

Abstract
The fungus Candida rugosa produces multiple lipase isoenzymes (CRLs) with distinct differences in substrate specificity, in particular with regard to selectivity toward the fatty acyl chain length. Moreover, isoform CRL3 displays high activity towards cholesterol esters. Lipase isoenzymes share over 80% sequence identity but diverge in the sequence of the lid, a mobile loop that modulates access to the active site. In the active enzyme conformation, the open lid participates in the substrate-binding site and contributes to substrate recognition. To address the role of the lid in CRL activity and specificity, we substituted the lid sequences from isoenzymes CRL3 and CRL4 in recombinant rCRL1, thus obtaining enzymes differing only in this stretch of residues. Swapping the CRL3 lid was sufficient to confer to CRL1 cholesterol esterase activity. On the other hand, a specific shift in the chain-length specificity was not observed. Chimeric proteins displayed different sensitivity to detergents in the reaction medium.
Keywords: Lipase, cholesterol esterase, lid, domain grafting, substrate recognition
Triacylglycerol lipases (EC 3.1.1.3) catalyse both the hydrolysis and the synthesis of ester groups in insoluble substrates. Both protein structure and kinetics of reaction reflect these unusual catalytic properties (for review, see Rubin and Dennis 1997). In aqueous medium, most lipases are activated by the presence of a water/lipid interface, a phenomenon known as interfacial activation, which involves the displacement of a surface structure named the lid. Although neither the presence of a lid structure nor the occurrence of interfacial activation is a strict requirement for an enzyme to be classified in this family (Verger 1997), this architecture and mechanism of activation are common to most lipases described to date. Interface-activated lipases occur in alternative conformational states endowed with different activity: In the closed conformation the lid covers the enzyme active site, making it inaccessible to the substrate molecules, whereas its transition to the open conformation opens the entrance of the catalytic tunnel. Lids are amphipathic structures; in the closed enzyme structure their hydrophilic side faces the solvent, whereas the hydrophobic one is directed towards the protein core. As the enzyme shifts to the open conformation, the hydrophobic face becomes exposed and contributes to the substrate-binding region. Therefore, not only the amphipathic nature of the lid but also its specific amino acid sequence might be of importance for activity and specificity of lipases, as has been demonstrated in both mammalian (Dugi et al. 1992, 1995; Carriére et al. 1998; Yang and Lowe 2000) and fungal lipases (Holmquist et al. 1995a,b; Martinelle et al. 1996).
In this work we sought to determine whether the lid might be responsible for different catalytic properties observed in isoforms of the same enzyme. The target of this study was the family of extracellular lipases produced by the Ascomycete Candida rugosa (CRLs). CRLs are the products of a family of related genes that code for proteins with over 80% identity in their amino acid sequence (Longhi et al. 1992; Lotti et al. 1993). The expression of lipase genes has been shown to be tuned by the conditions of growth and in particular by the carbon source present in the culture medium (Lotti et al. 1998; Lee et al. 1999). Crystallographic structures of CRL1 (Grochulski et al. 1993, 1994) and CRL3 (Ghosh et al. 1995) have been reported. Interestingly, and despite their close sequence relatedness, lipase isoforms purified by chromatography or expressed in heterologous systems have been shown to differ in substrate specificity, in particular with regard to substrate chain length (Rua et al. 1993; Brocca et al. 1998; Lopez et al. 2000; Tang et al. 2001; Lee et al. 2002). CRL3 displays high activity towards cholesterol esters and is therefore also known as cholesterol esterase (Ghosh et al. 1995). Comparison of the amino acid sequences reveals that variability among isoenzymes is clustered at the substrate binding sites and in the lid region, which interacts with the substrate molecules in the enzyme open conformation (Grochulski et al. 1993; Lotti et al. 1994).
In an effort to explore the contribution of the lid to the catalytic properties of lipase1 (CRL1, Brocca et al. 1998), we modified the recombinant enzyme by substituting its lid with the corresponding peptides from isoforms CRL3 and CRL4. Activity of the chimeric proteins was then investigated in the hydrolysis of soluble and insoluble substrates as well as in alcoholysis reactions in organic solvent (Carrea and Riva 2000). Activity towards cholesterol esters was conferred through CRL3 lid swapping. Moreover, chimeras were affected in their sensitivity to the reaction environment, suggesting that the lid may play a role in the dynamics of the transition between the closed and open enzyme conformations.
