

Abstract
The anti-17β-estradiol antibody 57-2 has been a subject for several protein engineering studies that have produced a number of mutants with improved binding properties. Here, we generated a set of 16 antibody 57-2 variants by systematically combining mutations previously identified from phage display–derived improved antibody mutants. These mutations included three point mutations in the variable domain of the light-chain and a heavy-chain variant containing a four-residue random insertion in complementarity determining region CDR-H2. The antibody variants were expressed as Fab fragments, and they were characterized for affinity toward estradiol, for cross-reactivity toward three related steroids, and for dissociation rate of the Fab/estradiol complex by using time-resolved fluorescence based immunoassays. The double-mutant cycle method was used to address the cooperativity effects between the mutations. The experimental data were correlated with structural information by using molecular modeling and visual analysis of the previously solved antibody 57-2 crystal structures. These analyses provided information about the steroid-binding mode of the antibody, the potential mechanisms of individual mutations, and their mutual interactions. Furthermore, several combinatorial mutants with improved affinity and specificity were obtained. The capacity of one of these mutants to detect estradiol concentrations at a clinically relevant range was proved by establishing a time-resolved fluorescence based immunoassay.
Keywords: Antibody, estradiol, mutation, site-directed mutagenesis, steroid
The capability of antibodies to specifically recognize an immense variety of other molecules is widely used in life sciences. For example, immunoassays that are commonly used for the quantitation of the concentrations of specific components in complex molecular mixtures such as blood are based on the recognition properties of antibodies. However, the generation of high-quality antibodies for some antigens is challenging. An example of this type of molecule is the steroid hormone 17β-estradiol (E2), which consists of a hydrophobic steroid skeleton containing only two functional hydroxyl groups (Fig. 1 ▶). Concentration of E2 in the blood is low, and a number of steroids with closely related structures are present in the circulation, many of them in much higher concentration than E2. Therefore, the antibody used as a binder in an E2 immunoassay needs to have high affinity and good specificity. Currently, sufficiently good monoclonal antibodies are not available, and the E2 immunoassays are based on polyclonal antibodies.
Figure 1.

Schematic structures of 17β-estradiol, testosterone, estrone, and 17β-estradiol-3-sulfate. The numbering of the carbon atom skeleton and the lettering of the steroid rings are shown for estradiol. The methyl group C19 missing from estrogens is indicated in testosterone.
We have previously cloned three murine antibodies raised against 17β-estradiol-6-CMO conjugate (Pajunen et al. 1997). One of these is known as antibody 57-2, and it has a moderate affinity toward E2, an excellent capacity to discriminate between E2 and estrone or estriol, but, unfortunately, a high cross-reactivity toward testosterone. Mainly because of the excellent selectivity among the members of the estrogen family, the antibody 57-2 was chosen as a starting point for antibody engineering with the aim of decreasing the testosterone cross-reactivity and increasing the affinity toward E2. Consequently, the antibody 57-2 was subjected to several random mutagenesis and phage display selection-based protein engineering trials that produced a number of different antibody variants with improved binding properties (Saviranta et al. 1998; Lamminmaki et al. 1999). However, variants with properties allowing the detection of E2 levels at the clinically relevant range were not obtained in these selections.
The steroid-binding mode of the antibody 57-2 has been studied by means of molecular modeling (Lamminmaki et al. 1997), and more recently, X-ray crystallography was used to determine the structure of a recombinant Fab fragment of the antibody in the presence and absence of the ligand E2 (Lamminmaki and Kankare 2001). From these studies, it is known that the antibody binds E2 in a deep hydrophobic pocket between VH and VL domains, and the excellent selectivity of the antibody between different estrogens originates from an intricate hydrogen bond arrangement involving a four-center hydrogen bond between the 17-OH group of E2 and three residues in the antibody. The crystallographic data also indicated that only small changes in the antibody structure occur during the ligand binding.
In the present study, we have analyzed the functional impacts and mechanisms of a set putative gain-of-function mutations in the antibody 57-2, and we systematically combined these mutations in order to generate improved antibody variants. The mutations studied originate from the clones isolated in previous phage display selections, and they include three light-chain point mutations as well as a high-affinity heavy-chain variant that differs from the wild-type heavy-chain primarily by having a four-residue insertion at the tip of complementarity determining region CDR-H2. All the possible combinations of the point mutations and the two heavy-chain variants (the wild type and the insertion mutant 4aa16) were generated, and the resulting antibody variants were experimentally characterized for various binding properties and analyzed by the means of molecular modeling. In addition to providing information on the mechanisms of the mutations, the systematic exploration of the mutation combinations produced several E2 binders with improved affinity and specificity.
Results
Three putative gain-of-function point mutations in the light-chain of the antibody 57-2, namely, Asn50L→Tyr, Phe83L→Ser, and Phe91L→Tyr, were identified by means of structure and sequence comparison from a set of multiply mutated clones originating from phage display selections of random mutation libraries (Saviranta 2001). The selection of the putative gain-of-function mutations was based on the criteria that a suitable mutation either existed in several different clones with improved binding properties or was located close to the antigen-combining site in one of the improved clones. These three light-chain mutations and all combinations of them were introduced in the wild-type antibody as well as in the heavy-chain mutant 4aa16 by means of site-directed mutagenesis. This approach resulted in a total of 16 antibody variants that were expressed as Fab fragments and analyzed for various binding properties by using time-resolved fluorescence based immunoassays. To address the structural basis of the functional mechanisms of the mutations, we modeled the light-chain mutations in the crystal structure of the Fab 57-2/E2 complex and inspected visually the crystal structure itself.
Affinity toward E2
Light-chain mutations
Mutation Asn→Tyr in residue 50 of the light-chain increased the affinity of the antibody toward E2 ∼3.7-fold (Table 1). The residue 50L is located at the mouth of the binding pocket and the side chain of the wild-type residue Asn does not contact the bound E2 (Fig. 2A ▶). However, according to the model of the mutant Asn50L→Tyr, the distal end of the Tyr50L side chain can directly interact with the A- and B-rings of E2 (Fig. 2C ▶), forming new van der Waals interactions between the antibody and the steroid.
