HCCH–TOCSY and 15N-edited TOCSY experiments (Marion et al. 1989; Bax et al. 1990). In fact, only seven residues were not assigned past the Cγ (or Hγ) positions for spin systems more complicated than AMX. The limited stability of the E:folate:NADP+ complex did not allow a full series of NMR experiments for extensive side-chain assignments. However, a three-dimensional C(CO)NH–TOCSY spectrum did afford assignments for most of the nonaromatic side chain 13C resonances belonging to residues that were not preceded by a proline. The assignments for E:folate:DHNADPH and E:folate:NADP+ have been deposited in BioMagResBank (accession nos. 5741 and 5740, respectively).
Figure 2.

1H-15N HSQC spectra of (A) E:folate:DHNADPH and (B) E:folate:NADP+ complexes of E. coli DHFR. Resonance assignments reported here using triple-resonance NMR techniques are shown.
Assignments for NADPH and folate binary complexes
Backbone resonance assignments have been reported previously for the E:NADPH binary complex at 9°C and pH 7.6 (Zaborowski et al. 2000). These conditions were required to minimize oxidation of NADPH during acquisition of triple-resonance NMR data. For purposes of comparison with chemical shifts of the E:folate:DHNADPH and E:folate: NADP+ complexes, assignments for the backbone 15N and 1HN resonances were extrapolated to 27°C by temperature titration; the extrapolated shifts were used to calculate the 15N and 1HN chemical shift differences. Published backbone resonance assignments for the E:folate complex (Falzone et al. 1994a) were used after correction for differences in chemical shift referencing.
Amide 15N and 1HN chemical shift changes
Pairwise differences in the chemical shifts of the 15N and 1HN resonances between the E:folate, E:NADPH, E:folate: DHNADPH, and E:folate:NADP+ complexes are plotted in Figures 3 ▶ and 4 ▶ as a function of residue number. Unlike the 13Cα chemical shifts, which show only minor differences between the complexes (see later) primarily because of the minimal differences in the backbone conformation except in the loop regions, the 1H and 15N shifts are highly sensitive to the oxidation state of the bound cofactor, the presence or absence of folate, and the conformation of the active site loop. By careful analysis of the patterns of chemical shift differences in Figures 3 ▶ and 4 ▶, a number of marker resonances can be identified that are diagnostic for binding of cofactor and binding of the folate substrate analog and for determining the conformation of the active site loops.
Figure 3.
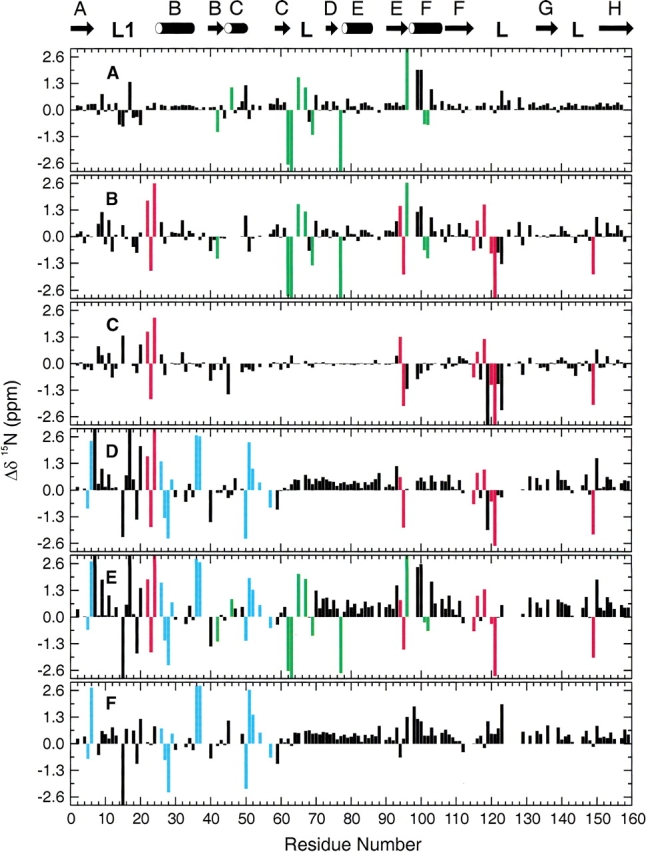
Backbone amide 15N chemical shift differences in DHFR complexes. (A) Chemical shifts in E:folate:DHNADPH (occluded) minus shifts in E:folate (occluded); (B) E:folate:NADP+ (closed) minus E:folate (occluded); (C) E:folate:NADP+ (closed) minus E:folate:DHNADPH (occluded); (D) E:NADPH (closed) minus E:folate:DHNADPH (occluded); (E) E:NADPH minus E:folate (occluded); (F) E:NADPH (closed) minus E:folate:NADP+ (closed). Marker resonances are colored green (cofactor binding), cyan (folate binding), and red (diagnostic of Met 20 loop conformation).