Results
Construction and expression of chimeric lipases
To evaluate the role of the lipase lid in the modulation of activity and specificity of CRL isoenzymes, we produced chimeras in which the lids of either CRL3 or CRL4 are swapped on the CRL1 scaffold. Significant substitutions differentiate lipase isoenzymes in such variable regions (Fig. 1 ▶). In particular, LID4 is characterized by an enhanced ratio of hydrophobic residues, yet maintains its amphipathic nature. Protein engineering was performed on plasmid pPICslip1 containing a synthetic sequence coding for CRL1 fused to the α-factor leader peptide (Brocca et al. 1998). To ease further manipulations, the coding sequence was transferred in pGEM-7Z because of the larger choice of unique restriction sites. To this purpose, a 1742-bp fragment encompassing the whole lip1 gene was excised by HindIII and BamH1 digestion and cloned in the polylinker sequence of plasmid pGEM7Z, yielding the construct pGEM-slip1. In this latter construct, the lid sequence is enclosed in a 541-bp fragment defined by two unique restriction sites for SmaI, located in the plasmid cloning sites and 42 bp downstream of the sequence of interest, respectively. Two cassettes were synthesized in order to replace the natural lid (LID1) with the corresponding sequences of isoform 3 (LID3) and 4 (LID4), using the experimental strategy described in Figure 2 ▶. The 541-bp sequence was subdivided into two regions: (1) a 5′ half (378 bp) containing a constant sequence which encompasses part of the cloning plasmid and the α-factor leader sequence; as this sequence did not need to be mutagenized, it was simply amplified by PCR, and (2) a 3′ moiety with sequences unique to either LID3 or LID4 (163 bp) was enzymatically synthesized by PCR overlap extension using overlapping oligonucleotides 88–93-bp long. Being the 3′-end of the invariant half complementary to the 5′-half of the lid-containing fragment, a final step of PCR overlap extension allowed the reconstruction of whole cassettes that could be easily inserted in pGEM-slip1 restricted with SmaI. Sequencing confirmed that chimeric lipases had been constructed on the CRL1 scaffold with the lid replaced by the corresponding sequence in CRL3 or CRL4.
Figure 1.

Comparison of sequences encompassing the lid in lipase isoforms CRL1, CRL3, and CRL4. Substitutions with respect to the CRL1 lid are shown in bold.
Figure 2.

Domain swapping strategy for the construction of lipase chimeras. Positions of external oligonucleotides used for amplification and restriction sites used for the cassette cloning are shown. (A) The invariable 378-bp cassette common to both CRL1LID3 and CRL1LID4 chimeras was amplified as described. Gray, sequence of pGEM plasmid; black, α-factor leader peptide; white, 5′-terminus of CRL1 coding sequence; arrows indicate primers position. The 163-bp cassette containing the lid and unique to each gene was synthesized by PCR from overlapping oligonucleotides. (B) Generation by PCR overlap extension of a 541-bp cassette containing the chimeric fragment to be replaced in the wtCRL1 sequence. Pins mark the overlap.
Sequences were replaced in the original pPICZ-slip1. In this expression vector, the lipase-encoding genes are cloned under the control of the methanol-inducible alcohol oxidase (AOX1) promoter and allow regulated expression in Pichia pastoris X33 as described (Brocca et al. 1998). Clones were screened for acquired resistance to zeocin and for the production of lipase, as revealed by the formation of clear halos surrounding colonies growing on tributyrin-containing plates. Colonies containing either CRL1wt or CRL1-LID3 formed comparable halos after a 3-d incubation at 30°C, whereas reduced clarification areas surrounded CRL1-LID4 transformants. A negative control (P. pastoris transformed with the empty expression vector) never formed halos, even after several days of incubation.
Four clones of each transformant type were grown on a small scale (25 mL) in YEPS medium. Cultures were induced with methanol, and the presence of lipase in the culture medium and within the cells was assessed by Western blotting with anti-lipase antibodies (Fig. 3 ▶). Whereas CRL1-LID3 was secreted in amounts comparable to wild-type (wt) CRL1 (0.2 mg/mL culture broth), CRL1-LID4 was hardly detectable in the culture medium (0.1 μg/mL). However, high amounts of CRL1-LID4 were revealed intracellularly (not shown).
Figure 3.

Western blotting analysis of recombinant lipases secreted by P. pastoris (30 μL culture medium). Lane 1, 5 μg commercial lipase; lane 2, CRL1LID4; lane 3, CRL1LID3; lane 4, wt CRL1.
Activity and specificity of lipase chimeras
Enzymes were recovered from the culture supernatants and characterized for activity in the hydrolysis of p-nitrophenylesters, triacylglycerols, and cholesterol esters and in reactions of alcoholysis in organic solvent. Activity of wt CRL1 and CRL1LID3 was investigated in any considered reaction. Despite its poor secretion, it was possible to obtain a small amount of the chimera CRL1-LID4 to use in the hydrolysis of p-nitrophenylesters.
Activity in aqueous medium
Hydrolytic activity on soluble substrates was measured on 1 mM p-nitrophenylesters of different chain length: p-n-phenylbutyrate (C4), p-n-phenylcaproate (C6), p-n-phenylcaprylate (C8), p-n-phenyllaurate (C12), p-n-phenylpalmitate (C16), and p-n-phenylstearate (C18). Reaction mixtures varied in their composition for the presence of different nonionic detergents (Table 1). Mixture 1 contained only the appropriate buffer, whereas mixture 2 was added with arabic gum and Triton X-100 well above its critical micellar concentration (CMC). In this latter case, only C4, C6, and C8 substrates were tested due to the poor solubility of longer-chain compounds in 0.5% Triton. In addition, three different detergents, namely Triton X-100 (3), Tween 20 (4), and Tween 80 (5) were used in concentrations below the respective CMC, as high detergent concentrations are often inhibitory for lipase activity (Helistö and Korpela 1998).
