

Abstract
The Notch receptor contains a conserved ankyrin repeat domain that is required for Notch-mediated signal transduction. The ankyrin domain of Drosophila Notch contains six ankyrin sequence repeats previously identified as closely matching the ankyrin repeat consensus sequence, and a putative seventh C-terminal sequence repeat that exhibits lower similarity to the consensus sequence. To better understand the role of the Notch ankyrin domain in Notch-mediated signaling and to examine how structure is distributed among the seven ankyrin sequence repeats, we have determined the crystal structure of this domain to 2.0 Å resolution. The seventh, C-terminal, ankyrin sequence repeat adopts a regular ankyrin fold, but the first, N-terminal ankyrin repeat, which contains a 15-residue insertion, appears to be largely disordered. The structure reveals a substantial interface between ankyrin polypeptides, showing a high degree of shape and charge complementarity, which may be related to homotypic interactions suggested from indirect studies. However, the Notch ankyrin domain remains largely monomeric in solution, demonstrating that this interface alone is not sufficient to promote tight association. Using the structure, we have classified reported mutations within the Notch ankyrin domain that are known to disrupt signaling into those that affect buried residues and those restricted to surface residues. We show that the buried substitutions greatly decrease protein stability, whereas the surface substitutions have only a marginal affect on stability. The surface substitutions are thus likely to interfere with Notch signaling by disrupting specific Notch-effector interactions and map the sites of these interactions.
Keywords: Notch signal transduction, ankyrin repeats, signaling mutants, X-ray crystallography, protein stability
The Notch signaling pathway mediates cell–cell signaling in metazoan development (Artavanis-Tsakonas et al. 1999). Notch signaling is mediated by a single-pass transmembrane receptor encoded by the Notch gene (Wharton et al. 1985; Kidd et al. 1986). Following receptor activation by extracellular ligands, the cytosolic portion of the Notch receptor (∼1000 residues in Drosophila melanogaster) is cleaved from the membrane and enters the nucleus, where it can participate in transcriptional activation of downstream targets (Jarriault et al. 1995; Schroeter et al. 1998; Struhl and Greenwald 1999). Notch signaling activity is modulated by interaction between the intracellular portion of the Notch receptor and a number of cytosolic and nuclear effector proteins, such as Suppressor of Hairless (Matsuno et al. 1997), Deltex (Diederich et al. 1994; Matsuno et al. 1995), EMB-5 (Hubbard et al. 1996), and Skip (Zhou et al. 2000). Many of these interactions appear to involve direct binding to a set of ankyrin repeats in the cytosolic portion of the Notch receptor. As would be expected of a region that facilitates multiple binding reactions, the Notch ankyrin repeats have been shown to be critical for Notch signaling (Rebay et al. 1993; Roehl and Kimble 1993), and a number of mutations that introduce residue substitutions within the Notch ankyrin domain disrupt Notch signaling (Kodoyianni et al. 1992; Diederich et al. 1994; Kopan et al. 1994; Joutel et al. 1996; Kurooka et al. 1998).
Ankyrin repeats are 33 residues in length and consist of two α-helices connected by a short loop. Adjacent ankyrin repeats are linked together via a long loop that terminates with a tight β-turn (Gorina and Pavletich 1996; Luh et al. 1997; Batchelor et al. 1998; Huxford et al. 1998; Jacobs and Harrison 1998; Russo et al. 1998; Venkataramani et al. 1998; Foord et al. 1999; Mandiyan et al. 1999; Sedgwick and Smerdon 1999). The sequence conservation among the seven ankyrin repeat sequences of the Notch ankyrin domain is modest (17% average pairwise identity; Zweifel and Barrick 2001a); however the sequence conservation of analogous repeats from different taxa is quite high (approximately 70% identity, see Fig. 1 ▶ and Stifani et al. 1992). The Drosophila Notch receptor contains six tandem ankyrin sequence repeats previously identified as closely matching the ankyrin consensus sequence (Bork 1993) and a putative seventh (C-terminal) repeat that exhibits lower similarity to the consensus (Zweifel and Barrick 2001a). Modeling the seventh repeat sequence as a C-terminal ankyrin repeat suggests that residues deviating from the consensus sequence would be exposed to solvent, providing a possible explanation for the lack of conservation of these residues (Zweifel and Barrick 2001a). A polypeptide containing all seven repeats was found to have a free energy of unfolding nearly twice that of a polypeptide lacking the C-terminal repeat (Zweifel and Barrick 2001b), suggesting that this putative seventh ankyrin repeat is an integral part of the Notch ankyrin domain.
Figure 1.

Sequence alignment of Notch ankyrin domains from different taxa. The bar above the alignment indicates the boundaries of the seven ankyrin sequence repeats. Insertions in the first repeat are indicated by plus symbols in the bar above the alignment. α-Helices are shown below the sequences as determined using DSSP (Kabsch and Sander 1983). Absolutely conserved residues are colored red, positions with greater than 50% conservation are colored green, and positions that contain conservative amino acid substitutions are colored yellow. The numbering refers to the Drosophila ankyrin construct studied here; residue 1 corresponds to residue 1902 of the full length Notch receptor (Wharton et al. 1985). This figure was made using the program ALSCRIPT (Barton 1993).
Despite its importance in metazoan development, there is little high-resolution structural information available for the Notch receptor. Although high-resolution structural information has been obtained for one important extracellular domain (Vardar et al. 2003), no high-resolution structural information has been obtained for the cytosolic portion of the Notch receptor. Here we report the 2.0 Å crystal structure of the Notch ankyrin domain. The structure provides insight into the means by which the seventh repeat contributes to the stability of the Notch ankyrin domain, helps define the terminal boundaries of the domain, and provides a model for potential homotypic interaction. The structure also provides a framework for interpreting the effects of mutations in the Notch ankyrin domain that disrupt signaling, and helps define the degree to which tertiary structural details are determined by primary sequence in modular ankyrin domains.