Table 1.
Binding data for interactions between the antibody 57-2 mutants and the steroids 17β-estradiol (E2), testosterone (TES), estrone (E1), and 17β-estradiol-3-sulfate (E2-3-SO4)
| E2 | Cross-reactivity (%) | ||||||
| Clonea | Ka (×108 M−1) | koff (×104 s−1) | kon (×105 M−1s−1) | Ka(TES)(× 107M− 1 | TES | E1 | E2-3-SO4 |
| wt/wt | 2.0 ± 0.1 | 39.6 ± 8.1 | 7.8 ± 1.9 | 6.1 ± 0.6 | 27.4 ± 3.8 | 0.1 ± 0.0 | 5.3 ± 0.1 |
| wt/N50Y | 7.3 ± 0.3 | 17.3 ± 0.1 | 12.7 ± 0.5 | 9.4 ± 0.0 | 11.8 ± 1.7 | 0.1 ± 0.0 | 5.5 ± 0.3 |
| wt/F83S | 2.8 ± 0.1 | 38.9 ± 1.0 | 11.0 ± 0.7 | 5.0 ± 1.0 | 15.1 ± 1.4 | 0.1 ± 0.0 | 5.0 ± 0.3 |
| wt/F91Y | 4.2 ± 0.2 | 21.6 ± 5.1 | 9.2 ± 2.7 | 9.3 ± 1.3 | 20.7 ± 0.7 | 0.1 ± 0.0 | 5.0 ± 0.2 |
| wt/N50Y + F83S | 8.9 ± 0.2 | 11.3 ± 3.3 | 10.0 ± 3.2 | 6.8 ± 0.5 | 7.2 ± 0.8 | 0.1 ± 0.0 | 5.3 ± 0.1 |
| wt/F83S + F91Y | 5.6 ± 0.3 | 13.5 ± 0.9 | 7.5 ± 0.9 | 8.1 ± 1.7 | 12.1 ± 0.9 | 0.1 ± 0.0 | 4.7 ± 0.2 |
| wt/N50Y + F91Y | 3.8 ± 0.3 | 20.8 ± 0.6 | 8.0 ± 0.8 | 5.7 ± 0.2 | 14.2 ± 1.4 | 0.1 ± 0.0 | 5.1 ± 0.0 |
| wt/N50Y + F83S + F91Y | 4.8 ± 0.6 | 19.8 ± 1.4 | 9.4 ± 1.9 | 4.6 ± 0.1 | 9.6 ± 2.1 | 0.1 ± 0.0 | 4.9 ± 0.3 |
| 4aa16/wt | 10.9 ± 0.5 | 10.4 ± 0.7 | 11.3 ± 1.2 | 0.5 ± 0.1 | 0.4 ± 0.0 | <0.1 ± 0.0 | 4.3 ± 0.2 |
| 4aa16/N50Y | 21.3 ± 0.9 | 4.4 ± 1.1 | 9.4 ± 2.8 | 0.3 ± 0.1 | 0.2 ± 0.0 | <0.1 ± 0.0 | 5.0 ± 0.4 |
| 4aa16/F83S | 16.2 ± 1.2 | 7.4 ± 1.6 | 11.9 ± 3.4 | 0.3 ± 0.1 | 0.2 ± 0.0 | <0.1 ± 0.0 | 4.0 ± 0.5 |
| 4aa16/F91Y | 20.0 ± 0.8 | 5.1 ± 1.8 | 10.2 ± 4.0 | 0.5 ± 0.0 | 0.2 ± 0.0 | <0.1 ± 0.0 | 4.5 ± 0.4 |
| 4aa16/N50Y + F83S | 34.7 ± 1.3 | 3.1 ± 0.3 | 10.8 ± 1.4 | 0.2 ± 0.1 | 0.1 ± 0.0 | <0.1 ± 0.0 | 4.8 ± 0.5 |
| 4aa16/F83S + F91Y | 30.2 ± 1.1 | 3.1 ± 0.2 | 9.3 ± 1.1 | 0.2 ± 0.1 | 0.1 ± 0.0 | <0.1 ± 0.0 | 4.5 ± 0.7 |
| 4aa16/N50Y + F91Y | 14.9 ± 0.8 | 5.8 ± 0.8 | 8.6 ± 1.7 | 0.2 ± 0.1 | 0.1 ± 0.1 | <0.1 ± 0.0 | 5.0 ± 0.7 |
| 4aa16/N50Y + F83S + F91Y | 23.7 ± 0.3 | 3.4 ± 1.2 | 8.0 ± 2.8 | 0.1 ± 0.1 | <0.1 ± 0.0 | <0.1 ± 0.0 | 4.8 ± 0.2 |
a Mutants are expressed in the format: heavy-chain/light-chain. wt indicates wild type.
Figure 2.

Antibody 57-2 mutations. In all the figures, the heavy-chain is colored purple; the light-chain, brown. The side chains of the important residues are drawn. For the other parts of the protein, only the Cα-trace or the backbone heavy atoms are shown, if not mentioned. (A) The variable domain and a part of the constant domain of the antibody 57-2. The estradiol molecule and the side chains of light-chain wild-type residues targeted by point mutations are shown. The tip of the heavy-chain CDR2 targeted by a four-amino-acid insertion is shown as cyan. (B) The insertion site in CDR2 of heavy-chain. The insertion was introduced between the residues labeled with purple balls. Only the Cα-atoms are shown, except for the tryptophans W50H and W95H, for which side chains are also shown. (C) Mutation N50LY. The side chain of the mutated residue is shown and the lettering of the steroid rings is indicated. (D) Mutation F91LY. The dotted lines represent hydrogen bonds. (E) Mutation F83LS. The van der Waals surface of the side chain of Phe83L replaced by the mutation is indicated with white dots. The blue surface shows the volume occupied by the hydrophobic side chains of the residues situated in the vicinity of the residue 83L. The light-chain constant domain atoms are drawn as CPK-models, whereas for the variable domain only Cα-trace or the backbone heavy atoms is shown. (F) The double mutation N50LY and F91LY. The white lines represent the close contacts (∼2.7 to 3.1 Å) between the ηOH of Tyr91L and the aromatic carbons of Tyr50L.