Table 1.
Composition of buffers used in the reactions of hydrolysis of p-nitrophenol esters
| 1 | 100mM Tris-HCl pH 7.5 | – | – |
| 2 | 100mM Tris-HCl pH 7.5 | 0.1% arabic gum | 0.5% (v/v) Triton X-100 |
| 3 | 100 mM Tris-HCl pH 7.5 | – | 0.0075% Triton X-100 |
| 4 | 100mM Tris-HCl pH 7.5 | – | 0.0035% Tween 20 |
| 5 | 100mM Tris-HCl pH 7.5 | – | 0.0005% Tween 80 |
Results obtained with wt CRL1 are quite consistent with data reported in the literature, stating for this enzyme a preference for medium-chain substrates (Brocca et al. 1998). Generally speaking, CRL1LID4 displayed a higher activity in most considered reactions, with the notable exception of long-chain substrates in Tween 80 containing reaction medium (Fig. 4 ▶, arrow). CRL1LID3 and CRL1LID4 both showed an increase in relative activity toward the C4 substrate compared to esters with longer acyl chain. However, no general trend in lid-dependent substrate preference was apparent. All lipases turned out to be extremely sensitive, but with varied responses, to the presence of detergents. Striking evidence of a differential effect of the detergent on enzymes differing in their lids emerged from the comparison of specific activities of mutant lipases under different experimental conditions, as presented in Figure 4 ▶. Due to the complexity of the results, and for the sake of clarity, we will point out only a few significant results. Thus, for example, from the comparison of the activity data obtained with and without detergents, it can be observed that Triton X-100 above its CMC inhibited wt CRL1 but activated the two chimeras on C8 substrates (Fig. 4 ▶, stars). In contrast, the activity of CRL1LID4 on the C18 substrate is drastically decreased by the presence of Tween 80 (Fig. 4 ▶, arrows).
Figure 4.
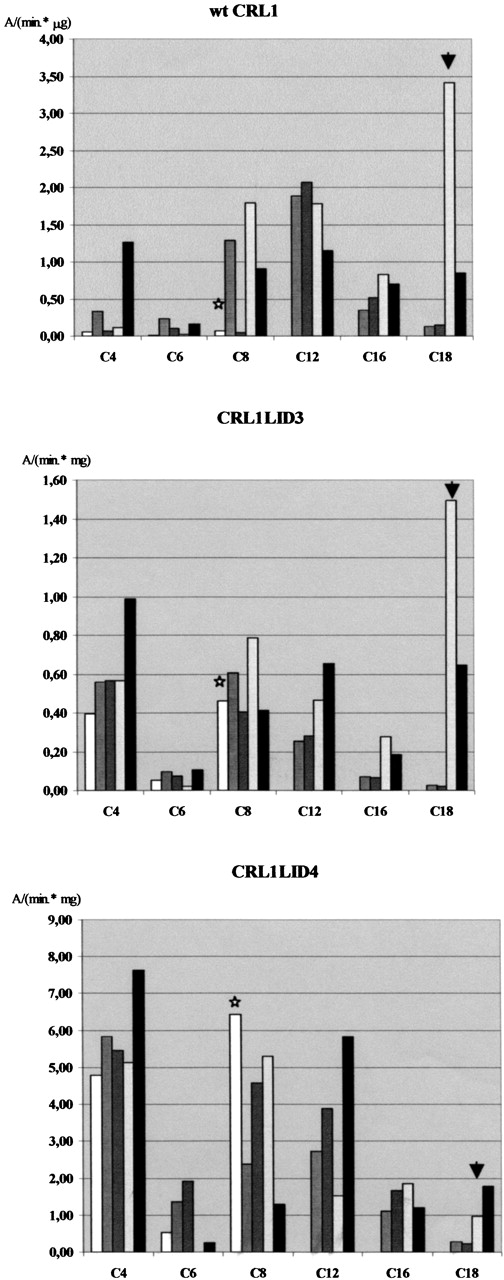
Activity of wt CRL1 and chimera lipases in the hydrolysis of p-nitrophenylesters of different chain length as measured in different reaction conditions. White bars, 0.5% Triton X-100, 0.1% arabic gum; medium gray, 0.0075% Triton X-100; dark gray, 0.0035% Tween 20; light gray, 0.0005% Tween 80; black, buffer only. Note the change of scale. Stars and arrows mark changes in activity discussed in the text.
Activity towards triacylglycerols was tested only for wt CRL1 and CRL1LID3 because of the low secretion of the CRL1LID4 mutant, showing a similar C4:C18 ratio activity for the two enzymes (Table 2).
Table 2.
Hydrolytic activity of CRL1, CRL1LID3, cholesterol esterase on triglyceride substrates, and cholesteryl linoleate
| Tributyrina (μmole/min) | Triolein (65%)a (μmole/min) | Cholesteryl linoleatea (μmole/min) | |
| CRL1 | 2270 | 180 | 0.1 |
| CRL1LID3 | 2900 | 230 | 19 |
| Cholesterol esterase | 120b | n.d. | 35b |
a Initial rate of hydrolysis per mg of lipase protein.
b Initial rate of hydrolysis per mg of total protein.