Results
Notch ankyrin domain structure
We obtained crystals of a polypeptide that contains all seven ankyrin sequence repeats. These seven repeats include the six N-terminal sequence repeats identified through sequence analysis (residues 1902–2115 of the Notch receptor) and the putative seventh repeat (residues 2116–2139). As there are three polypeptide chains in the asymmetric unit, we are able to examine the degree to which the structure of this modular domain is influenced by environmental differences in the crystal lattice. From these crystals, we determined the three-dimensional structure of the Notch ankyrin domain to 2.0 Å resolution. The resulting structures have good geometry and statistics (Table 1). The overall architecture of the domain consists of an elongated array of antiparallel α-helices, consistent with an ankyrin repeat structure. Moreover, the seventh C-terminal ankyrin repeat of all three chains clearly adopts an ankyrin fold (repeat seven, violet; Fig. 2A ▶), consistent with previous thermodynamic and solution structural studies (Zweifel and Barrick 2001a,b). The charged and polar residues in repeat seven that deviate from the ankyrin consensus are solvent exposed (Fig. 2B ▶; compare Arg 221, Asp 222, and Ser 225 of repeat seven with the analogous residues Leu 188, Phe 189, and Ala 192 of repeat six). In contrast, the N-terminal sequence repeat (repeat one, red; Fig. 2A ▶) does not adopt a regular ankyrin fold. This repeat is variably disordered among the three chains in the asymmetric unit, and shows significant conformational heterogeneity when the three chains are superposed over repeats two through seven (Fig. 3A ▶). Only the C-terminal portion of the first repeat of each chain is visible in the electron density (from residues 29, 36, and 47 in chains A, B, and C, respectively; see Fig. 1 ▶ for numbering), and it adopts a different conformation in each of the three chains.
Table 1.
Summary of data collection, phasing, and refinement statistics
| Data set | Native 1 | Native 2 | SeMet (λ1) | SeMet (λ2) | SeMet (λ3) |
| X-ray source | Rigaku CuKα | NSLS X25 | NSLS X4-A | NSLS X4-A | NSLS X4-A |
| Wavelength (Å) | 1.5418 | 1.1000 | 0.9791 | 0.9786 | 0.9686 |
| Resolution (Å) | 30–2.8 | 60–2.0 | 30–2.8 | 30–2.8 | 30–2.8 |
| Completenessa (%) | 87.2 (82.9) | 96.9 (93.2) | 92.1 (96.1) | 92.4 (96.3) | 91.0 (95.0) |
| Rmergeb | 0.034 (0.062) | 0.048 (0.138) | 0.090 (0.235) | 0.097 (0.259) | 0.103 (0.266) |
| Mean FOM (30–3.8 Å) | 0.435 | ||||
| Refinement against Native 2 | |||||
| Rcryst/Rfreec | 0.180/0.201 | ||||
| Resolution range (Å) | 20–2.0 | ||||
| RMS deviations | |||||
| Bonds (Å) | 0.005 | ||||
| Angles (°) | 1.43 | ||||
| B-factors (Å2) | 1.11/3.66d | ||||
| Ramachandran plot | |||||
| Most favored regions | 90.2% | ||||
| Additionally allowed regions | 9.8% | ||||
| Generously allowed/disallowed | 0.0% |
a The numbers in parentheses describe the relevant values for the last resolution shell.
bRmerge = ∑|Ii − ≤I>|/∑Ii, where Ii is the intensity of the ith observation and ≤I> is the mean intensity of the reflection.
cR = ∑|Fobs − Fcalc|/∑|Fobs|. Rcryst and Rfree were calculated from the working and test sets, respectively. The test set contained 10% of the total reflections.
dThe two values represent average root mean square deviations in B-factors of adjacent main-chain and side-chain atoms, respectively.
Figure 2.
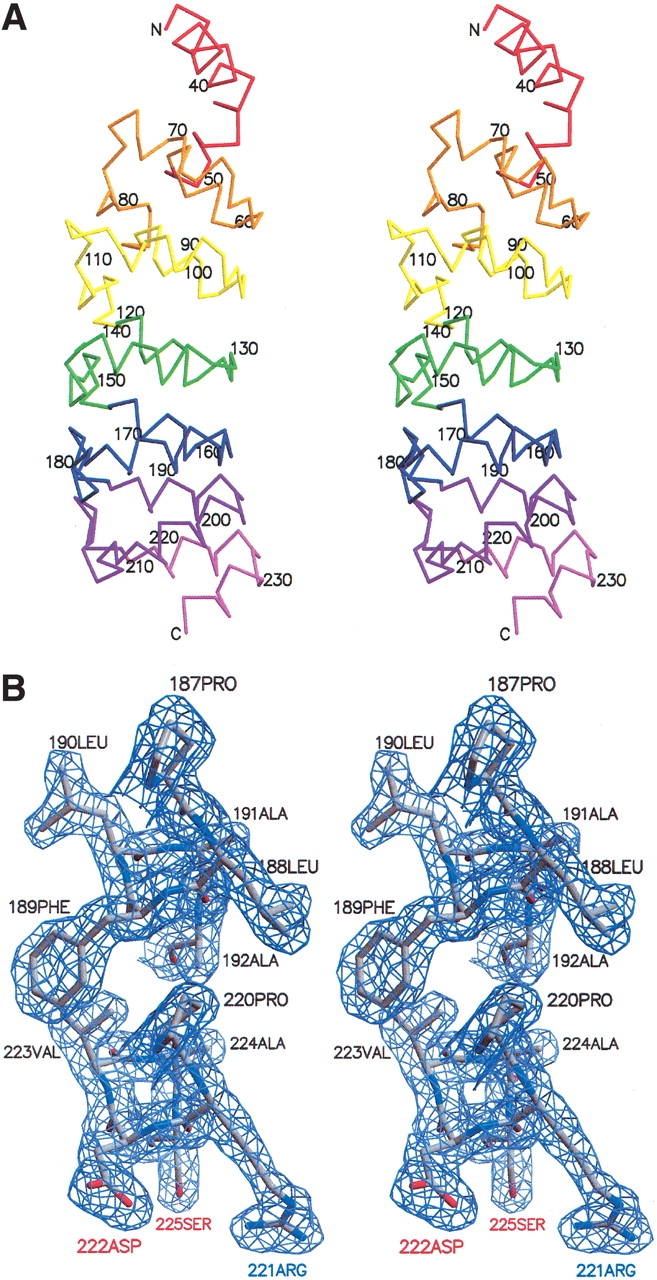
Crystal structure of the ankyrin repeat domain of the Drosophila Notch receptor. (A) Stereoview of the Cα trace of a single copy (chain A) of the ankyrin repeat domain. Individual ankyrin repeats are identified in different colors as in Figure 1 ▶. (B) Stereoview of representative portion of the electron density (2Fo-Fc) contoured at 1.2 sigma, showing the interface between the first helices of repeats 6 and 7. This figure was made using the programs MOLSCRIPT (Kraulis 1991), BOBSCRIPT (Esnouf 1997), and Raster3D (Merritt and Bacon 1997).