Figure 4.
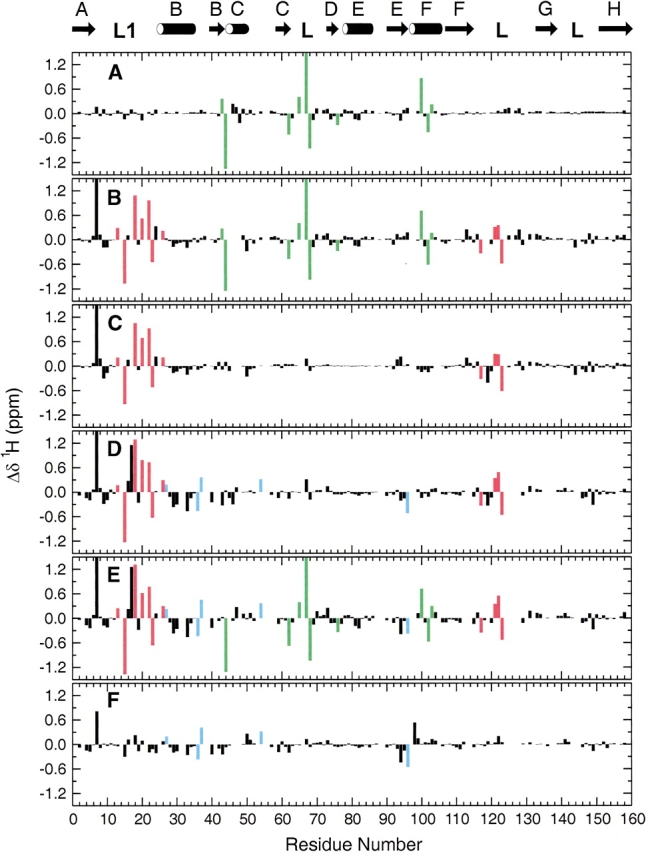
Backbone amide 1HN chemical shift differences in DHFR complexes. (A) Chemical shifts in E:folate:DHNADPH (occluded) minus shifts in E:folate (occluded); (B) E:folate:NADP+ (closed) minus E:folate (occluded); (C) E:folate:NADP+ (closed) minus E:folate:DHNADPH (occluded); (D) E:NADPH (closed) minus E:folate:DHNADPH (occluded); (E) E:NADPH minus E:folate (occluded); (F) E:NADPH (closed) minus E:folate:NADP+ (closed). Marker resonances are colored as in Figure 3 ▶.