In sharp contrast, a dramatic effect was observed in the hydrolysis of cholesteryl linoleate. In this reaction, CRL1LID3 specific activity was almost 200-fold higher than that of the wt enzyme (Table 2). This result clearly points to the lid as a major functional determinant of the cholesterol esterase activity reported for CRL3.
Activity in organic solvent
Table 3 reports the initial rates of alcoholysis of wt CRL1 and CRL1LID3 in toluene using n-octanol as the nucleophile and different activated acyl donors. The specific activity of wt CRL1 was higher than that of CRL1LID3 with most tested acyl donors. We observed that the rates of the two enzymes were affected in the same way by the structure and chain length of the acyl donor used. The highest alcoholysis rate was obtained with activated esters carrying a butyryl moiety. Interestingly, activity on TFE octanoate was higher (about five- and threefold, respectively, for wt CRL1 and CRL1LID3) than on TFE hexanoate. This observation is in good agreement with the low activity on C6 substrates obtained in aqueous medium reactions. These results suggest that in organic solvents, replacement of the lid did affect the specific activity of the enzyme but not its specificity towards activated monoesters employed as acyl donors in synthetic reactions.
Table 3.
Initial alcoholysis rate of CRL1 and CRL1LID3 in toluene using n-octanol and different acyl donors
| Acyl donor | Initial rate CRL1 (nmole/min)a | Initial rate CRL1LID3 (nmole/min)a |
| Tributyrin | 100 | 17 |
| Tricaprilin | 27 | 3 |
| Triolein (99%) | No measurable activity | No measurable activity |
| Vinyl acetate | 72 | 48 |
| Vinyl butyrate | 580 | 272 |
| Vinyl laurate | 60 | 15 |
| TFE butyrate | 757 | 165 |
| TFE hexanoate | 11 | 13 |
| TFE octanoate | 54 | 40 |
| TFE dodecanoate | 17 | 7 |
| TFE hexadecanoate | 5 | 3 |
a Activity per mg of lipase
Discussion
A number of studies report the different substrate and reaction specificity of CRL isoenzymes. As CRLs can be hardly separated from each other and characterized as pure proteins, detailed information is available for only some of them. CRL1 overexpressed in P. pastoris was found to exert highest activity on medium-chain substrates (C8–C10) in the hydrolysis of both triglycerides and methyl-esters (Brocca et al. 1998), whereas recombinant CRL4 and CRL2 overexpressed in P. pastoris acted preferentially on long-chain molecules (C16–C18, Tang et al. 2001; Lee et al. 2002). A third isoform, CRL3, is characterized by its significant activity on short-chain soluble substrates (Rua et al. 1993) and by its ability to hydrolyse cholesterol esters of long-chain fatty acids (Ghosh et al. 1995). Cholesterol esterase activity was also demonstrated more recently in CRL2 (Lee et al. 2002).
Here we describe the effect of swapping the lid domains among lipase isoforms. This approach results in the production of chimeric proteins differing in only a few of 534 residues, all of them clustered in the same structural and functional region. However, the lid is the most variable region in sequences which are otherwise very tightly related (Lotti et al. 1994) and is therefore an interesting target for investigating the reported catalytic differences of CRL isoenzymes. Information provided by X-ray analysis supports this choice, indicating that the lid is an important functional structure (Cygler and Schrag 1999). Not only does the lid’s conformation modulate accessibility of the catalytic machinery to substrates, but the lid also contributes to the substrate-binding surface and is likely to be directly involved in substrate recognition. In a number of lipases, but not in CRL, movement of the lid to the open conformation generates the correct positioning of the oxyanion hole, a constellation of amino acids responsible for the stabilization of the reaction tetrahedral intermediate.
Surprisingly, whereas CRL1LID3 was secreted by P. pastoris at levels comparable to those of the wt, the CRL1LID4 mutant was almost fully retained within the cells. This result was unexpected, because this fusion protein was originated by the swapping of a domain from a natural enzyme (CRL4) that can be correctly expressed, folded, and transported by P. pastoris cells (Tang et al. 2001). However, we also observed in other cases that a single mutation in CRL may result in defects of its transport (S. Brocca et al. 2000, unpubl.). A recent series of papers by Sagt and colleagues put emphasis on the sensitivity of yeast expression systems to changes in the sequence of heterologous proteins. Those authors showed that introduction of hydrophobic patches in a cutinase enzyme resulted in retention of the protein in the endoplasmic reticulum (ER), association with chaperones, and impaired secretion (Sagt et al. 1998) and evoked the unfolded protein response (UPR) with consequent proliferation of ER membranes, oxidative stress, and proteasomal degradation of the modified recombinant enzyme (Sagt et al. 2002). It might be hypothesized that the observed block in protein secretion depends on the increased hydrophobicity introduced in the CRL1-based protein by the LID4 peptide. As recombinant CRL4 can be correctly transported (Tang et al. 2001), one should assume that the lid features are counterbalanced by other as yet unknown properties of the wt CRL4 protein, that make it compatible with the P. pastoris secretion machinery.