Figure 3.

Superposition of ankyrin repeat structures. (A) Cα trace of a superposition of the three copies of the Notch ankyrin domain present in the asymmetric unit. The superposition was made from repeat two to repeat seven. Chain A is red, chain B is green, and chain C is blue. (B) Cα trace of superpositions of individual ankyrin repeats (two through seven) from the Notch ankyrin domain (chain A). Individual ankyrin repeats are identified using different colors as in Figure 1 ▶. (C) Superposition of the Notch ankyrin repeat domain (chain A, red) with GABPβ (PDB ID 1AWC; purple) and IκBα (PDB ID 1NFI; cyan). This figure was made using the programs MOLSCRIPT (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
With the exception of the high structural heterogeneity in the first repeat, the structures of the three ankyrin polypeptides of the asymmetric unit are very similar (Fig. 3A ▶). Repeats two through seven superpose with an average pairwise root mean square deviation (RMSD) of 0.17 Å (over 184 Cα atoms of each of the three polypeptides in the asymmetric unit). This close structural similarity is significantly higher than that among individual 33 residue Notch ankyrin repeats, which superpose with an average pairwise RMSD on Cα of 0.60 Å (Fig. 3B ▶).
The structural similarity between the three Notch ankyrin polypeptides of the asymmetric unit is also significantly higher than that between the Notch ankyrin domain and ankyrin domains from other proteins. Using the program VAST (Madej et al. 1995), the highest structural similarity between the Notch ankyrin domain and another ankyrin structure, obtained by aligning repeats three through seven of the Notch ankyrin domain with the five ankyrin repeats of GABPβ (Batchelor et al. 1998), produces an RMSD of 1.1 Å over 149 Cα atoms (Fig. 3C ▶). The longest alignment, involving the six repeats of IκBα and the Notch ankyrin domain (Jacobs and Harrison 1998), produces an RMSD of 1.9 Å over 188 Cα atoms (Fig. 3C ▶). These comparisons indicate that although the tertiary structures of different ankyrin repeat domains show significant variability, tertiary structure is precisely specified by amino acid sequence in spite of the modular, linear architecture of these domains.
Contacts between ankyrin repeat polypeptides in the crystal lattice
Although our previous hydrodynamic studies at pH 8.0 indicate that the Notch ankyrin domain is monomeric, two-hybrid and other indirect interaction studies have suggested a direct homotypic interaction involving the ankyrin domain of the Notch receptor (Roehl et al. 1996; Matsuno et al. 1997; Kurooka et al. 1998). To identify potential modes by which the Notch ankyrin domain may self-associate, perhaps with the assistance of specific dimerization partners in trans, we examined interactions between Notch ankyrin polypeptides within the crystal lattice.
Although most interchain contacts within the crystal lattice appear to be rather modest, we find one extensive interface, represented three times among the three polypeptides of the asymmetric unit. Within the crystal lattice, each polypeptide in the asymmetric unit is related to its neighbors by a noncrystallographic threefold screw axis that runs perpendicular to the long axis of each ankyrin domain. This threefold screw is continued by the adjacent polypeptide of the next unit cell. Along this screw axis, each polypeptide forms two substantial pseudosymmetric interfaces, one each with each of its two nearest neighbors. Interchain contacts are made between repeats two, three, and four on the convex surface of one polypeptide and repeats four, five, and six of the concave surface of the neighboring polypeptide (Fig. 4A ▶). Each interface buries roughly 1500 Å2 of total surface area per contact (∼750 Å2 per polypeptide) and yields a relatively high shape correlation parameter (Lawrence and Colman 1993; average Sc = 0.74 for the three interfaces). This shape correlation value, which is a measure of the proximity and the shape similarity of contacting surfaces, is as large as values for stable, well-characterized protein oligomer and protease/protein inhibitor complexes (Lawrence and Colman 1993).
Figure 4.

Interactions among Notch ankyrin repeat polypeptides within the crystal lattice. (A) Molecular surface representation of the three polypeptides present in the asymmetric unit (chain A, red; chain B, green; and chain C, blue) and chain B′ (light green) from the adjacent unit cell. The approximate threefold screw axis runs in the vertical direction on the page. (B) GRASP (Nicholls et al. 1991) potential surface depicting the interface between chains B′ (upper right) and A (lower left). The magnesium ion associated with Asp 136 of chain B′ and located at the interface between these two chains is colored yellow. (C) Location of interchain salt bridges at the interface between ankyrin chain A (repeats 5–7, gray) and repeats 2–4 of chain B′ (repeat 2, orange; repeat 3, yellow; repeat 4, green), oriented as in B. This figure was made using MOLSCRIPT (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
The structural features of this interface (buried surface area and shape complimentarity) are consistent with the formation of a stable complex. These interfaces contain multiple interactions between oppositely charged residues deriving largely from the second helix and tight turn of repeats two and three (at the base of the convex surface), and residues distributed along the first helix and β-turn of repeats six and seven (along the concave surface; Fig. 4B,C ▶). At one edge of each interface (near repeat four of the convex surface), a feature in the electron density is seen that appears to be a magnesium ion, with octahedral coordination to five waters and a single aspartic acid (Asp 136) from the convex surface of repeat four. The residues surrounding this magnesium ion include several acidic residues; thus this region has a high negative electrostatic potential (Fig. 4B,C ▶). The presence of this divalent ion may be required to neutralize the negative charge that is juxtaposed at this interface, consistent with the high (200 mM) magnesium concentrations required for crystallization.