Because the E:folate and E:folate:DHNADPH complexes both contain bound folate and are both in the occluded conformation, the chemical shift differences between them (Figs. 3A ▶ and 4A ▶) can be attributed to interactions with co- factor. A number of resonances are highly sensitive to binding of the adenosine moiety of the cofactor. These include the 1HN resonances of residues 43, 44, 62, 65, 67, 68, 76, 100, 102, and 103 and the 15N resonances of residues 42, 46, 62, 63, 65, 67, 69, 77, 96, 101, and 102. Importantly, this set of resonances exhibits shifts of similar magnitude and sign between the E:folate and E:folate:NADP+ complexes (Figs. 3B ▶ and 4B ▶) and between the E:folate and E:NADPH binary complexes (Figs. 3E ▶ and 4E ▶) indicating similar mechanisms affect these residues, namely presence or absence of cofactor. Many additional resonance perturbations are observed in these cases but arise because of differences in the Met 20 loop conformation and the presence of folate in the substrate binding site (see below). Inspection of Figures 3A ▶ and 4A ▶ reveals a few additional resonances that also appear to reflect binding of NADPH; however, the magnitude of the shift changes is not consistent across all complexes and these resonances are therefore not reliable markers of cofactor binding. All of the marker resonances are associated with residues that form the adenosine binding pocket or are on a flexible loop immediately adjacent to it (residues 67, 68, 69; Fig. 5 ▶). The backbone amide protons of Arg 44, His 45, and Thr 46 form hydrogen bonds to phosphate oxygens of the adenosine moiety, as do the backbone amides of Ser 64 and Gly 96 and the side chain of Arg 98, and Gln 102 hydrogen bonds directly to the adenine ring (Bystroff et al. 1990).
Figure 5.

Location of chemical shift marker residues in the DHFR structure. Residues whose 1HN or 15N resonances reflect cofactor binding are shown in green; those that indicate folate binding are blue; and markers of the Met 20 loop conformation are shown in red. The figure was made using MOLMOL (Koradi et al. 1996) from the coordinates of the E:folate:NADP+ complex (1rx2). The NADP+ cofactor is yellow, and the folate is shown in pale blue.
Marker resonances that are diagnostic for folate binding can be identified by comparison of spectra of the E:NADPH binary complex and the E:folate:NADP+ ternary complex, both of which adopt a closed Met 20 loop conformation (Figs. 3F ▶ and 4F ▶). Of course, to be useful as markers these resonances should be insensitive to loop conformation: The selected marker resonances exhibit shifts of similar magnitude and sign between the E:NADPH and E:folate: DHNADPH complexes and between the E:NADPH and E:folate complexes (Figs. 3, D and E ▶ and 4, D and E ▶). The resonances that undergo significant shifts and are reliable markers of folate binding are the 1HN resonances of residues 27, 36, 37, 54, 96 and the 15N resonances of residues 5, 6, 26, 27, 28, 29, 36, 37, 50, 51, 52, 54, 57. All of these residues cluster around the substrate binding site (Fig. 5 ▶).
Resonances that reflect the active site loop conformation can be identified from conserved patterns of chemical shift differences in Figures 3B–E ▶ and 4B–E ▶, all of which summarize shift changes between closed and occluded states (E:folate:NADP+–E:folate; E:folate:NADP+–E:folate:- DHNADPH; E:NADPH–E:folate:DHNADPH; E:NADPH–E:folate). Significant 1HN chemical shift differences are observed for all closed–occluded pairs for resonances of the following residues: 13, 15, 18, 20, 22, 23, 26, 117, 121, 122, 123. For 15N, resonances of residues 22, 23, 24, 94, 95, 115, 116, 118, 120, 121, and 149 are consistently and significantly shifted between the closed and occluded conformations. Most of these residues are located in the Met 20 and FG loops (Fig. 5 ▶), the regions that undergo the largest conformational changes between the closed and occluded forms. Substantial changes in hydrogen bonding interactions between these loops are evident in the crystal structures (Sawaya and Kraut 1997; Fig. 1 ▶) and these undoubtedly contribute to the observed chemical shift changes. Backbone resonances of residues 148 and 149 are also sensitive to the closed–occluded conformational switch, presumably reflecting the formation of new hydrogen bonds between Ser 148 and the backbone amide of Asn 23 and carbonyl oxygen of Leu 24 in the occluded conformer. The 15N resonances of Ile 94 and Gly 95 also appear to report upon the closed–occluded conformational change, although it is not clear from the available structures just what interactions lead to the observed chemical shift changes.
The 15N resonances of Gly 15 and Met 16 exhibit a characteristic pattern of shifts in the E:NADPH complexes (Fig. 3D ▶–F). Inspection of the X-ray structures suggests the probable origin of these shift perturbations. In the NADPH complex (1rx1), a pair of hydrogen bonds is formed between the backbone amide and carbonyl groups of Gly–15 and Thr–123; these hydrogen bonds are not formed in the E:folate binary or E:folate:NADP+ complexes (Sawaya and Kraut 1997).
