

Abstract
Amphipathic helices in membrane proteins that interact with the hydrophobic/hydrophilic interface of the lipid bilayer have been difficult to structurally characterize. Here, the backbone structure and orientation of an amphipathic helix in the full-length M2 protein from influenza A virus has been characterized. The protein has been studied in hydrated DMPC/DMPG lipid bilayers above the gel to liquid-crystalline phase transition temperature by solid-state NMR spectroscopy. Characteristic PISA (Polar Index Slant Angle) wheels reflecting helical wheels have been observed in uniformly aligned bilayer preparations of both uniformly 15N labeled and amino acid specific labeled M2 samples. Hydrogen/deuterium exchange studies have shown the very slow exchange of some residues in the amphipathic helix and more rapid exchange for the transmembrane helix. These latter results clearly suggest the presence of an aqueous pore. A variation in exchange rate about the transmembrane helical axis provides additional support for this claim and suggests that motions occur about the helical axes in this tetramer to expose the entire backbone to the pore.
Keywords: Influenza A, membrane protein structure, amphipathic helices, M2 proton channel, solid-state NMR, PISEMA, PISA wheel, hydrogen–deuterium exchange
Numerous amphipathic helical peptides have been structurally characterized and associated with the interfacial region in lipid bilayers (Bechinger et al. 1998; Marassi et al. 1999; Huang 2000; Han et al. 2001; Chou et al. 2002), but few protein structural characterizations have resolved such helical domains on the surface of a membrane mimetic environment. Rhodopsin, the KcsA, and KirBac1.1 K+ channels, and the Escherichia coli outer membrane protein, PagP, are exceptions (Palczewski et al. 2000; Cortes et al. 2001; Hwang et al. 2002; Kuo et al. 2003). Indeed, modeling the membrane environment for structural studies is very challenging. Not only is there a hydrophobic domain that must be generated, but also a planar interfacial region, which has been shown to have a very substantial thickness (Wiener and White 1992). Here, using uniformly aligned lipid bilayers, an amphipathic helix in M2 protein from Influenza A virus has been initially characterized by solid-state NMR spectroscopy using PISA (polar index slant angles) wheels (Marassi and Opella 2000; Wang et al. 2000).
M2 protein occurs as a terameric protein (Holsinger and Lamb 1991; Pinto et al. 1997; Sakaguchi et al. 1997) in the viral coat where it is a pH activated H+ channel. This 97- residue protein has a 24-residue N-terminal segment, a 54-residue C-terminal domain, and a single transmembrane 19- residue α-helix. The backbone structure of a 25-residue tetrameric peptide from M2, spanning the hydrophobic domain has recently been solved (Nishimura et al. 2002), and an initial PISA wheel analysis of the transmembrane helix in the full-length protein has also been achieved (Tian et al. 2002). In this article it was shown that structurally homogeneous preparations of the M2 protein was achieved in both detergent and lipid bilayer environments by solution and solid-state NMR of amino acid specific-labeled M2 protein in which single site resonances were observed. In particular, the results from solid-state NMR of uniformly aligned samples were important, because conformations with orientations for specific 15N sites that differed by as little as 3 or 4° would give rise to multiple resonances. The native tetrameric structure is either a pair of disulfide-linked (Cys17, Cys19) dimers or a disulfide-linked tetramer, but here and in previous studies Cys 19 has been mutated to a serine such that disulfide-linked tetramers are not possible. However, a noncovalent tetrameric structure has been shown by gel electrophoresis (Tian et al. 2002), and recent solution NMR data shows both amantadine and Cu2+ binding to our protein construct (F. Gao, C. Tian, and T. Cross, unpubl.). The M2 protein facilitates the uncoating of the endocytosed virus, and later in the life cycle it functions to modulate the pH of the trans-Golgi network (Lamb et al. 1985; Grambas et al. 1992). Electrophysiological evidence for the ion channel activity has been obtained by expression in Xenopus laevis oocytes (Lamb et al. 1985; Holsinger et al. 1994), in mammalian cells (Wang et al. 1994; Chizhmakov et al. 1996), and through reconstitution in synthethic bilayers (Tosteson et al. 1994; D. Busath, V. Vijayvergiya, F. Gao, and T. Cross, unpubl.).