To test whether stable homomeric complexes form at lower pH values (the nominal pH during crystallization was 6.8, whereas our previous hydrodynamic studies were done at pH 8.0) or are stabilized by magnesium ions, we performed multi-angle static light scattering over a range of pH values in the presence and absence of magnesium. At 150 mM NaCl, the Notch ankyrin domain remained monomeric over the examined pH range (6.5–8.0), both in the absence and presence of MgCl2 (Fig. 5 ▶, Table 2). Thus, the interface observed within the lattice alone is not sufficient to promote tight association of the Notch ankyrin domain.
Figure 5.

Molecular weight of the Notch ankyrin domain polypeptide at pH 6.5 determined by multiangle static light scattering. Static light scattering measurements were made in the presence (solid line, plus symbols) and absence (dashed line, open circles) of 10 mM MgCl2. The chromatographic peaks (lines) were monitored by refractometry (right axis) and the weight-averaged molecular weights (left axis) are represented as points within the chromatogram. The downward curvature of the molecular weight distribution is likely to result from peak broadening between the scattering and refractive index detectors.
Table 2.
Effects of pH and magnesium ion concentration on the apparent molecular weight of the Notch ankyrin domain
| pH | Magnesium concentration (mM) | Mw,obsa (daltons) |
| 6.5 | 0 | 28,100 ± 140 |
| 10 | 26,900 ± 720 | |
| 200 | 25,700 ± 180 | |
| 7.0 | 0 | 30,400 ± 340 |
| 10 | 28,500 ± 610 | |
| 7.4 | 0 | 26,800 ± 820 |
| 10 | 25,100 ± 450 | |
| 8.0 | 0 | 25,700 ± 240 |
| 10 | 26,700 ± 160 | |
| Mw,calcb | 25,935 |
All samples contained 150 mM NaCl and 25 mM buffer as described in Materials and Methods.
aMolecular weight determined by multi-angle static light scattering, as described in Materials and Methods. Reported errors are the root mean square deviation from at least three separate measurements.
bMolecular weight calculated from primary sequence.
Stability of signaling-deficient point substitutions within the Notch ankyrin domain
Several point mutations that disrupt various aspects of Notch signaling fall within the ankyrin domain of Notch receptors from various taxa (Kodoyianni et al. 1992; Diederich et al. 1994; Kopan et al. 1994; Joutel et al. 1996; Kurooka et al. 1998). These include a mutation that leads to a single alanine-to-valine substitution in the fifth ankyrin repeat of Drosophila Notch (Su42c; Diederich et al. 1994), and three mutations in the ankyrin domains of vertebrate Notch receptors. One of these vertebrate mutations results in a single alanine-to-threonine substitution in the second repeat of the human Notch3 receptor (F18; Joutel et al. 1996), and has been associated with CADASIL, a late-onset neurodegenerative disorder. The other two vertebrate mutations introduce combinations of two- and three-residue substitutions in ankyrin repeat four of the mouse Notch1 receptor (M2 and M1, respectively; Kopan et al. 1994). These substitutions in the vertebrate Notch genes occur in regions of the ankyrin repeats that share high sequence identity with the corresponding region of the Drosophila Notch ankyrin domain (Fig. 1 ▶), and thus can be mapped onto the structure of the Drosophila Notch ankyrin domain with reasonable certainty (Fig. 6 ▶).
Figure 6.

Location of point substitutions within the Notch ankyrin domain that disrupt Notch signaling. The F18 substitution (repeat 2) is colored blue, the M1 and M2 substitutions (repeat 4) are colored orange and red, respectively, and the Su42c substitution (repeat 5) is colored green.
To investigate the properties of the signaling-deficient substitutions described above, we introduced these substitutions into the Drosophila Notch ankyrin domain and determined their effects on structural stability. The far-UV circular dichroism (CD) spectra of ankyrin domain variants containing surface substitutions (Su42C and F18) closely match the spectrum of the parent construct (Nank1–7*, Fig. 7A ▶), indicating that these surface substitutions do not greatly perturb the Notch ankyrin fold. In contrast, the spectra of proteins containing substitutions within the core of the domain (M1 and M2) differ significantly from that of the parent construct, showing a substantial loss of α-helical structure.
Figure 7.
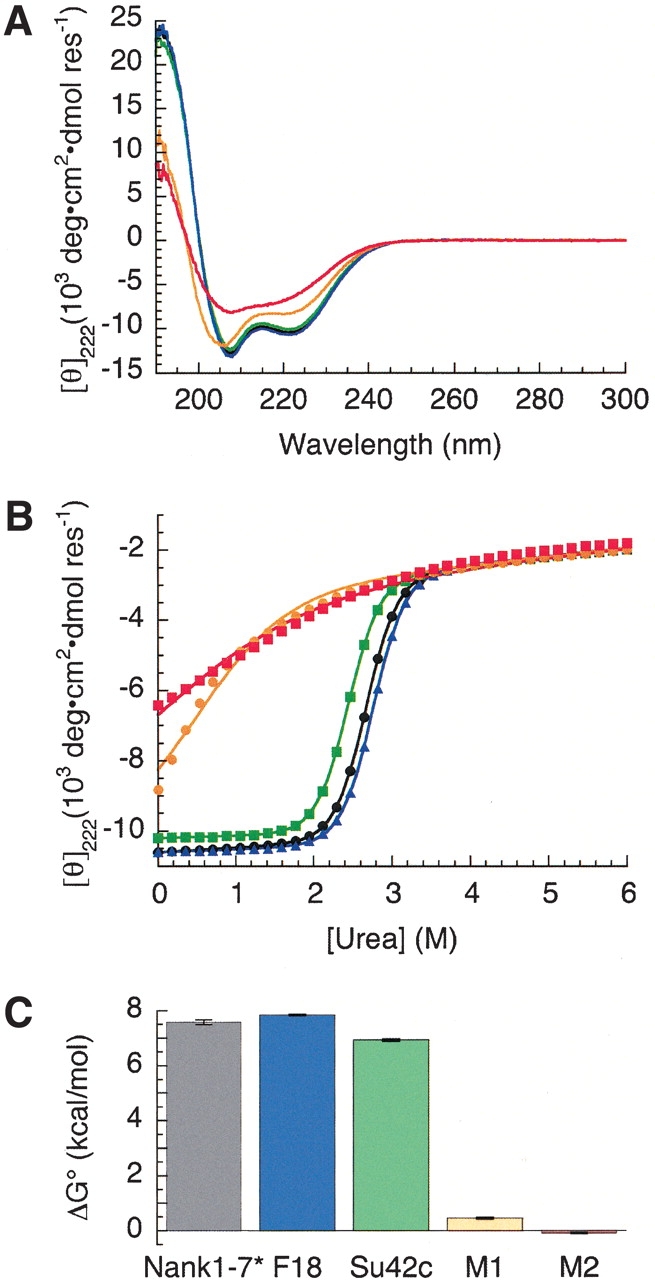
Structural stabilities of Notch variants. (A) Circular dichroism spectra of the Drosophila Notch ankyrin domain (black) and variants F18 (blue), Su42C (green), M1 (orange), and M2 (red). (B) Urea unfolding of the wild-type Drosophila Notch ankyrin domain (black circles) and variants F18 (blue triangles), Su42c (green squares), M1 (orange circles), and M2 (red squares). (C) Free energies of unfolding of the ankyrin domain variants. Colors are as in A.