The residue 91L was targeted by a conserved mutation Phe→Tyr that caused a 2.1-fold affinity improvement toward E2 at 36°C (Table 1). According to the modeling results, Tyr91L most likely adopts the same orientation as the wild-type Phe91L, the side chain of which makes extensive contacts with the C- and D-rings of the steroid (Fig. 2A,D ▶). In the model, the ηOH of Tyr91L is situated within a hydrogen bonding distance from the backbone N of the residue Asn50L (3.0 Å) as well as from the side chain δ1O of the same residue (2.5 Å; Fig. 2D ▶).
The replacement of Phe by Ser at the position 83L had a slight but systematic positive impact on the E2-binding affinity of the antibody, increasing it ∼1.4-fold (Table 1). The residue 83L is located >20 Å away from the binding site in the loop connecting two β-strands at the bottom of the VL domain. The aromatic side chain of the wild-type Phe83L is largely buried in the hydrophobic core of the domain (Fig. 2A,E ▶), and therefore, the replacement of the bulky phenylalanine by a small serine residue generates a sizable cavity in the interior of the molecule. To analyze the effects of the mutation Phe83L→Ser on the stability of the antibody, the steroid-binding affinities of the mutant wild type/F83S and the wild-type antibody were determined in various temperatures within the temperature range of 15°C to 35°C. For both antibodies, the binding affinity decreased along the increase of the temperature in a very similar manner (data not shown).
Heavy-chain insertion
The heavy-chain mutant 4aa16 showed ∼5.5-fold higher affinity toward E2 at 36°C than did the wild-type antibody (Table 1). The mutant 4aa16 was previously isolated by phage display selection from a library of antibody 57-2 mutants with four-residue random insertions between the residues Thr52aH and Gln53H at the tip of the CDR-H2 loop (Lamminmaki et al. 1999). In addition to the insertion itself, the residues neighboring the insertion site were partially randomized in this library, and in the mutant 4aa16, the insertion Ser-Trp-Leu-Arg is surrounded by the mutations Thr52aH→Ala and Gln53H→His. In addition, the VH of 4aa16 contains two other amino acid replacements outside the randomized loop region, namely, Pro57HSer and Asn82bH→Asp. The former one is located in the stem of the CDR-H2 loop away from the binding site and can thus be considered as part of the major modification of CDR-H2. The mutation Asn82bH→Asp, in turn, targets a solvent-exposed residue located ∼25 Å away from the binding site at the bottom of the VH domain and is therefore unlikely to cause any detectable changes in the binding properties of the antibody.
Combinations of mutations
The double-mutant cycle method (Carter et al. 1984; Horovitz 1987; Horovitz and Fersht 1990) was used to investigate effects of the combinations of the mutations on the capacity of the antibody 57-2 to bind E2. Free energies of binding were deduced from the measured affinity constants by using the equation ΔG = −RTlnKa, and these were used to calculate interaction energies, ΔGint, for every pair of mutations by using the following equation expressed here for hypothetical mutations A→a and B→b: ΔGint(A→a,B→b) = ΔG(A,B) − ΔG(A→a,B) − ΔG(A,B→b) + ΔG(a,b). The magnitude of the difference of ΔGint value from zero reflects the strength of the coupling effects between the two mutations examined, and the positive and negative values of ΔGint-term indicate the negative and the positive cooperativity between the mutations, respectively. Coupling effects between a double mutation A→a, B→b and a third mutation C→c can be analyzed by calculating the difference of the ΔGint values for the double-mutant in the presence and the absence of the mutation C→c, that is, ΔΔGint(A→a,B→b;C→c) = ΔGint(A→a,B→b;C) − ΔGint(A→a,B→b;C→c). For this type of triple mutation system, corresponding thermodynamic cycles can be expressed in a form of a box (Horovitz and Fersht 1990) in which possible coupling effects between any single mutation C→c and a double-mutant A→a,B→b are reflected as a difference between the ΔGint values obtained from the mutation cycles consisting the two opposite faces of the box, that is ΔΔGint(A→a, B→b; C→c) ≠ 0 (Fig. 3 ▶).
Figure 3.
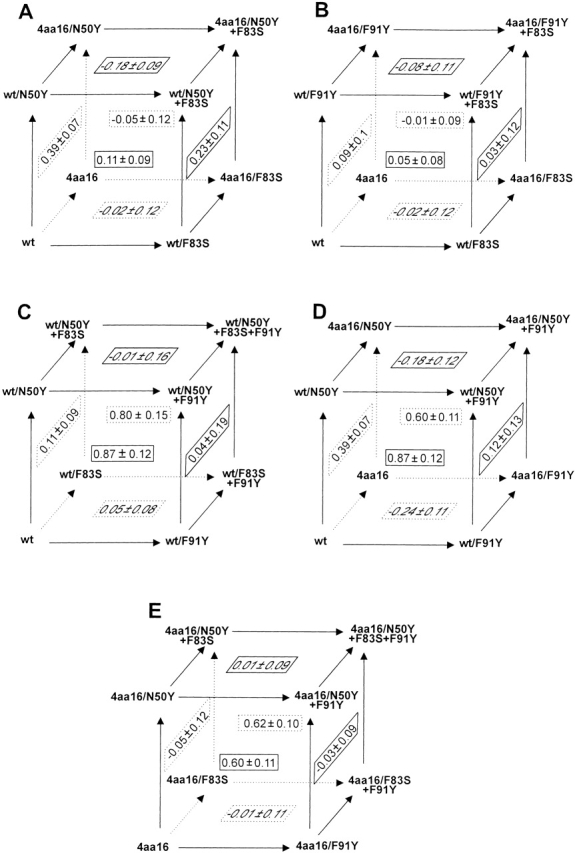
Thermodynamic cycles used to analyze the interactions between the antibody 57-2 mutations. Each face of the boxes forms a double-mutant cycle and the corresponding interaction energy (ΔGint in kcal/mole) is shown in the middle of the face.