Finally, the backbone 1HN and 15N resonances of Ala 7 are shifted significantly to low field in the closed complexes. These chemical shift changes can be attributed to formation of a hydrogen bond between the Ala 7 amide and the carboxamide oxygen atom of the nicotinamide moiety of the cofactor, as observed in the X-ray structures of the E:NADPH and E:folate:NADP+ complexes (Bystroff et al. 1990; Sawaya and Kraut 1997). The Ala 7 resonances are therefore exquisitely sensitive to binding of the nicotinamide ring in the active site pocket.
13Cα Chemical shift differences
Deviations of 13Cα chemical shifts from random coil values (Wishart et al. 1995a) for the E:folate:NADP+ and E:folate: DHNADPH complexes are shown as black bars in Figure 6 ▶, a and b, respectively. For comparison, the corresponding 13Cα shift deviations for the E:NADPH and E:folate complexes (calculated from published data of Falzone et al. 1994a and Zaborowski et al. 2000) are also included as red bars in Figure 6 ▶, A and B. The complexes are paired according to their Met 20 loop conformation, with the data for the two closed complexes in Figure 6A ▶ and for the occluded complexes in Figure 6B ▶. The pattern of deviations of the 13Cα shifts from the random coil values is very similar for all complexes, although some resonances do reflect the closed–occluded conformational transition (Fig. 6C ▶). Because 13Cα chemical shifts are highly sensitive to backbone φ,Ψ dihedral angles (Spera and Bax 1991; Wishart et al. 1991), the 13Cα shifts confirm that the secondary structure observed in the crystal structures is conserved in solution. Furthermore, it is clear that the polypeptide backbone adopts a common structure in all of the complexes except for localized ligand-dependent conformational changes in the Met 20 loop and part of the neighboring FG loop. In particular, the 13Cα resonances of residues 17, 19, 21, 22, 119, 120, 122, and 124 are sensitive to the conformational changes that accompany the closed–occluded transition.
Figure 6.
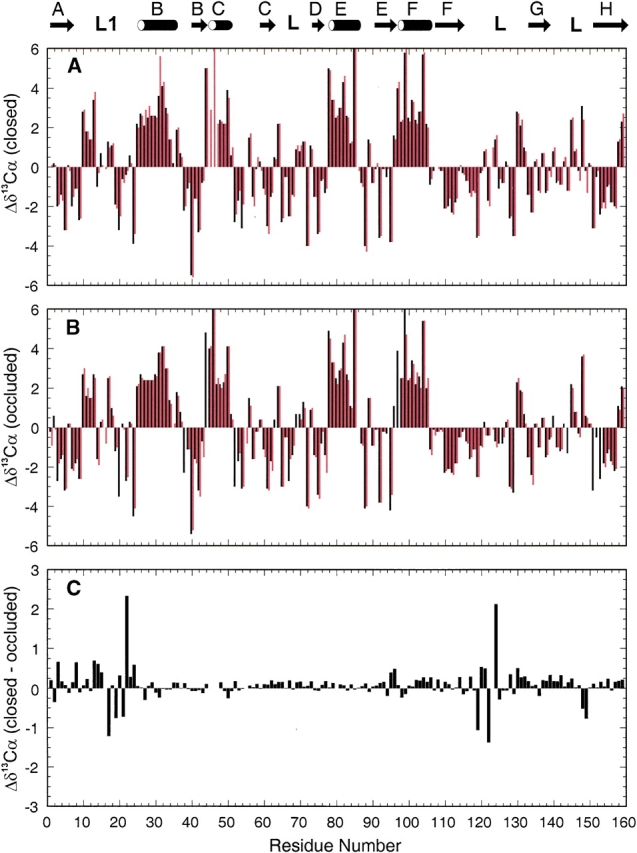
Variations in 13Cαchemical shifts in as a function of residue number in DHFR complexes. The location of the regular secondary structural elements defined from E. coli X-ray structures are denoted by the cartoon at the top of the figure. (A) Deviations from random coil shifts for complexes in which the Met 20 loop adopts the closed conformation. Shift differences for the E:folate: NADP+ complex (this study) and the E:NADPH complex (Zaborowski et al. 2000) are represented by black and red bars, respectively. (B) Deviations from random coil shifts for DHFR complexes in which the Met 20 loop is in the occluded configuration; data for the E:folate:DHNADPH (this study) and E:folate (Falzone et al. 1994a) complexes are plotted with black and red bars, respectively. (C) Changes in 13Cα chemical shift between closed and occluded Met 20 loop conformations. The histogram shows 13Cα chemical shifts of the E:folate:NADP+ complex (closed) minus those of the E:folate:DHNADPH complex (occluded) plotted versus residue number.