Solid-state NMR is the spectroscopy of samples that do not undergo isotropic motions on the spin interaction timeframe (e.g., 15N-1H dipolar, 50 μsec). Here, such NMR studies were carried out on hydrated lipid bilayer preparations in the Lα liquid crystalline phase. This spectroscopy is unique among structural methods in being able to characterize membrane proteins in a native-like environment.
There are numerous ways in which structural restraints can be obtained from this form of spectroscopy including distance, torsional, and orientational restraints (Fu and Cross 1999). These latter restraints are obtained from samples that are uniformly aligned with respect to the magnetic field. A film of hydrated bilayers approximately 100 μm thick can be annealed above the gel to liquid crystalline phase transition to form extensive bilayers with uniform alignment. The mosaic spread based on the spectroscopy is on average less than 3°, and in the best of cases, less than a few tenths of a degree (Hu et al. 1993). Alignment is needed only with respect to one axis of the laboratory frame, the magnetic field direction. With such alignment, anisotropic spin interactions having a P2cosθ orientational dependence with respect to the field can be assessed. Because 15N-1H, 13C-1H, 15N-13C dipolar, and 15N and 13C chemical shift interactions have well-known tensor element magnitudes and tensor orientations to the covalent geometry of the polypeptide backbone (Teng et al. 1992; Mai et al. 1993; Wang et al. 2000), it is possible to convert these NMR observables into structural restraints (Cross and Quine 2000; Quine and Cross 2000). In other words, the observed resonance frequencies are consistent with a limited range of orientations for the observed atomic sites. The complete 3D structure of gramicidin A has been solved using orientational restraints (Ketchem et al. 1993, 1997; PDB #1MAG), and likewise the structure of the polypeptide backbone of the transmembrane peptide of M2 protein has also been achieved (Wang et al. 2001; PDB #1MP6).
Moreover, resonance patterns are evident in the solid-state NMR spectra of aligned α-helices when 15N chemical shift/15N-1H dipolar correlated PISEMA (Polarization Inversion Spin Exchange at the Magic Angle) spectra are obtained (Marassi and Opella 2000; Wang et al. 2000). The resonance patterns for helices tilted by 10° to 50° with respect to the bilayer normal are very similar to helical wheels, because the orientation dependence for the spin interaction tensor elements is the same for the chemical shift and dipolar interactions and because the tensors in the peptide plane are not collinear. The position, size, and shape of these patterns within the spectrum are all dependent on the tilt of the helix. Consequently, the tilt angle can be determined without the need for resonance assignments. In addition, the rotational orientation of the helix can be determined with only minimal resonance assignments.
Here, PISA wheels are used for nontransmembrane α-helices that have larger tilt angles with respect to the bilayer normal. Because there is a plane of reflection for the P2cosθ terms perpendicular to the field, the PISA wheel patterns fold over, such that at a 90° tilt angle a full rotation around the pattern is achieved by half a turn about the helical wheel. At 80° this symmetry is broken, forming a complex pattern that is readily modeled for characterizing the helical tilt and rotation angles from the observed resonances for surface bound amphipathic helices.
The recognizable presence of PISA wheels in PISEMA spectra occurs only when helices are very regular in structure. Fortunately, the low dielectric environment and lack of a significant water concentration results in enhanced electrostatic interactions leading to shortened hydrogen bonds and nearly ideal α-helical structures (Kim and Cross 2002), at least for the transmembrane helices. In addition, amphipathic helices that lie approximately parallel to the plane of the bilayer may be positioned at various depths within the bilayer depending on the degree of hydrophobicity and the nature of the hydrophilic residues.