The effects of the signaling mutations on the stability of the Notch ankyrin domain were assessed by measuring urea-induced unfolding curves (Fig. 7B ▶). The two surface substitutions (Su42c and F18) have only modest effects on the unfolding transitions. In contrast, substitutions that include buried residues (M1 and M2) result in major shifts in the unfolding transitions to lower urea concentrations. In addition, the observed structural transitions of the M1 and M2 variants are less cooperative, and appear to be underway in the absence of urea. In terms of unfolding free energy, the surface substitutions (Su42c and F18) have only minor effects on unfolding free energy, whereas the substitutions that include buried residues (M1 and M2) decrease unfolding free energy by 7–8 kcal mole−1 (Fig. 7C ▶). Thus, the Su42c and F18 substitutions are likely to disrupt signaling by directly perturbing interactions with specific components of the pathway, whereas the M1 and M2 substitutions are likely to disrupt the overall fold of the ankyrin domain, indirectly disrupting interactions with multiple interacting partners in a nonspecific manner.
Discussion
Boundaries of the Notch ankyrin domain
The ankyrin domain of the Drosophila Notch receptor contains six consensus ankyrin sequence repeats and a putative seventh C-terminal sequence repeat that is of lower sequence similarity to the ankyrin consensus. We have shown previously that a polypeptide containing all seven repeats has a free energy of unfolding nearly twice that of a polypeptide lacking the seventh sequence repeat (Zweifel and Barrick 2001b). The crystal structure of the ankyrin repeat domain of the Drosophila Notch receptor reveals that this stabilizing C-terminal repeat adopts a regular ankyrin fold, enhancing stability by extending the ankyrin domain. These results demonstrate the importance of terminal repeats in linear repeat protein stability, and highlight the sequence differences that may be expected for terminal repeats (Bork 1993; Zweifel and Barrick 2001a).
In contrast, the first ankyrin sequence repeat appears to be largely disordered. The visible portion of this repeat adopts a different conformation in each of the three polypeptides present in the asymmetric unit. This heterogeneity is consistent with the observation that a core mutation in the first Notch ankyrin sequence repeat has very little effect on stability, whereas analogous mutations in repeats two through seven are highly destabilizing (Bradley and Barrick 2002). It is unlikely that the lack of order in the first repeat can be attributed to N-terminal sequence outside the ankyrin domain that is absent from our construct, because inclusion of additional N-terminal sequence (135 residues) enhances neither helix formation nor stability of the Notch ankyrin domain (M.E. Zweifel and D. Barrick, unpubl.). Furthermore, the elimination of the N-terminal sequence that extends between the Notch transmembrane and the ankyrin repeat domains does not alter the observed homotypic association of the Notch ankyrin domain as determined using a yeast two-hybrid assay (Matsuno et al. 1997).
Although the two helices of the first sequence repeat show a close match to the ankyrin consensus sequence, an obvious difference between the first sequence repeat and the other ankyrin repeats is that there is an insertion of 15 residues between the two consensus helical regions of the first repeat (Fig. 1 ▶), whereas the two helical segments are connected by a short two-residue turn in other ankyrin sequences. This insertion may contribute to the instability and/or heterogeneity of the first sequence repeat, either through strain or through an entropic penalty for loop closure. Similar insertions are seen in the first sequence repeat of Notch ankyrin domains from a range of species (Fig. 1 ▶, see + signs). Although these inserted sequences vary in length, they have a high proportion of acidic (D, E) and polar (S, T, N, G) residues. This conservation suggests that the insertion may play a functional role in Notch signaling, and may undergo a functionally important disorder–order transition upon the binding of one or more effector proteins, as has been seen in several other systems (Daughdrill et al. 1997; Dyson and Wright 2002). One functional role for this acidic sequence may be in transcriptional activation (Giniger and Ptashne 1987; Gill and Ptashne 1988), given that the intracellular region of the Notch receptor acts as a cotranscription factor in the activated state.
Potential interactions between ankyrin repeat polypeptides
Indirect interaction studies have suggested a homotypic interaction of the Notch receptor involving the ankyrin domain (Roehl et al. 1996; Matsuno et al. 1997; Kurooka et al. 1998). The extensive contact surface identified between each of the three polypeptides of the asymmetric unit are consistent with such an interaction, although association via the crystallographically observed interface in solution would be expected to lead the formation of helical fibers, rather than to a closed complex of discrete stoichiometry. Despite this large crystallographic interface, we find the Notch ankyrin domain to remain monomeric under conditions where it is soluble from pH 6.5 to 8.0, in 150 mM NaCl. Furthermore, the Notch ankyrin domain remains monomeric under these conditions in the presence of high MgCl2 concentrations, indicating that the interfacial magnesium ion seen in the crystal structure is not, on its own, sufficient to mediate oligomerization under the conditions examined here. Thus, if this mode of interaction is relevant in solution, it must be rather weak, and may be enhanced by secondary binding either to effector proteins or through indirect interactions with DNA. One system in which weak association plays a role in function is the regulation of transcription in Escherichia coli by GalR. Although GalR is largely dimeric in solution (Majumdar et al. 1987), it is thought to mediate DNA looping through weak tetramerization, facilitated by HU protein (Semsey et al. 2002). Weak binding interactions have the potential to enhance allosteric responses, maximizing the degree to which populations of various noncovalent complexes can be modulated by changes in the concentrations of the polypeptides from which they are formed.