Conclusion
By analysis of backbone amide 1HN and 15N chemical shifts for four complexes of E. coli DHFR, namely the E:folate and E:NADPH binary complexes and the E:folate: DHNADPH and E:folate:NADP+ ternary complexes, we have identified a number of marker resonances that are reliable indicators of binding of substrate and cofactor, and of the conformation of the Met 20 and neighboring active site loops. It is notable that the chemical shift data for the various complexes of DHFR studied in the present work indicate that the enzyme populates two dominant structural states in solution, with the active site loops in either the closed or occluded conformations observed crystallographically. There is no evidence that the open conformation observed in the X-ray structures of DHFR complexes in certain space groups is populated to a significant extent in solution and it seems likely that this conformation is stabilized in the crystalline state by crystal contacts involving Met 20 loop residues, as suggested earlier by Sawaya and Kraut (1997). The identification of marker resonances and the demonstration that only two conformational states predominate in solution provide a basis for direct conformational analysis of the various intermediates formed in the DHFR catalytic cycle (Fierke et al. 1987), for structural characterization of kinetically impaired mutants, and for analysis of slow time scale conformational fluctuations observed using NMR relaxation measurements. Such analyses are in progress in our laboratory.
Materials and methods
Sample preparation
The E:folate:DHNADPH and E:folate:NADP+ complexes were prepared as described previously (Osborne and Wright 2001; Osborne et al. 2001). Briefly, uniformly 15N/13C-labeled proteins were produced from cells grown on minimal media in which 15N-ammonium sulfate and 13C-glucose were the sole sources of nitrogen and carbon, respectively. For NMR studies the complexes were exchanged into a buffer previously purged with argon gas comprising of 0.1 M KCl, 1 mM EDTA, 1 mM [2H]DTT, 50 mM potassium phosphate in 7% D2O at pH 6.8. DHFR concentrations ranged from 1 to 1.7 mM. Substrate and cofactor analogs were present in ~12-fold excess.
A sample of E:NADPH was prepared at a DHFR concentration of 2 mM in NMR buffer (70 mM potassium phosphate in 7% D2O, pH 7.6, containing 25 mM KCl and 0.02% sodium azide). NADPH was added in twofold excess and was kept enzymatically reduced by addition of 30 units of glucose-6-phosphate dehydrogenase and 1 mM glucose-6-phosphate to the NMR buffer.
NMR spectroscopy
Resonance assignments for the E:folate:DHNADPH and E:folate: NADP+ complexes were obtained at 306.4 K (calibrated with neat methanol) on Bruker AMX-500, AMX-600, and DRX-600 spectrometers equipped with a triple-resonance probe and triple-axis pulsed-field gradients. Coherence selection was achieved using pulsed-field gradients. Backbone assignments were obtained from three-dimensional HNCA (Grzesiek and Bax 1992b), HNCACB (Wittekind and Mueller 1993), and CBCA(CO)NH (Grzesiek and Bax 1992a) experiments on 13C/15N double-labeled samples. Side-chain 1H and 13C assignments were made for the majority of the resonances of the E:folate:DHNADPH complex using three-dimensional HBHA(CBCACO)NH (Grzesiek and Bax 1993), C(CO)NH–TOCSY (Ikura et al. 1991), and HCCH–TOCSY (Bax et al. 1990) spectra in addition to 15N-edited 3D HSQC–TOCSY and HSQC–NOESY experiments on doubly and singly labeled samples. Side-chain 13C assignments for the E:folate:NADP+ complex were obtained from a three-dimensional C(CO)NH–TOCSY experiment.