Structural characterization for amphipathic helices that interact with the lipids in the plane of the bilayer has been more commonly achieved for peptides. Peptide toxins are typically amphipathic helices that under specific conditions of pH, concentration, voltage, lipid composition, etc., are induced to form pores (Huang 2000). Models of such pores include the barrel stave and toroidal models, where the former stabilizes the planar lipid bilayer environment and the latter stabilizes a highly curved lipid pore (Yang et al. 2001). Amphipathic helices are also associated with fusion peptides (Han et al. 2001). In membrane proteins, amphipathic helices can be associated with the formation of channels, although the hydrophobicity moment is quite low compared to amphipathic helices that lie approximately parallel to the bilayer surface at the boundary between the hydrophobic domain and the interfacial region. Few such helices have been characterized, in part due to the difficulty in crystallizing them (Cortes et al. 2001), although a couple of bilayer surface amphipathic helices have been crystallized from Rhodopsin (Palczewski et al. 2000) and the KirBac1.1 K+ channel (Kuo et al. 2003). For rhodopsin this helix (VIII) is weakly hydrophobic as an isolated peptide and only forms a helix in the presence of a bilayer environment (Krishna et al. 2002), a property that may not be uncommon (Oh et al. 1999; Zakharov et al. 1999). In addition to binding transducin, this amphipathic helix acts as a membrane-dependent conformational switch domain. Other amphipathic helices have been characterized from solid-state NMR (Bechinger et al. 1998; Marassi et al. 1999), solution NMR (Han et al. 2001; Chou et al. 2002; Hwang et al. 2002) and EPR spectroscopy (Oh et al. 1996; Altenbach et al. 2001; Cortes et al. 2001).
Results and Discussion
Simulated PISA wheel patterns for α-helices at different tilt angles (τ) to the external magnetic field direction, B0, and the bilayer normal are shown in Figure 1 ▶. These simulations are achieved by calculating the 15N anisotropic chemical shift and the 15N-1H dipolar observables for different positions around an α-helical axis (ρ). Helices have a repeat unit of a single amino acid residue, and the transformation between these repeat units includes a rotation by 100° and a translation in the helical axis direction of approximately 1.5Å. The 15N chemical shift and 15N-1H dipolar interaction tensors are fixed with respect to the molecular frame, and therefore, a pattern is generated in the PISEMA spectra for regular helical structures in uniformly aligned samples. These PISA wheels reflect helical wheels with 3.6 resonances per turn, and hence, the 100° rotation between sequential resonances. The position of the pattern, its shape, and size are all dependent on the tilt angle (Marassi and Opella 2000; Wang et al. 2000). The PISA wheels for the two seperate dipolar transitions form mirror images about the νobs = 0 kHz axis. Because of this symmetry only half of the PISEMA spectrum will be shown in later figures, but here a series of PISA wheels for tilt angles of 5°, 25°, 45°, 65°, and 90° are shown including both transitions. Note that the two different dipolar transitions give rise to wheels of opposite handedness, because the two halves of the spectrum represent a mirror image. Therefore, in viewing just half of the spectrum, the handedness for the PISA wheels at opposite ends of the chemical shift range will be different, because the dipolar transitions displayed at the two ends of the chemical shift range are different.
Figure 1.

PISA wheels. (A) Definitions of the tilt angle τ, and rotational angle ρ, for an α-helix are shown, as well as a simulated PISA wheel for a 25° tilt angle that shows a right-handed pattern of correlations between sequential resonances. The tilt angle τ = 0° is defined when the helix axis (hz) is parallel to the static magnetic field B0 direction. The rotational angle ρ = 0° is defined when the Cα atom of a residue is in the B0/hz plane. (B) Simulated PISA wheels of ideal α-helices with a series of tilt angles: 5°, 25°, 45°, 65°, and 90°.