We did not notice a specific shift in the chain-length specificity as a consequence of the lid swapping. Accordingly, a recent site-directed mutagenesis study unambiguously demonstrated the role of specific residues located in the substrate-binding tunnel of CRL1 (Schmitt et al. 2002). Subtle changes—if any—related to the substrate–lid interaction might be more difficult to assess.
On the other hand, replacement of the loop building the lid with the LID3 peptide was sufficient to confer to CRL1, a protein completely inactive towards cholesterol esters, activity in the hydrolysis of cholesteryl linoleate. This observation is of interest, as it confronts the issue of how CRL1 and CRL3, which share 89% sequence identity and are very similar in their structure, can display completely different behaviors in the hydrolysis of cholesteryl esters. Complexes of CRL1 with substrate-like inhibitors and of linoleate-bound CRL3 disclosed molecular details of substrate–enzyme interactions (Grochulski et al. 1994; Ghosh et al. 1995). CRL1 and CRL3 differ in 55 amino acids, 23 of them located near the active site and at the active-site gorge region, to which the lid contributes. The structural analysis reported by Ghosh and colleagues (1995), besides focussing on substitutions within the substrate-binding pocket, suggested a specific role of the lid residues Phe69, Gly74, Thr76, and Gln88 (Tyr69, Pro74, Ala76, and Glu88 in CRL1) in enhancing hydrophobicity at the entrance of the substrate-binding pocket. This effect should favor binding of very hydrophobic substrates as cholesteryl esters. We have shown that the LID3 sequence alone was sufficient to obtain activity toward cholesteryl linoleate. This result therefore identifies the lid as one of the major structural and functional determinants of the cholesterol esterase activity typical of CRL3. However, the information obtained does not allow us to unambiguously conclude whether specificity is conferred by specific amino-acid side chains, as suggested by Ghosh et al. (1995). Lid-dependent conformational effects as well as the contribution of other protein regions cannot be excluded. We addressed, by site-directed mutagenesis, position 344 within the substrate-binding pocket (Phe in CRL1), because it was suggested as a possible critical residue (Ghosh et al. 1995). However, the mutant did not display any activity on cholesteryl linoleate (data not shown).
Lid replacement was found to produce a striking and substrate-independent change in the enzyme response to detergents added to the reaction mixture in reactions of hydrolysis of p-nitrophenolesters carried out under different experimental conditions. Because three nonionic detergents were used, a differential action on proteins differing from each other in only a 26-residue stretch was hardly to be expected. A clue to this observation is provided by a recent study by Gonzalez-Navarro and colleagues (2001), who addressed the issue of how the conformational state of CRL contributes to substrate preference. In that work, different enzyme conformations present in aqueous solution in the presence of detergents/interfaces were trapped by freeze-drying and used for reactions in a water-restricted environment. It was shown that different conformational states are endowed with different activity on substrates of growing chain length. These results were interpreted as the lipase existing in solution as a continuum of conformations concerning the position of the lid, whose equilibrium can be shifted by the environment (i.e., the detergent concentration). Accordingly, different opening of the lid would make the enzyme accessible to substrates of different sizes. In this conceptual frame, the effects of detergents on enzymes that differ only in their lid structures might be interpreted in terms of specific lid/detergent interactions as well as of a modulation of the conformational flexibility introduced by lid swapping. LID3 differs from the CRL1 sequence in six residues, of which tyrosine 69 forms a hydrogen bond with the sugar linked to Asn 351, which has been shown to stabilize the lid in its open conformation (Grochulski et al. 1993; Brocca et al. 2000). The substitution of Tyr with Phe removes the hydroxyl function and exposes a hydrophobic side chain to the solvent. Moreover, replacement of Pro 74 by Gly might enhance the lid conformational flexibility. The LID4 sequence differs markedly from the wt, with 16 residues substituted. Besides the significant increase in the hydrophobicity of this sequence, replacement of a few residues appears worthy of discussion. In LID4, Glu66, which was recognized by Grochulski et al. (1994) as one of the hinge points for the swinging of the lid, is replaced by leucine. Trp and Asp also replace, respectively, Tyr 69 and Glu70, which have been shown to stabilize the open lid by interactions with sugars. In particular, the lack of the Tyr hydroxyl function and the presence of a bigger side chain might be of some hindrance for the closing of the lid. This might contribute to making the open conformation more populated. In both mutants, the two substitutions Ser91Leu and Ser93Gln/Asn locate larger side chains at the two sides of Pro92, whose cis-trans isomerization determines the opening of the lid (Grochulski et al. 1993).
In conclusion, the role of the lid in affecting CRL activity appears to be very complex and might involve both specific interactions with the substrate molecules and the equilibrium between the active and inactive enzyme conformations.
Materials and methods
Enzymes and chemicals
Restriction enzymes used in this study were obtained from New England Biolabs. p-nitrophenyl esters, triglycerides, arabic gum, and glycerol (99%) were obtained from Sigma.