As shown in Figure 3 ▶, the ΔGint values of the double-mutants wild type/N50Y+F83S (front face of box A) and wild type/F83S+F91Y (front face of box B) were small, indicating that the mutation Phe83L→Ser behaved additively with both other point mutations in the light-chain. In other words, the mechanism of the mutation Phe83L→Ser was not coupled with that of either of the two other point mutations.
For the variants containing both Asn50L→Tyr as well as Phe91L→Tyr mutation, the ΔGint values were significantly positive (Fig. 3C ▶,E—front and back faces of the boxes), which showed that these two binding site mutations were negatively cooperative.
The light-chain point mutations Phe83L→Ser and Phe91L→Tyr did not show coupling with the structural modifications present in the 4aa16 heavy-chain as reflected by very small ΔGint values observed for the combinations of the position 83L and 91L mutations and the heavy-chain variant, respectively. (Fig. 3B ▶, see the left and the bottom faces). However, in the cases of the combinations of the mutation Asn50L→Tyr and the 4aa16 heavy-chain, moderately positive ΔGint values were obtained (Fig. 3A ▶, left and right faces of the box).
In general, the mutants showing highest affinity toward E2 consisted of the combinations of the 4aa16 heavy-chain and the light-chain point mutations. The strongest binding mutant was the combination of the light-chain mutations Asn50L→Tyr and Phe83L→Ser and the 4aa16 type heavy-chain, and it showed ∼17.5-fold increased affinity toward E2 at 36°C as compared with that of the wild-type antibody.
Binding kinetics
To address the binding kinetics of the antibody variants, we determined the dissociation rates of the antibody/E2 complexes by using immunoassay and calculated the corresponding association rates from the equation kon = Ka × koff. The measured off-rate constants (koff) for the dissociation of the complexes varied widely and were generally longer in the higher-affinity mutants, whereas the variation of the calculated on-rate constants (kon) was much more limited among the mutants (Table 1).
Cross-reactivity
The antibody variants were also characterized for binding toward three other steroids: testosterone, estrone, and estradiol-3-sulfate (chemical structures in Fig. 1 ▶). As shown in Table 1, none of the point mutations in the light-chain caused significant changes in the cross-reactivity of either estrone or E2-3-sulfate, and the 4aa16 heavy-chain produced only a very marginal decrease of the estrone cross-reactivity compared with that of the wild type (Table 1).
Relative binding affinity of testosterone was, in turn, affected by all the mutations studied (Table 1). The 4aa16 heavy-chain alone produced ∼68-fold decrease in the testosterone cross-reactivity compared with that of the wild-type antibody, and the light-chain point mutations Asn50L→Tyr, Phe91L→Tyr, and Phe83L→Ser reduced the relative affinity toward testosterone ∼ 2.3-, 1.8-, and 1.3-fold, respectively. The best combinatorial mutants 4aa16/N50Y+F83S and 4aa16/N50Y+F83S+F91Y altogether showed 400- to 500-fold reduced cross-reactivity to testosterone.
Estradiol assay
Time-resolved fluorescence-based competitive immunoassays were established by using the mutant 4aa16/N50Y+F83S and the wild-type Fab. A series of E2 dilutions over a concentration range 50 to 5000 pM were used as calibration standards. The assay performed with the mutant 4aa16/N50Y+F83S gave a good response over the whole E2 concentration range used and resulted in the ED50 value of 270 pM (Fig. 4 ▶). The detection limit of the assay was 8 pM if the lowest detectable concentration was defined as the concentration corresponding to a signal 3 SD below the mean of the zero calibrators. CV values of the calibration standards varied between 1% and 2.4%. In the case of the wild-type antibody, no response was observed, even at the highest E2 concentrations (data not shown).
Figure 4.

Standard curve for estradiol assay. The competitive immunoassay was performed with the mutant 4aa16/N50Y+F83S.
Discussion
In this study, we systematically explored the individual and combinatorial impacts of a set of putative gain-of-function mutations in the variable domain of the anti-estradiol antibody 57-2 with the aims to understand the mechanism of the mutations and to generate antibody mutants with improved binding properties. All of the studied mutations, including three point mutations in the light-chain and a heavy-chain insertion variant, turned out to improve the affinity of the antibody toward E2, showing that the sequence and structural analysis based identification of the putative gain-of-function mutations from the multiple mutated parental clones was successful.
The previously determined crystal structure of the antibody 57-2/E2 complex (Lamminmäki and Kankare 2001) provided good basis for the modeling and visual observation–based structural analysis of functional mechanisms of the mutations. According to the three-dimensional models, the mutations Asn50L→Tyr and Phe91L→Tyr result in new steroid-antibody contacts between the mutated side chains and the steroid. Thus, their positive impact on the binding affinity is probably largely related to increased complementarity of the antibody for the antigen. This view is consistent with the fact that, for all of the mutations, the increased affinity appears to be primarily defined by the lowered dissociation rate of the antibody/antigen complex. However, the mutations can also induce changes in the structure and dynamics of the combining site in their surroundings. Thus, they might contribute to the improved affinity, for example, by stabilizing the structure of the binding site. In the case of the mutation Asn50L→Tyr, the introduction of a Tyr in the position 50L appears to result in an aromatic–aromatic stacking interaction between Tyr50L and the neighboring Tyr49L. This interaction can affect the position or mobility of the side chain of Tyr49L, which contacts widely the α-side of the A-ring of the bound steroid as well as interacts with the CDR-H3 loop through a hydrogen bond in the presence of the bound E2 (Lamminmaki and Kankare 2001). On the other hand, mutations in the surface residues likely result in solvent reorganization in the binding site, and these changes can be reflected in the binding properties of the mutants.