15N and 13C chemical shifts were referenced indirectly from the proton frequency for DSS (Wishart et al. 1995b). NMR spectra were processed using FELIX MSI or NMRPipe (Delaglio et al. 1995) and analyzed using FELIX or NMRView (Johnson and Blevins 1994). In general, time domain data in the indirect dimension were zero-filled once and apodized with cosine, cosine-squared, or Lorentzian-to-Gaussian window functions after application of mirror-image linear prediction. Assignment of resonances was achieved using the software packages FELIX95 and NMRView. Backbone assignments were made in a semi-automated fashion using an in-house strip-manipulation tool. Assignments have been deposited in the BioMagResBank.
Assignments for other DHFR complexes used in this analysis
The appearance of the 1H-15N HSQC spectra of DHFR complexes is susceptible to changes in experimental conditions (data not shown). Backbone assignments for two other E. coli DHFR complexes have been reported from our group and are used for comparison. The published assignments for the E:folate complex (Falzone et al. 1994a) were acquired at slightly lower ionic strength and temperatures. Thus, the 1HN and 15N shifts used for comparison in the present work were measured from a 1H-15N HSQC spectrum recorded under identical conditions to those used for the two ternary complexes. Assignments for the E:NADPH complex (Zaborowski et al. 2000) were originally made under substantially different conditions (pH 7.6 and 9°C) to prevent oxidation of NADPH during acquisition of triple-resonance NMR data. Consequently, the assignments were extrapolated to a temperature (27°C) closer to that at which data for the E:folate:DHNADPH and E:folate:NADP+ complexes was recorded. This was accomplished by recording a series of 1H-15N HSQC spectra at temperatures of 9°C, 14°C, 19°C, 24°C, and 27°C at pH 7.6.
Acknowledgments
We thank Dr. John Chung for assistance with NMR experiments, Dr. Eduardo Zaborowski, Dr. David Epstein, and Dr. Stephen Benkovic for valuable discussions, and Linda Tennant for expert technical assistance. This work was supported by grant GM 56879 from the NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
NMR, nuclear magnetic resonance
HSQC, heteronuclear single quantum coherence
DHFR, dihydrofolate reductase
NADP+, nicotinamide adenine dinucleotide phosphate
NADPH, reduced nicotinamide adenine dinucleotide phosphate
DHF, 7,8-dihydrofolate
THF, 5,6,7,8-tetrahydrofolate
DHNADPH, 5,6-dihydro NADPH
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03219603.
References
- Bax, A., Clore, G.M., and Gronenborn, A.M. 1990. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson. 88 425–431. [Google Scholar]
- Blakley, R.L. 1969. The biochemistry of folic acid and related pteridines. Elsevier, Amsterdam.