Strains, plasmids, and growth conditions
Escherichia coli strain DH5α (Promega) was used as the host for plasmid amplification, and Pichia pastoris X-33 (Cregg et al. 1993; Invitrogen) was used for the expression of recombinant lipases. The plasmids used were pPICαB (Invitrogen) for expression in P. pastoris and pGEM-7Zf(+) for intermediate cloning steps (Promega). E. coli was grown at 37°C in low-salt Luria-Bertani (LB) medium (DIFCO) containing either 50 μg/mL ampicillin for selection of clones containing pGEM-7Z or 25 μg/mL zeocin for selection of clones carrying pPICZαB. P. pastoris was grown in shaking flasks at 30°C in buffered YEPS medium containing 1% LB yeast extract, 2% peptone, and 1% sorbitol. Selection of transformants was on the appropriate medium containing 25 μg/mL zeocin.
DNA synthesis and generation of chimeric lipases
Plasmid DNA was purified using either the alkaline lysis procedure for mini-preparations (Birnboim and Doly 1979) or kits for mini- or maxi-preparations (QIAGEN). Other standard DNA manipulations were carried out according to Sambrook et al. (1989).
The parent plasmid used for the generation of chimeric lipases was pPICslip1 (Brocca et al. 1998). A 541-bp sequence containing the lid region and delimited by two SmaI restriction sites was replaced with the corresponding sequence of the CRL3 or CRL4 isoenzymes obtained as described in Results. PCR was performed in an automated DNA Thermal Cycler (Perkin Elmer) using 50 pmoles of each primer and 2.5 Units of Pfu Turbo™ Polymerase (Stratagene) in a total volume of 100 μL. Samples were subjected to 25 cycles of 1-min denaturation at 94°C, 1-min annealing at 77°C, and 1-min extension at 72°C, followed by a final step of extension of 10 min at 72°C. After validation by sequencing (M-Medical), synthetic sequences were used to replace the corresponding SmaI-SmaI fragment in pPICslip1.
Expression of recombinant lipases
Pichia pastoris X-33 cells were transformed by the Bicine method (Klebe et al. 1983) with 10 μg of SacI-linearized plasmid DNA carrying wt or mutated lipase-encoding genes. Freshly transformed cells were plated onto solid YEPS medium containing 25 μg/mL zeocin. Positive transformants were checked for lipase activity by transferring colonies onto YEPS plates containing 1% tributyrin. After overnight incubation at 30°C, lipase activity was detected by the formation of a clear halo on the opaque tributyrin emulsion. Recombinant cultures were grown in flasks (shaking rate 150 rpm) at 30°C in buffered YEPS medium at pH 6.0 for 5 d, reaching an OD600 of 60–80. The cultures were maintained at constant pH by adding 1 M phosphate buffer pH 6.0 and fed by the addition of 1% v/v methanol twice a day. Wt and chimeric lipases were recovered from the culture supernatants by centrifugation at 4000 rpm. Concentration was carried out by tangential flow filtration against 4–5 vol of 5 mM Tris-HCl pH 7.5 with a Minitan system (Millipore) using filter plates at 60 kD exclusion.
The wild-type and mutant enzymes were identified by electrophoresis on 10% SDS-polyacrylamide gel (Laemmli 1970), with the wild-type LIP1 as a molecular mass reference. The purity degree of the different enzymes was assessed by Coomassie Blue staining of SDS-polyacrylamide gels after electrophoresis. Culture supernatant samples and rough cellular extracts were subjected to Western blotting analysis with a polyclonal antibody raised against G. candidum lipase (GCL). Lipase was quantified by densitometry analysis.
Activity assays
Activity on p-nitrophenyl-esters was determined by following the increase in absorbance at 410 nm due to the release of p-nitrophenol, with a Jasco V-530 UV/Vis spectrophotometer. In 1 mL final volume, 100 μL of 10 mM substrates dissolved in isopropanol were mixed with 100 mM Tris-HCl pH 7.5 and detergents as specified. Nonionic detergents used were Triton X-100 (polyoxyethylene mono p-tert-octylphenylether, CMC 0.015%), Tween 20 (polyoxyethylene sorbitan monolaurate, CMC 0.007%), and Tween 80 (polyoxyethylene sorbitan monooleate, CMC 0.001%).
Activity on triglycerides was determined by titrating released fatty acids with 0.01 N sodium hydroxide using a STAT TITRINO (Metrohm). Emulsions of 20 mM triacylglycerols with 2% arabic gum were used as a substrate in 20 mM potassium phosphate buffer, pH 7.5, in a volume of 20 mL.
Cholesterol esterase activity was determined as follows: 5.3 mg of solid cholesteryl linoleate was dissolved in 0.2 mL polyoxyethylene 9 lauryl ether (Sigma) and mixed under heating with 0.8 mL 0.9 % (w/w) sodium chloride in 50 mM potassium phosphate buffer, pH 7.2 (substrate concentration 8.2 mM). Hydrolysis of cholesteryl linoleate was carried out at 25°C, adding a proper amount of lipase to 1 mL of substrate solution, and the reaction progress was monitored by measuring the release of cholesterol by GLC (HP-1 Crosslinked Methyl Silicone Gum, 25 m, 0.32 mm ID, Hewlett-Packard) under the following operating conditions: oven temperature from 270°C (initial time 5 min) to final temperature 278°C with a heating rate of 0.5°C/min, H2 as the carrier gas. Control CRL3 was the commercial cholesterol esterase, from Roche (n.393916).