The fact that mutations in the variable domain but outside CDRs, which are primarily responsible for the contacts with the antigen, can affect binding properties of antibodies has been proved in several studies (Foote and Winter 1992; ; Hawkins et al. 1993; Patten et al. 1996). The effect of the mutation Phe83LSer, located >20Å from the antigen-binding site, adds to this data. The mutation Phe83LSer is particularly interesting in a sense that the replacement of the aromatic phenylalanine by a small serine residue generates a sizable cavity in the interior of the molecule. Cavities inside protein cores are generally considered to be structure-destabilizing (Eriksson et al. 1992; Matthews 1995), which can be reflected, for example, in increased sensitivity for elevated temperature or chaotropes. However, when analyzed for E2 binding, the mutant wild type/F83LS showed very similar exothermic behavior within the temperature range 15°C to 35°C as the wild-type antibody, indicating that the stability of the mutant wild type/F83LS is not considerably compromised at least within this relatively narrow, but in practical-sense important, temperature range.
The prediction of the functional mechanism of the mutation Phe83LSer is very difficult due to small magnitude of its effect on affinity and the distal location of the residue F83L from the binding site. It is possible that the mechanism of this mutation is based on the fact that the cavity forming mutations in the protein core typically cause compensating structural rearrangements (Eriksson et al. 1992; Xu et al. 1998). Such modifications might be reflected further in the protein structure and, in the case of the mutation Phe83LSer, affect the binding site region of the antibody. On the other hand, the crystal structure of the antibody 57-2 shows that residue 83L interacts with Val106L that seems to be one of the residues intermediating contacts between the VL and CL domains (Fig. 2E ▶). It is possible that the mutation Phe83LSer causes changes in the interaction of CL and VL, which in turn may affect the properties of the antibody in a similar manner as mutations in the ball-and-socket joint in between VH and CH (Lesk and Chothia 1988; Landolfi et al. 2001).
It is not straightforward to assess the structural basis of the 5.5-fold improved affinity of the variant 4aa16 because the stretch of four additional residues at the tip of the β-hairpin type loop, lined with another two mutated residues, can in theory adopt numerous different conformations, and the reliability of the structural predictions for this type of segments is fairly limited. Nevertheless, the visual analysis of the crystal structure of the Fab 57-2/E2 complex indicated that the bulky side chain of Trp50H in the stem of CDR-H2 probably prevents direct interactions between the inserted residues and the steroid (Fig. 2A,B ▶). Therefore, the positive impact of the extended CDR-H2 on the affinity is most likely mediated through the original contact residues, especially Trp95H, which alone accounts for the majority of contacts from the VH to the β-side of the steroid (Fig. 2B ▶).
If combined, mutations can behave in either an additive or nonadditive manner; that is, the functions of the individual mutations are either independent or coupled. Nonadditive effects between mutations can result from two types of mechanisms: (1) The mutated residues interact with each other by direct contacts or indirectly through structural perturbations or electrostatic effects, and (2) the mutations cause changes in the reaction mechanism of the protein (Wells 1990). On the basis of the double-mutant cycle analysis, most of the combined mutations studied were directly additive. However, a clear coupling effect was observed between the mutations Asn50L→Tyr and Phe91L→Tyr. This phenomenon can be explained by modeling data that indicate that the most favorable side-chain rotamers of Tyr50L and Tyr91L clash each other (Fig. 2F ▶). Moreover, the mutation Asn50L→Tyr eliminates the proton acceptor group (Asn50L δ1O) for the ηOH of Tyr91L, abolishing the hydrogen bonds that the ηOH of Tyr91L, according to the modeling, forms with both δ1O as well as the backbone N of the residue Asn50L when the mutation Phe91L→Tyr exists alone (Fig. 2D,F ▶). In the absence of these hydrogen bonds, the side chain of Tyr91L cannot adopt the same rotamer as the wild-type Phe91L because of the too short interatomic distances (∼3 Å) between its ηOH and the main chain of residue 50L. Thus, the antibody cannot adapt to the double mutation by changing the rotamer of Tyr50L, but changes in the position of Tyr91L, making important contacts with the steroid, take place in any case.
In addition to the combination of the mutations Asn50L→Tyr and Phe91L→Tyr, a moderate interference between the 4aa16 heavy-chain and the mutation Asn50L→Tyr was observed. According to the three-dimensional structure of the antibody, Tyr50L and the extended CDR-H2 cannot contact each other, and therefore, the coupling effect is probably mechanism-based, being possibly caused by slight changes in the position of the steroid as a consequence of the mutations.
To probe the effects of the mutations on the binding specificity, we analyzed the cross-reactivities of the antibody variants toward three steroids: estrone, E2-3-sulfate, and testosterone. Estrogens have two functional groups attached to carbons 17 (C17) and 3 (C3) of steroid skeleton, and estrone and E2-3-sulfate differ from E2 in these positions, respectively (Fig. 1 ▶). The previous crystal structure-based analysis revealed that the low cross-reactivity to estrone could be attributed to the 17-keto group which, unlike 17-OH in estradiol, could not form a strong four-center hydrogen bond with the antibody (Lamminmäki and Kankare 2001). The rather high cross-reactivity to E2-3-sulfate (∼5%), in turn, was possibly because the orientation of the steroid in the binding pocket allowed the bulky sulfate group to protrude out into the solvent. For all antibody variants studied here, the relative binding affinities toward both estrone and E2-3-sulfate were almost the same as in the case of the wild-type antibody. This is not surprising because the modeling indicates that any of the mutations does not directly contact either the position C3 or C17 of the steroid. In concert with the fact that these two steroids differ from E2 in the opposite ends of the steroid skeleton, these data imply that none of the mutations causes large changes in the position of the steroid. Considering the use of antibody in clinical E2-immunoassay applications, its selectivity between E2 and estrone (as well as estriol) is excellent; however, the poor discrimination capacity around the position C3 might be a potential source for specificity problems because the position C3-attached sulfates and glucuronides are natural metabolites of E2.