Stability of Notch ankyrin domain variants
We have examined all reported mutations that can be mapped to the Drosophila Notch ankyrin domain. Analysis of the effect of these substitutions on stability reveals that the two single-residue substitutions (Su42c and F18) located at surface-exposed positions do not significantly perturb the overall stability of the domain, whereas the two multiresidue substitutions (M1 and M2) that include one or more buried positions greatly diminish the overall stability of the domain, resulting in significant disruption of structure. Given the location of these substitutions within the structure of the Notch ankyrin domain and their effects on stability, the substitutions that include buried positions (M1 and M2) seem likely to block Notch signaling activities through general disruption of the fold of the Notch ankyrin domain, whereas the surface substitutions (Su42c and F18) seem likely to block signaling through disruption of direct interactions with a limited set of effector proteins.
Although not directly comparable, the severity of the effects of these substitutions on Notch signaling is consistent with the effects of these substitutions on the structural stability of the Notch ankyrin domain. Consistent with this proposal, the Su42c mutation is phenotypically similar to mutations in the Deltex gene, the product of which has been shown to interact with the Notch ankyrin domain in a two-hybrid interaction assay (Diederich et al. 1994). Thus, this surface substitution may interfere with the Notch–Deltex interaction. Su42c, a surface substitution that has only a minor effect on stability, produces relatively mild, nonlethal changes in the adult body plan (Diederich et al. 1994). Similarly, the effects of the F18 surface substitution, which also has only minor effects on stability, do not appear until relatively late in adult life (Joutel et al. 1996). The location of these two surface substitutions may define the site of interaction with specific effectors of Notch signaling. In contrast, mutations that result in core substitutions (M1 and M2) abolish MyoD/RBP-J-mediated transcriptional activity in cultured cells, a central activity in the Notch signaling pathway (Kopan et al. 1994). Although the effects of these multisite substitutions on normal development have not been determined, disruption of Notch-mediated transcriptional activation would likely produce severe developmental defects. Disruption of signaling as a result of substitutions at buried positions within an ankyrin domain has also been seen in the tumor suppressor p16INK4A, for which structurally disruptive substitutions have been identified in several cancer cell lines (Tevelev et al. 1996; Tang et al. 1999).
Materials and methods
Protein purification and crystallization
A polypeptide containing the seven ankyrin repeat sequences (Nank1–7&Δ [Bradley and Barrick 2002] encoding residues 1902–2139 of the Drosophila Notch receptor) was expressed in E. coli strain BL21(DE3) as described previously (Zweifel and Barrick 2001a). Selenomethionine was incorporated by expression in E. coli strain B834(DE3) (a methionine auxotroph) grown in minimal media supplemented with seleno-L-methionine. Protein was purified from the soluble fraction of a bacterial lysate using a Ni•NTA affinity column. The protein was then dialyzed into buffer containing 150 mM sodium chloride and 25 mM Tris-HCl (pH 8.0) and then treated with thrombin to remove the 6xHis-tag. The protein was further purified by ion-exchange and gel-filtration chromatography, concentrated to 10–15 mg/ml, and dialyzed into buffer containing 150 mM sodium chloride and 25 mM Tris-HCl (pH 8.0). Crystals were grown by hanging drop vapor diffusion with 15%–20% (v/v) MPD, 200 mM magnesium acetate, and 100 mM sodium cacodylate (pH 6.5–7.0). Crystals grew in 1 to 2 weeks to a size of approximately 0.1 × 0.1 × 0.05mm, belonging to the tetragonal space group P43212, with a = b = 73.6 and c = 341.1 Å.
Structure determination and refinement
Crystals were soaked in a cryoprotectant solution consisting of 25% (v/v) MPD, 200 mM magnesium acetate, and 100 mM sodium cacodylate at pH 7.0 for 30 sec and then flash-frozen at −175°C. A 2.8 Å native data set was collected at this temperature using a Rigaku RU-300HR X-ray generator with an R-AxisIV detector. Crystals diffracted to a resolution of 2.0 Å; however, at resolutions higher than 2.8 Å, reflections could not be resolved because of the long (c = 341.1 Å) axis of the unit cell. Diffraction data from the selenomethionine derivative was collected at National Synchrotron Light Source (NSLS) beamline X-4A and processed using DENZO/SCALEPACK (Otwinowski and Minor 1997). Initial phases were calculated using SOLVE/RESOLVE (Terwilliger and Berendzen 1999) to produce an interpretable map. An initial model was constructed using the program O (Jones et al. 1991). Refinement of the initial model against the 2.8 Å native data set was performed using CNS (Brunger et al. 1998). To extend resolution, a second native data set (2.0 Å) was collected at NSLS beamline X-25 to improve separation of higher resolution reflections. The 2.8 Å model was refined against the 2.0 Å native data set using REFMAC (Murshudov et al. 1997) to Rworking = 18.0 and Rfree = 20.1. The final model contained 5042 atoms including 471 solvent molecules. The quality of the final model was assessed using the program PROCHECK (Laskowski et al. 1993). Over 90% of residues were found to be in the most favored regions of the Ramachandran plot, and showed good stereochemistry (Table 1). Buried surface area was calculated using the method of Lee and Richards (1971) as implemented in the program CNS (Brunger et al. 1998). The shape correlation parameter was calculated using the program SC (Lawrence and Colman 1993) from the CCP4 suite (Collaborative Computational Project 1994).
Measurement of the conformational stability of Notch mutants
Notch mutants were constructed by site-directed mutagenesis using the QuikChange Kit (Stratagene, La Jolla, CA). Mutations were verified by sequencing and were purified as described previously (Zweifel and Barrick 2001a). Unfolding free energies were estimated by urea denaturation in 25 mM Tris-HCl, 150 mM NaCl (pH 8.0) at 20°C. Unfolding transitions were detected by monitoring the CD signal at 222 nm using an Aviv model 62DS CD spectrometer as described (Zweifel and Barrick 2001b). Free energies of unfolding were extrapolated to zero denaturant concentration assuming a linear dependence on denaturant (Pace 1986; Santoro and Bolen 1988) as described previously (Zweifel and Barrick 2001b). For the unfolding curves for the M1 and M2 variants, the native CD signal was assumed to be the same as that for the parent construct. This assumption was necessary because these two variants only show partial unfolding transitions. Thermodynamic analysis of partial unfolding transitions of the Notch ankyrin domain has previously been shown to provide reasonable estimates of unfolding free energies (Mello and Barrick 2003).