Activity in toluene was determined by measuring the initial rate of alcoholysis by n-octanol of vinyl esters or trifluorethylesters or triglycerides. The lipase samples were prepared by co-lyophilizing 0.03 mg of lipase protein with 5 mg PEG (Secundo et al. 1999). Before reaction, solvent, reagents, and enzyme samples were brought separately to the water activity value of 0.11 by equilibration (for at least 48 h, at 25°C) in sealed vessels, with the vapor phase of a saturated LiCl solution. In a typical experiment, a sample of lipase was added to 1 mL toluene containing 19 mM n-octanol and 39 mM activated ester or 20 mM triglycerides, and the reaction mixture was shaken at 150 rpm, at 25°C. The reaction progress was monitored by GLC as described above, but with an oven temperature from 35°C (initial time 10 min) to final temperature 300°C (depending on the chain length of the obtained ester) with a heating rate of 15°C/min.
Acknowledgments
This work was supported by grants from the Progetto Finalizzato Biotecnologie of the Italian National Research Council and the Project COFIN 2002 to L.A. and by a grant from F.A.R. to M.L. We thank Luca DeGioia for assistance in designing chimeric proteins and Chiara Tarabiono for skillful technical assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CMC, critical micellar concentration
CRLs, Candida rugosa lipases
TFE, 2,2,2-tifluoroethyl
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0304003.
References
- Birnboim, H.C. and Doly, J. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca, S., Schmidt-Dannert, C., Lotti, M., Alberghina, L., and Schmid, R.D. 1998. Design, total synthesis, and functional overexpression of the Candida rugosa lip1 gene coding for a major industrial lipase. Protein Sci. 7 1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca, S., Persson, M., Wehtje, E., Adlercreutz, P., Alberghina, L., and Lotti, M. 2000. Mutants provide evidence of the importance of glycosydic chains in the activation of lipase 1 from Candida rugosa. Protein Sci. 9 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrea, G. and Riva, S. 2000. Properties and synthetic applications of enzymes in organic solvents. Angew. Chem. Int. Ed. Engl. 39 2226–2254. [PubMed] [Google Scholar]
- Carriére, F., Withers-Martinez, C., van Tilbeurgh, H., Roussel, A., Cambillau, C., and Verger, R. 1998. Structural basis for the substrate selectivity of pancreatic lipases and some related proteins. Biochim. Biophys. Acta 1376 417–432. [DOI] [PubMed] [Google Scholar]
- Cregg, J.M., Vedvick, T.S., and Raschke, W.C. 1993. Recent advances in the expression of foreign genes in Pichia pastoris. Biotechnology 11 905–910. [DOI] [PubMed] [Google Scholar]
- Cygler, M. and Schrag, J.D. 1999. Structure and conformational flexibility of Candida rugosa lipase. Biochim. Biophys. Acta 1441 205–214. [DOI] [PubMed] [Google Scholar]
- Dugi, K.A., Dichek, H.L., Talley, G.D., Brewer Jr., H.B., and Santamarina-Fojo, S. 1992. Human lipoprotein lipase: The loop covering the catalytic site is essential for interaction with lipid substrates. J. Biol. Chem. 267 25086–25091. [PubMed] [Google Scholar]
- Dugi, K.A., Dichek, H.L., and Santamarina-Fojo, S. 1995. Human hepatic and lipoprotein lipase: The loop covering the catalytic site mediates substrate specificity. J. Biol. Chem. 270 25396–25401. [DOI] [PubMed] [Google Scholar]
- Ghosh, D., Wawrzak, Z., Pletnev, V.Z., Li, N., Kaiser, R., Pangborn, W., Jörnvall, H., Erman, M., and Duax, W.L. 1995. Structure of uncomplexed and linoleate-bound Candida cylindracea cholesteol esterase. Structure 3 279–288. [DOI] [PubMed] [Google Scholar]
- Grochulski, P., Li, Y., Schrag, J.D., Douthillier, F., Smith, P., Harrison, D., Rubin, B., and Cygler, M. 1993. Insights into interfacial activation form an open structure of Candida rugosa lipase. J. Biol. Chem. 268 12843–12847. [PubMed] [Google Scholar]
- Grochulski, P., Li, Y., Schrag, J.D., and Cygler, M. 1994. Two conformational states of Candida rugosa lipase. Protein Sci. 3 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helistö, P. and Korpela, T. 1998. Effects of detergents on activity of microbial lipases as measured by the nitrophenyl alkanoate esters method. Enzyme Microb. Technol. 22 113–117. [Google Scholar]
- Holmquist, M., Clausen, I.G., Patkar, S., Svendsen, A., and Hult, K. 1995a. Probing a functional role of Glu87 and Trp89 in the lid of Humicola lanuginosa lipase through transesterification reactions in organic solvent. J. Protein Chem. 14 217–224. [DOI] [PubMed] [Google Scholar]