All the mutations decreased cross-reactivity of the antibody toward testosterone that differs from E2 mainly in the A-ring. As in the case of E2 affinity, most of the mutations were positively cooperative when combined, and dramatically decreased cross-reactivities, being, at best, 450-fold lower than that of the wild type, were observed for the combinatorial mutants. Very low testosterone cross-reactivity is essential for an anti-E2 antibody intended for clinical immunoassays because, even in female in fertile age, the concentration of testosterone is generally significantly higher than that of E2. The inspection of the affinity constants for the antibody/testosterone interaction showed that the mutations Asn50L→Tyr and Phe91L→Tyr actually somewhat improved the absolute affinity of the antibody toward testosterone when combined with the wild-type heavy-chain (Table 1). When either of these two mutations was combined with the 4aa16 type heavy-chain, affinity improvement (compared with 4aa16) was not observed or it was very slight, indicating negative cooperativity in the testosterone recognition between the 4aa16 heavy-chain and the point mutations Asn50L→Tyr and Phe91L→Tyr. A possible explanation for this behavior is that in the presence of the light-chain point mutations (Asn50L→Tyr and Phe91L→Tyr) or the heavy-chain 4aa16 alone, there is still some extra space in the binding pocket that facilitates the accommodation testosterone by allowing small changes in the position/orientation of the steroid. However, when both the light-chain and the heavy-chain are mutated, the binding pocket is tightened on two sides and the movement-aided binding of testosterone is impeded. The large decrease in the testosterone cross-reactivity by the 4aa16 heavy-chain is interesting because, according to crystal structure of the antibody/E2 complex, the extended CDR-H2 cannot reach to contact the A-ring of the steroid. These data also support the surmise that the effects of the modified CDR-H2 loop of the 4aa16 variant are largely indirect and mediated through other binding site residues.
We analyzed the capability of the highest affinity mutant 4aa16/N50Y+F83S to detect low E2 concentrations in a time-resolved fluorescence-based immunoassay. The assay performed with the mutant produced a good response over the E2 concentration range, corresponding to that generally used in the current clinical E2-immunoassays, whereas the wild type failed to respond even to the calibration standard of the highest concentration. These results clearly demonstrate the improved performance of the modified antibody over the wild type. The combinatorial mutants of the antibody 57-2 with improved affinity and considerably decreased testosterone cross-reactivity generated in this study can be useful for the applications that aim to the measurement of E2 concentrations, and in any case, they provide for good basis for the further protein engineering experiments aiming to the development of high quality monoclonal anti-E2 antibodies.
Materials and methods
Strains and plasmids
The strain Escherichia coli XL-1 Blue (recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1, lac [F′ proAB, lacIqZΔM15, Tn10 {Tetr}]; Stratagene) was used as a host for cloning as well as for the recombinant Fab expressions. The plasmid pCombIII (Barbas et al. 1991), originally intended for the phage display usage but from which the phage pIII-gene had been removed, was used as an expression vector.
DNA manipulation
The light-chain point mutations were introduced in both the wild-type Fab and the Fab variant 4aa16 containing a mutated heavy-chain by using the commercial site-directed mutagenesis kit QuikChange (Stratagene) according to the manufactorer’ instructions. The mutagenizing primers were purchased from MedProbe or were synthesized in the in-house facility by using Applied Biosystems (ABI) 392 DNA/RNA Synthesizer. The sequences of the primers were as follows: for the mutation Asn50L→Tyr, 5′-CAGCTCCTGGTCTATTATGCAAAAACCTTAGCAG-3′ and 5′-CTGCTAAGGTTT-TTGCATAATAGACCAGGAGCTG-3′; for the mutation Phe83L→Ser, 5′-AATAGTCTGC-AGCCTGAAG ATTCTGGGACTTATTATTGTC-3′ and 5′-GACAATAATAAG TC-CCAGAATCTTCAGGCTGCAGACTATT-3′; and for the mutation Phe91L→Tyr, 5′-CTTATTA-TTGTCACCATTATTGG AGTACTCCGTG-3′ and 5′-CACGGAGTACTCCAATA-ATG GTGACAATAATAAG-3′. The success of the mutagenesis was verified by DNA sequencing by using ABI PRISM 377 dye terminator cycle sequencer. In some cases, the desired mutations appeared to be present only either in the wild-type antibody or in the variant 4aa16. Here, the missing mutants were generated by cutting loose the successively mutated light-chain from the plasmid with the restriction enzymes SacI and XbaI (Promega) and ligating then the fragment, after gel purification, to the desired vector digested with the corresponding restriction enzymes.
Molecular modeling
The light-chain point mutations were introduced in the crystal structure of the Fab 57-2/estradiol complex (Lamminmäki and Kankare 2001) by replacing the original amino acid by the mutated one using the program InsightII (Accelrys). For each of the mutated residues, several different side-chain rotamers were considered, and the energetically most favorable one was adopted in the model (in all cases, this was same rotamer as in the wild-type residue). The program InsightII was also used for the visual inspection of the structures and for the generation of Figure 2 ▶.
Fab production
The antibody mutants were expressed as Fab fragments into the periplasmic space of the E. coli cells in small-scale cultures. The cells were grown in 4 mL of SB medium (30 g/L Tryptone, 20 g/L yeast extract, and 10 g/L, 4-(N-Morphdino) butane sulfonic acid (MOPS)-buffer at pH 7.0) supplemented with 0.2% glucose, 100 μg/mL ampicillin, and 10 μg/mL tetracycline at 37°C, 300 rpm. When an O.D.600 of 0.8 was achieved, the expression of the recombinant protein was induced by the addition of 100 μM isopropyl-β-D-thio-galactopyranoside (IPTG), and after that, the cells were grown for additional 5 h at 30°C and 240 rpm. The cells were harvested by centrifugation (4000g, 10 min at 4°C), 1 mL ice cold Delfia assay (Perkin-Elmer Wallac) buffer was added, and the cells were broken by sonication to release the Fab from the periplasm. The cell extract was divided in 50 μL aliquots, which were stored at −20°C.