Static light scattering measurements
Static light scattering measurements were performed at 25°C using an HPLC system equipped with a TosoHaas G3000PWXL HPLC column, a three-angle light scattering detector (Wyatt MiniDAWN; Wyatt Technologies, Santa Barbara, CA) and a differential refractive index detector (Wyatt Optilab DSP; Wyatt Technologies). Samples were dialyzed overnight at 4°C into running buffer (150 mM NaCl, either 25 mM Tris at pH 8.0 or 25 mM HEPES at pH 7.4, 7.0, and 6.5, and 0.01% sodium azide) with or without 10 mM MgCl2. Chromatographic separation was performed at a flow rate of 0.5 ml/min. 50 μl samples were injected onto a preequilibrated column at a loading concentration between 1.5 and 3.0 mg/ml (approximately 60 to 120 μM protein). Data acquisition and analysis were performed using ASTRA 4.0 software (Wyatt Technologies).
Coordinates
The atomic coordinates of the Notch ankyrin domain have been deposited in the Protein Data Bank (PDB ID 1OT8).
Acknowledgments
Data for this study were collected at beamlines X-4A and X-25 of the National Synchrotron Light Source, which is funded by the NIH National Center for Research Resources and the Department of Energy Office of Biological and Environmental Research. This work is also based on research conducted at the Joint Crystallography Facility at Johns Hopkins University, which is funded by Grant 9970207 from the NSF. We thank the staff at NSLS beamlines X4-A (C. Ogata and R. Abramowitz) and X-25 (M. Becker and L. Berman), and Apostolos Gittis (Johns Hopkins University) for assistance with the collection of diffraction data. We thank Dr. Spyros Artavanis-Tsakonis for providing the cDNA for Drosophila Notch. We also thank Christina Bradley for providing the vector used to express Nank1–7Δ. This research was supported by a Beckman Young Investigator award to D.B., and by NIH grant GM60001.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RMSD, root mean square deviation
CD, circular dichroism spectroscopy
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03279003.
References
- Artavanis-Tsakonas, S., Rand, M.D., and Lake, R.J. 1999. Notch signaling: Cell fate control and signal integration in development. Science 284 770–776. [DOI] [PubMed] [Google Scholar]
- Barton, G.J. 1993. ALSCRIPT—A tool to format multiple sequence alignments. Protein Eng. 6 37–40. [DOI] [PubMed] [Google Scholar]
- Batchelor, A.H., Piper, D.E., de la Brousse, F.C., McKnight, S.L., and Wolberger, C. 1998. The structure of GABPα/β: An ETS domain-ankyrin repeat heterodimer bound to DNA. Science 279 1037–1041. [DOI] [PubMed] [Google Scholar]
- Bork, P. 1993. Hundreds of ankyrin-like repeats in functionally diverse proteins: Mobile modules that cross phyla horizontally? Proteins 17 363–374. [DOI] [PubMed] [Google Scholar]
- Bradley, C.M. and Barrick, D. 2002. Limits of cooperativity in a structurally modular protein: Response of the Notch ankyrin domain to analogous alanine substitutions in each repeat. J. Mol. Biol. 324 373–386. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR System (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Daughdrill, G., Chadsey, M., Karlinsey, J., Hughes, K., and Dahlquist, F. 1997. The C-terminal half of the anti-sigma factor, FlgM, becomes structured when bound to its target, sigma 28. Nat. Struct. Biol. 4 285–291. [DOI] [PubMed] [Google Scholar]
- Diederich, R.J., Matsuno, K., Hing, H., and Artavanis-Tsakonas, S. 1994. Cytosolic interaction between Deltex and Notch ankyrin repeats implicates Deltex in the Notch signaling pathway. Development 120 473–481. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2002. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12 54–60. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of molscript that includes generally enhanced colouring capabilities. J. Mol. Graphics Modelling 15 132–134. [DOI] [PubMed] [Google Scholar]
- Foord, R., Taylor, I.A., Sedgwick, S.G., and Smerdon, S.J. 1999. X-ray structural analysis of the yeast cell cycle regulator Swi6 reveals variations of the ankyrin fold and has implications for Swi6 function. Nat. Struct. Biol. 6 157–165. [DOI] [PubMed] [Google Scholar]
- Gill, G. and Ptashne, M. 1988. Negative effect of the transcriptional activator GAL4. Nature 334 721–724. [DOI] [PubMed] [Google Scholar]
- Giniger, E. and Ptashne, M. 1987. Transcription in yeast activated by a putative amphipathic α helix linked to a DNA binding unit. Nature 330 670–672. [DOI] [PubMed] [Google Scholar]
- Gorina, S. and Pavletich, N.P. 1996. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 274 1001–1005. [DOI] [PubMed] [Google Scholar]
- Hubbard, E.J., Dong, Q., and Greenwald, I. 1996. Evidence for physical and functional association between EMB-5 and LIN-12 in Caenorhabditis elegans. Science 273 112–115. [DOI] [PubMed] [Google Scholar]
- Huxford, T., Huang, D.B., Malek, S., and Ghosh, G. 1998. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell 95 759–770. [DOI] [PubMed] [Google Scholar]
- Jacobs, M.D. and Harrison, S.C. 1998. Structure of an IκBα/NF-κB complex. Cell 95 749–758. [DOI] [PubMed] [Google Scholar]
- Jarriault, S., Brou, C., Logeat, F., Schroeter, E.H., Kopan, R., and Israel, A. 1995. Signalling downstream of activated mammalian Notch. Nature 377 355–358. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Joutel, A., Corpechot, C., Ducros, A., Vahedi, K., Chabriat, H., Mouton, P., Alamowitch, S., Domenga, V., Cecillion, M., Marechal, E., et al. 1996. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383 707–710. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kidd, S., Kelley, M.R., and Young, M.W. 1986. Sequence of the Notch locus of Drosophila melanogaster: Relationship of the encoded protein to mammalian clotting and growth factors. Mol. Cell. Biol. 6 3094–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodoyianni, V., Maine, E.M., and Kimble, J. 1992. Molecular basis of loss-of-function mutations in the glp-1 gene of Caenorhabditis elegans. Mol. Biol. Cell 3 1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan, R., Nye, J.S., and Weintraub, H. 1994. The intracellular domain of mouse Notch: A constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development 120 2385–2396. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystal. 24 946–950. [Google Scholar]