- Holmquist, M., Martinelle, M., Berglund, P., Clausen, I.G., Patkar, S., Svendsen, A., and Hult, K. 1995b. Lipases from Rhizomucor miehei and Humicola lanuginosa: modification of the lid covering the active site alters enantioselectivity. J. Protein Chem. 12 749–757. [DOI] [PubMed] [Google Scholar]
- Klebe, R.J., Harriss, J.V., Sharp, Z.D., and Douglas, M.G. 1983. A general method for polyethylene-glycol induced genetic transformation of bacteria and yeast. Gene 25 333–341. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lee, G.C., Tang, S.J., Sun, K.H., and Shaw, J.F. 1999. Analysis of the gene family encoding lipases in Candida rugosa by competitive reverse transcription-PCR. Appl. Environ. Microbiol. 65 3888–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G.C., Lee, L.C., Sava, V., and Shaw, J.F. 2002. Multiple mutagenesis of non-universal serine codons of the Candida rugosa LIP2 gene and biochemical characterization of purified recombinant LIP2 lipase overexpressed in Pichia pastoris. Biochem. J. 366 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhi, S., Lotti, M., Fusetti, F., Pizzi, E., Tramontano, A. and Alberghina, L. 1992. Homology-derived three-dimensional structure prediction of Candida cylindracea lipase. Biochim. Biophys. Acta 1165 129–133. [DOI] [PubMed] [Google Scholar]
- Lopez, C., Guerra, N.P., and Rua, M.L. 2000. Purification and characterization of two isoforms form Candida rugosa lipase B. Biotech. Lett. 22 1291–1294. [Google Scholar]
- Lotti, M., Grandori, R., Fusetti, F., Longhi, S., Brocca, S., Tramontano, A., and Alberghina, L. 1993. Cloning and analysis of Candida cylindracea lipase sequences. Gene 124 45–55. [DOI] [PubMed] [Google Scholar]
- Lotti, M., Tramontano, A., Longhi, S., Fusetti, F., Brocca, S., Pizzi, E., and Alberghina, L. 1994. Variabilità within the Candida rugosa lipases family. Protein Eng. 7 531–534. [DOI] [PubMed] [Google Scholar]
- Lotti, M., Monticelli, S., Montesinos, J.L., Brocca, S., Valero, F., and Lafuente, J. 1998. Physiological control on the expression and secretion of Candida rugosa lipase. Chem. Phys. Lipids 93 143–148. [DOI] [PubMed] [Google Scholar]
- Martinelle, M., Holmquist, M., Clausen, I.G., Patkar, S., Svendsen, A., and Hult, K. 1996. The role of Glu87 and Trp89 in the lid of Humicola lanuginosa lipase. Protein Eng. 9 519–524. [DOI] [PubMed] [Google Scholar]
- Rua, M.L., Diaz-Maurino, T., Fernandez, V.M., Otero, C., and Ballesteros, A. 1993. Purification and characterization of two distinct lipases from Candida cylindracea. Biochim. Biophys. Acta 1156 181–189. [DOI] [PubMed] [Google Scholar]
- Rubin, B. and Dennis, E.A., eds. 1997. Lipases. Methods in enzymology, Vol. 284. Academic Press, New York.
- Sagt, C.M.J., Mueller, W.H., Boonstra, J., Verkleij, A.J., and Verrips, C.T. 1998. Impaired secretion of a hydrophobic cutinase by Saccharomyces cerevisiae cells correlates with an increased association with immunoglobulin heavy-chain binding protein (BiP). Appl. Environ. Microbiol. 64 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagt, C.M.J., Mueller, W.H., van der Heide, L., Boonstra, J., Verkleij, A.J., and Verrips, C.T. 2002. Impaired cutinase secretion in Saccharomyces cerevisiae induces irregular endoplasmic reticulum (ER) membrane proliferation, oxidative stress, and ER-associated degradation. Appl. Environ. Microbiol. 68 2155–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, K.J., Fritsch, E.F., and Maniatis T. 1989. Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schmitt, J., Brocca, S., Schmid, R.D., and Pleiss, J. 2002. Blocking the tunnel: Engineering of Candida rugosa lipase mutants with short chain length specificity. Protein Eng. 15 595–601. [DOI] [PubMed] [Google Scholar]
- Secundo, F., Spadaro, S., Carrea, G., and Overbeeke, P.L.A. 1999. Optimization of Pseudomonas cepacia lipase preparations for catalysis in organic solvents. Biotechnol. Bioeng. 62 554–561. [DOI] [PubMed] [Google Scholar]
- Tang, S.J., Shaw, J.F., Sun, K.H., Sung, G.H., Chang, T.Y., Lin, C.K., Lo, Y.C., and Lee, G.C. 2001. Recombinant expression and characterization of the Candida rugosa lip4 lipase in Pichia pastoris: Comparison of glycosylation, activity and stability. Arch. Biochem. Biophys. 387 93–98. [DOI] [PubMed] [Google Scholar]
- Verger, R. 1997. “Interfacial activation” of lipases: Facts and artifacts. Trends Biotechnol. 15 32–38. [Google Scholar]
- Yang, Y. and Lowe, M.E. 2000. The open lid mediates pancreatic lipase function. J. Lipid Res. 41 48–57. [PubMed] [Google Scholar]