Europium-labeled estradiol derivatives
Measurement of the concentrations of the expressed Fab fragments and analysis of binding properties of the Fabs were performed with DELFIA technology (Perkin-Elmer Wallac) based time-resolved immunofluorometric microplate assays. Two different labeled E2 derivatives were used as tracers in the assays: E2-6-CMO-N1[Eu] was used for the concentration measurements; E2-4-CET-N1[Eu], for the determination of the binding properties of the Fabs. The tracers were obtained as gifts from the H. Mikola (Perkin-Elmer Wallac). E2-6-CMO-N1[Eu] was made by labeling the 6-oxoestradiol 6-(O-carboxymethyl)-oxime derivative with the europium chelate N1-(p-aminobenzoyl)-diethyleneamine-N1,N2,N3,N3-tetraacetic acid (Mukkala et al. 1989), using the method described by Mikola et al. (1993). The analog E2-4-CET-N1[Eu] was synthesized as described in the study by Meltola et al. (1999).
Fab determination
Concentrations of the Fab fragments in the crude bacterial extracts were determined with immunoassays by using a purified wild-type Fab 57-2 as a calibration standard and the labeled estradiol E2-6-CMO-N1[Eu] as a tracer. First, 2 nM tracer E2-6-CMO-N1[Eu] in the volume of 200 μL was pipetted into rabbit antimouse IgG-coated microtiter wells (RAM) followed by the addition of 2 μL of the sample or standard. The reactions were incubated for 60 min with shaking 900 rpm at room temperature, and after washing the wells, 200 μL DELFIA Enhancement solution was added. After shaking the reactions for 15 min, the fluorescence was measured by using a plate fluorometer Victor 1420 Multilabel Counter (Perkin-Elmer Wallac) controlled by the program Multicalc, version 2.6, which automatically produced a spline-fitted standard curve and calculated the Fab concentrations in the samples.
Ka determinations for tracer and free steroids
The affinities of the antibody mutants toward the tracer E2-4-CET-N1[Eu] were determined with a time-resolved fluoroimmunoassay by using a hot saturation assay procedure. First, each Fab fragment was immobilized on RAM microtiter strips by incubating 2 ng of Fab in 200 μL DELFIA assay buffer in the wells for 2 h at 25°C. Then the wells were washed, and the tracer was added in several different concentrations for each Fab, ranging from 100 to 5000 pM for the Fabs having the wild-type heavy-chain and from 20 to 1000 pM for the Fabs having the 4aa16 heavy-chain. After that, the wells were washed, enhancement solution was added, and the fluorescence was measured as described above. The measured signals were converted to the molar concentrations of the bound tracer, and the tracer binding affinities were calculated with the program RADLIG of the KELL package (version 5.0.2, Biosoft).
The influence of the temperature on the steroid-binding affinity was analyzed by using the same labeled steroid (E2-4-CET-N1[Eu]) and similar hot saturation procedure as above with the following differences: MOPS buffer (pH 7.2) was used instead of Tris-based DELFIA assay buffer in order to avoid the large temperature dependence of pH in Tris buffer, and the incubations for the antibody-steroid binding reaction were performed at five different temperatures (15°C, 20°C, 25°C, 30°C, and 35°C). The measured Ka were used to construct a van’t Hoff plot; that is, the lnKa was plotted against 1 / T.
For the affinities of unlabeled steroids, a competitive assay was used. The unlabeled steroids were repeatedly diluted in a 2 nM tracer (E2-4-CET-N1[Eu]) solution to give a range of final concentrations of the unlabeled steroid and a fixed concentration of the labeled E2. The ranges of the unlabeled steroid concentrations used were the following: from 0 to 20 nM estradiol, from 0 to 0.9 nM testosterone for Fabs with wild-type heavy-chain and 0 to 9 nM testosterone for the Fabs with 4aa16 VH, from 0 to 1.5 μM E2-3-SO4, and from 0 to 50 μM estrone. Each member of the series was assayed for the binding to the Fabs preimmobilized in the RAM wells as described above. The steroids were allowed to react with the antibodies for 4 h under shaking of 900 rpm. After the incubation, the strips were washed, and the fluorescence was measured as above. The program RADLIG was used for the analyses of the steroid-binding affinities. The cross-reactivity was calculated by dividing the Ka for the cross-reacting steroid by the Ka for E2.
Determination of off-rates for Fab/E2 complexes
To measure the dissociation rates for Fab/E2 complexes, the Fab fragments were first immobilized on the RAM strips as described above. After washing the strips, 200 μL of 20 nM E2 solution was added, and the strips were incubated for 30 min at 36°C with shaking 900 rpm to saturate the steroid-binding sites. To start the off-rate detection reaction, the strips were washed again, and 200 μL of 5 nM tracer solution that was prewarmed to 36°C was added. The reactions were stopped at varying time points by removing individual strips of wells and washing them. The incubation times depended on the Fab variant and varied from 2 to 128 min. The fluorescence signals were measured from the individual strips as described above and were converted to the molar concentrations of the bound tracer. The dissociation rate constants were calculated with the program Excel 97 (Microsoft) and were obtained from the slopes of the following plots: the values from the equation ln(1 − B / Bmax) versus time. The B stands for the concentration of the bound E2, and Bmax indicates the total concentration of binding sites obtained from the reaction from which free E2 was omitted.