- Bystroff, C., Oatley, S.J., and Kraut, J. 1990. Crystal structures of Escherichia coli dihydrofolate reductase: The NADP+ holoenzyme and the folate. NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry 29 3263–3277. [DOI] [PubMed] [Google Scholar]
- Cameron, C.E. and Benkovic, S.J. 1997. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry 36 15792–15800. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Falzone, C.J., Cavanagh, J., Cowart, M., Palmer, A.G., Matthews, C.R., Benkovic, S.J., and Wright, P.E. 1994a. 1H,15N and 13C resonance assignments, secondary structure, and the conformation of substrate in the binary folate complex of Escherichia coli dihydrofolate reductase. J. Biomol. NMR 4 349–366. [DOI] [PubMed] [Google Scholar]
- Falzone, C.J., Wright, P.E., and Benkovic, S.J. 1994b. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry 33 439–442. [DOI] [PubMed] [Google Scholar]
- Fierke, C.A., Johnson, K.A., and Benkovic, S.J. 1987. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26 4085–4092. [DOI] [PubMed] [Google Scholar]
- Gekko, K., Kunori, Y., Takeuchi, H., Ichihara, S., and Kodama, M. 1994. Point mutations at glycine-121 of Escherichia coli dihydrofolate reductase: Important roles of a flexible loop in the stability and function. J. Biochem. 116 34–41. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., Lesk, A.M., and Chothia, C. 1994. Structural mechanisms for domain movements in proteins. Biochemistry 33 6739–6749. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992a. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114 6291–6293. [Google Scholar]
- ———. 1992b. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96 432–440. [Google Scholar]
- ———. 1993. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR 3 185–204. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S., Anglister, J., and Bax, A. 1993. Correlation of backbone amide and aliphatic side chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B 101 114–119. [Google Scholar]
- Hitchings Jr., G.H. 1989. Nobel lecture in physiology or medicine—1988. Selective inhibitors of dihydrofolate reductase. In Vitro Cell Dev. Biol. 25 303–310. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., and Bax, A. 1991. Improved three-dimensional 1H-13C-1H correlation spectroscopy of a 13C-labeled protein using constant-time evolution. J. Biomol. NMR 1 299–304. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Li, L., Falzone, C.J., Wright, P.E., and Benkovic, S.J. 1992. Functional role of a mobile loop of Escherichia coli dihydrofolate reductase in transition-state stabilization. Biochemistry 31 7826–7833. [DOI] [PubMed] [Google Scholar]
- Marion, D., Kay, L.E., Sparks, S.W., Torchia, D.A., and Bax, A. 1989. Three-dimensional heteronuclear NMR of 15N-labeled proteins. J. Am. Chem. Soc. 111 1515–1517. [Google Scholar]
- Matthews, D.A., Alden, R.A., Bolin, J.T., Freer, S.T., Hamlin, R., Xuong, N., Kraut, J., Poe, M., Williams, M., and Hoogsteen, K. 1977. Dihydrofolate reductase: X-ray structure of the binary complex with methotrexate. Science 197 452–455. [DOI] [PubMed] [Google Scholar]
- Miller, G.P. and Benkovic, S.J. 1998a. Deletion of a highly motional residue affects formation of the Michaelis complex for Escherichia coli dihydrofolate reductase. Biochemistry 37 6327–6335. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. Strength of an interloop hydrogen bond determines the kinetic pathway in catalysis by Escherichia coli dihydrofolate reductase. Biochemistry 37 6336–6342. [DOI] [PubMed] [Google Scholar]
- Miller, G.P., Wahnon, D.C., and Benkovic, S.J. 2001. Interloop contacts modulate ligand cycling during catalysis by Escherichia coli dihydrofolate reductase. Biochemistry 40 867–875. [DOI] [PubMed] [Google Scholar]
- Osborne, M.J. and Wright, P.E. 2001. Anisotropic rotational diffusion in model-free analysis for a ternary-DHFR complex. J. Biomol. NMR 19 209–230. [DOI] [PubMed] [Google Scholar]
- Osborne, M.J., Schnell, J., Benkovic, S.J., Dyson, H.J., and Wright, P.E. 2001. Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry 40 9846–9859. [DOI] [PubMed] [Google Scholar]
- Radkiewicz, J.L. and Brooks III, C.L. 2000. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J. Am. Chem. Soc. 122 225–231. [Google Scholar]
- Sawaya, M.R. and Kraut, J. 1997. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 36 586–603. [DOI] [PubMed] [Google Scholar]
- Spera, S. and Bax, A. 1991. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 113 5490–5492. [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S., and Sykes, B.D. 1995a. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids: I. Investigations of nearest neighbor effects. J. Biomol. NMR 5 67–81. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995b. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wittekind, M. and Mueller, L. 1993. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α- and β-carbon resonances in proteins. J. Magn. Reson. 101 201–205. [Google Scholar]
- Zaborowski, E., Chung, J., Kroon, G.J.A., Dyson, H.J., and Wright, P.E. 2000. Backbone HN, N, Cα, C′ and Cβ assignments of the 19 kDa DHFR/NADPH complex at 9°C and pH 7.6. J. Biomol. NMR 16 349–350. [DOI] [PubMed] [Google Scholar]