- Kurooka, H., Kuroda, K., and Honjo, T. 1998. Roles of the ankyrin repeats and C-terminal region of the mouse Notch1 intracellular region. Nucleic Acids Res. 26 5448–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystal. 26 283–291. [Google Scholar]
- Lawrence, M.C. and Colman, P.M. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234 946–950. [DOI] [PubMed] [Google Scholar]
- Lee, B. and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Luh, F.Y., Archer, S.J., Domaille, P.J., Smith, B.O., Owen, D., Brotherton, D.H., Raine, A.R., Xu, X., Brizuela, L., Brennner, S.L., et al. 1997. Structure of the cyclin-dependent kinase inhibitor p19Ink4d. Nature 389 999–1003. [DOI] [PubMed] [Google Scholar]
- Madej, T., Gibrat, J.-F., and Bryant, S.H. 1995. Threading a database of protein cores. Proteins 23 356–369. [DOI] [PubMed] [Google Scholar]
- Majumdar, A., Rudikoff, S., and Adhya, S. 1987. Purification and properties of Gal repressor:pL-galR fusion in pKC31 plasmid vector. J. Biol. Chem. 262 2326–2331. [PubMed] [Google Scholar]
- Mandiyan, V., Andreev, J., Schlessinger, J., and Hubbard, S.R. 1999. Crystal structure of the ARF-GAP domain and ankyrin repeats of PYK2-associated protein β. EMBO J. 18 6890–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno, K., Diederich, R.J., Go, M.J., Blaumueller, C.M., and Artavanis-Tsakonas, S. 1995. Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development 121 2633–2644. [DOI] [PubMed] [Google Scholar]
- Matsuno, K., Go, M.J., Sun, X., Eastman, D.S., and Artavanis-Tsakonas, S. 1997. Suppressor of Hairless-independent events in Notch signaling imply novel pathway elements. Development 124 4265–4273. [DOI] [PubMed] [Google Scholar]
- Mello, C.C. and Barrick, D. 2003. Measuring the stability of partly folded proteins using TMAO. Protein Sci. 12 1522–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Rebay, I., Fehon, R.G., and Artavanis-Tsakonas, S. 1993. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell 74 319–329. [DOI] [PubMed] [Google Scholar]
- Roehl, H. and Kimble, J. 1993. Control of cell fate in C. elegans by a GLP-1 peptide consisting primarily of ankyrin repeats. Nature 364 632–635. [DOI] [PubMed] [Google Scholar]
- Roehl, H., Bosenberg, M., Blelloch, R., and Kimble, J. 1996. Roles of the RAM and ANK domains in signaling by the C. elegans GLP-1 receptor. EMBO J. 15 7002–7012. [PMC free article] [PubMed] [Google Scholar]
- Russo, A.A., Tong, L., Lee, J.O., Jeffrey, P.D., and Pavletich, N.P. 1998. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395 237–243. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schroeter, E.H., Kisslinger, J.A., and Kopan, R. 1998. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393 382–386. [DOI] [PubMed] [Google Scholar]
- Sedgwick, S.G. and Smerdon, S.J. 1999. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 24 311–316. [DOI] [PubMed] [Google Scholar]
- Semsey, S., Geanacoloulos, M., Lewis, D.E.A., and Adhya, S. 2002. Operator-bound GalR dimers close DNA loops by direct interaction: Tetramerization and inducer binding. EMBO J. 21 4349–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stifani, S., Blaumueller, C.M., Redhead, N.J., Hill, R.E., and ArtavanisTsakonas, S. 1992. Human homologs of a Drosophila enhancer of split gene product define a novel family of nuclear proteins. Nat. Genet. 2 119–127. [DOI] [PubMed] [Google Scholar]
- Struhl, G. and Greenwald, I. 1999. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398 522–525. [DOI] [PubMed] [Google Scholar]
- Tang, K.S., Guralnick, B.J., Wang, W.K., Fersht, A.R., and Itzhaki, L.S. 1999. Stability and folding of the tumour suppressor protein p16. J. Mol. Biol. 285 1869–1886. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tevelev, A., Byeon, I.J., Selby, T., Ericson, K., Kim, H.J., Kraynov, V., and Tsai, M.D. 1996. Tumor suppressor p16INK4a: Structural characterization of wild-type and mutant proteins by NMR and circular dichroism. Biochemistry 35 9475–9487. [DOI] [PubMed] [Google Scholar]
- Vardar, D., North, C.L., Sanchez-Irizarry, C., Astor, J.C., and Blacklow, S.C. 2003. Nuclear magetic resonance structure of a prototype Lin12-Notch repeat module from human Notch1. Biochemistry 42 7061–7067. [DOI] [PubMed] [Google Scholar]
- Venkataramani, R., Swaminathan, K., and Marmorstein, R. 1998. Crystal structure of the CDK4/6 inhibitory protein p18INK4c provides insights into ankyrin-like repeat structure/function and tumor-derived p16INK4 mutations. Nat. Struct. Biol. 5 74–81. [DOI] [PubMed] [Google Scholar]
- Wharton, K.A., Johansen, K.M., Xu, T., and Artavanis-Tsakonas, S. 1985. Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 43 567–581. [DOI] [PubMed] [Google Scholar]
- Zhou, S., Fujimuro, M., Hsieh, J.J.-D., Chen, L., Miyamoto, A., Weinmaster, G., and Hayward, S.D. 2000. SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC to facilitate NotchIC function. Mol. Cell. Biol. 20 2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel, M.E. and Barrick, D. 2001a. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 1. Solution conformational and hydrodynamic properties. Biochemistry 40 14344–14356. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 2. Solution stability and cooperativity of unfolding. Biochemistry 40 14357–14367. [DOI] [PubMed] [Google Scholar]