Estradiol immunoassay
To test the performance of the mutated antibody 57-2 in an immunoassay-based estradiol measurement, a competitive immunoassay was set up by using the high-affinity mutant 4aa16/N50Y+F83S as well as the wild-type antibody. Two nanograms of antibodies in volume of 100 μL were immobilized into RAM strips in 2-h incubation at 25°C with shaking at 900 rpm. The wells were washed, and a series of E2 dilutions, including of the concentrations 0, 0.05, 0.15, 0.5, 1.5, and 5 ng/mL in DELFIA assay buffer, were added in volume of 100 μL, followed by a 4-h incubation at 25°C. Then, the wells were washed again and 100 μL of 5 nM tracer E2-4-CET-N1[Eu] was added, followed by a 15-min incubation at 25°C, 900 rpm, and another wash. Enhancement solution was added, and the fluorescence signal was measured as described above. Measured signals were converted to the molar concentrations of the bound tracer, and a sigmoidal standard curve was fitted to the data by using the program Excel 97 (Microsoft) expanded with the Life Science Workbench Data Analysis Toolbox (MDL) and using the algorithm y = (a − d) / [1 + (x / c)b] + d, where a and d correspond to the maximum and the minimum values of the response, respectively; b is the slope; and c is the ED50 value.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
E2, 17β-estradiol
CDR, complementarity determining region
CL, light-chain constant domain
VH, heavy-chain variable domain
VL, light-chain variable domain
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0353903.
References
- Barbas III, C.F., Kang, A.S., Lerner, R.A., and Benkovic, S.J. Assembly of combinatorial antibody libraries on phage surfaces. The gene III site. Proc. Natl. Acad. Sci. 88 7978–7982. [DOI] [PMC free article] [PubMed]
- Carter, P.J., Winter, G., Wilkinson, A.J., and Fersht, A.R. 1984. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38 835–840. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Foote, J. and Winter, G. 1992. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 224 487–499. [DOI] [PubMed] [Google Scholar]
- Hawkins, R.E., Russell, S.J., Baier, M., and Winter, G. 1993. The contribution of contact and non-contact residues of antibody in the affinity of binding to antigen: The interaction of mutant D1.3 antibodies with lysozyme. J. Mol. Biol. 234 958–964. [DOI] [PubMed] [Google Scholar]
- Horovitz, A. 1987. Non-additivity in protein–protein interactions. J. Mol. Biol. 196 733–735. [DOI] [PubMed] [Google Scholar]
- Horovitz, A., and Fersht, A.R. 1990. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214 613–617. [DOI] [PubMed] [Google Scholar]
- Lamminmaki, U. and Kankare, J.A. 2001. Crystal structure of a recombinant anti-estradiol Fab fragment in complex with 17β-estradiol. J. Biol. Chem. 276 36687–36694. [DOI] [PubMed] [Google Scholar]
- Lamminmaki, U., Villoutreix, B.O., Jauria, P., Saviranta, P., Vihinen, M., Nilsson, L., Teleman, O., and Lovgren, T. 1997. Structural analysis of an anti-estradiol antibody. Mol. Immunol. 34 1215–1226. [DOI] [PubMed] [Google Scholar]
- Lamminmaki, U., Pauperio, S., Westerlund-Karlsson, A., Karvinen, J., Virtanen, P.L., Lovgren, T., and Saviranta, P. 1999. Expanding the conformational diversity by random insertions to CDRH2 results in improved anti-estradiol antibodies. J. Mol. Biol. 291 589–602. [DOI] [PubMed] [Google Scholar]
- Landolfi, N.F., Thakur, A.B., Fu, H., Vasquez, M., Queen, C., and Tsurushita, N. 2001. The integrity of the ball-and-socket joint between V and C domains is essential for complete activity of a humanized antibody. J. Immunol. 166 1748–1754. [DOI] [PubMed] [Google Scholar]
- Lesk, A.M. and Chothia, C. 1988. Elbow motion in the immunoglobulins involves a molecular ball-and-socket joint. Nature 335 188–190. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1995. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 46 249–278. [DOI] [PubMed] [Google Scholar]
- Meltola, N., Jauria, P., Saviranta, P., and Mikola, H. 1999. Synthesis of novel europium-labeled estradiol derivatives for time-resolved fluoroimmunoassays. Bioconjug. Chem. 10 325–331. [DOI] [PubMed] [Google Scholar]
- Mikola, H., Sundell, A.C., and Hanninen, E. 1993. Labeling of estradiol and testosterone alkyloxime derivatives with a europium chelate for time-resolved fluoroimmunoassays. Steroids 58 330–334. [DOI] [PubMed] [Google Scholar]
- Mukkala, V.M., Mikola, H., and Hemmila, I. 1989. The synthesis and use of activated N-benzyl derivatives of diethylenetriaminetetraacetic acids: Alternative reagents for labeling of antibodies with metal ions. Anal. Biochem. 176 319–325. [DOI] [PubMed] [Google Scholar]
- Pajunen, M., Saviranta, P., Jauria, P., Karp, M., Pettersson, K., Mantsala, P., and Lovgren, T. 1997. Cloning, sequencing, expression and characterization of three anti-estradiol-17β Fab fragments. Biochim. Biophys. Acta 1351 192–202. [DOI] [PubMed] [Google Scholar]
- Patten, P.A., Gray, N.S., Yang, P.L., Marks, C.B., Wedemayer, G.J., Boniface, J.J., Stevens, R.C., and Schultz, P.G. 1996. The immunological evolution of catalysis. Science 271 1086–1091. [DOI] [PubMed] [Google Scholar]
- Saviranta, P. 2001. Immunoassay improvement by molecular engineering. In Annales Universitatis Turkuensis (series). Turku University, Turku, Finland.
- Saviranta, P., Pajunen, M., Jauria, P., Karp, M., Pettersson, K., Mantsala, P., and Lovgren, T. 1998. Engineering the steroid-specificity of an anti-17β-estradiol Fab by random mutagenesis and competitive phage panning. Protein Eng. 11 143–152. [DOI] [PubMed] [Google Scholar]
- Wells, J.A. 1990. Additivity of mutational effects in proteins. Biochemistry 29 8509–8517. [DOI] [PubMed] [Google Scholar]
- Xu, J., Baase, W.A., Baldwin, E., and Matthews, B.W. 1998. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 7 158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]