PISEMA spectra of uniformly 15N-labeled M2 in aligned DMPC/DMPG planar bilayers are shown in Figure 2 ▶. The protein has a single transmembrane helix generating the majority (but not all) of the resonances in the 140–200 ppm chemical shift range. These resonances suggest a PISA wheel representing a helical tilt of 25 ± 3° with respect to the bilayer normal (Tian et al. 2002). Although these resonances do not lie exactly on the theoretical wheel, there are numerous reasons for minor variations in the chemical shift (±10 ppm) and dipolar interaction (±1 kHz) that include local variations in structure and variations in tensor element magnitudes and orientations to the molecular frame. Moreover, several of the resonances are likely not to be part of the transmembrane helix, but are resonances from residues in loops and 15N sites in side chains.
Figure 2.

PISEMA spectra obtained at 30°C and 40 MHz for 15N before and after exposure of a uniformly 15N labeled M2 protein sample in uniformly aligned DMPC/DMPG lipid bilayers to a D2O atmosphere. (A) Spectrum of an M2 protein (12 mg) sample before hydrogen-deuterium exchange. (B) Spectrum of the same M2 protein sample after exposure to a D2O atmosphere at 43°C for 8 h. (C) Spectrum of the M2 protein sample after exposure of the sample to a D2O atmosphere for an additional 16 h at 43°C and following the direct addition of a D2O aliquot to the sample chamber. Both 25° and 80° PISA wheels have been superimposed on the spectra where the contours are separated on an exponential scale.
Because this protein is largely α-helical (CD spectra indicate 67% in micelles; Tian et al. 2002), many of the resonances in the 40–100 ppm region will be associated with helices having tilt angles near 90°. These could be amphipathic helices in the bilayer interfacial region. There is also likely to be some intensity in this region of the spectrum from portions of the sample that are not uniformly aligned with respect to B0.
Spectra in Figure 2 ▶, B and C, reflect residual resonances following hydrogen/deuterium exchange. The sample used for the spectrum in Figure 2A ▶ was opened and exposed to a saturated D2O atmosphere for 8 h before being resealed for recording the spectrum presented in Figure 2B ▶. Similarly, for Figure 2C ▶ the sample was again opened, extensively exposed to a D2O atmosphere, and 20 μL of D2O was directly added to the sample chamber before resealing. Interestingly, the transmembrane helical amide protons (resonances in the region of 140 to 210 ppm) completely exchange with D2O before a number of amide sites represented by resonances in the 40 to 100 ppm region of the spectrum exchange. Clearly, the transmembrane helix is exposed to water; if not, the exchange rate for the transmembrane region would be very slow due to the low dielectric of this environment and very low concentration of water in the membrane interstices. This structure is known to be tetrameric in the membranes of oocytes (Sakaguchi et al. 1997) and also known to be tetrameric in our preparations through tricine gels (Tian et al. 2002). Our preparations have also been shown in electrophysiological experiments to form conducting channels (D. Busath, V. Vijayvergiya, F. Gao, and T. Cross, unpubl.). Furthermore, structural studies of the transmembrane peptide have shown it to form a tetrameric bundle and an aqueous pore (Nishimura et al. 2002). However, an aqueous pore alone does not expose the entire transmembrane helix to water.
The distribution of resonances in the transmembrane region following partial exchange (Fig. 2B ▶) suggests that the resonances in the “top half” of the PISA wheel have exchanged more rapidly than those in the “bottom half.” In other words, there appears to be preferential exchange of amides from a particular surface of the transmembrane helix. Recall that the observed PISA wheel reflects a helical wheel, such that a helical surface can be associated with a sector of the PISA wheel. Based on homology with the well-characterized tetrameric structure of the M2 transmembrane peptide (M2-TMP; Nishimura et al. 2002) the resonances at the “top” of the PISA wheel represent residues that face toward the pore and are, therefore, likely to exchange more rapidly than those at the bottom of the wheel representing residues that face the lipid environment. The observation of differential exchange occurring in the sectors of the PISA wheel is supporting evidence for pore formation in the protein preparations. Such evidence from protein samples is important, even though the peptide and protein samples have both been shown to conduct protons (Duff and Ashley 1992; Schroeder et al. 1994) the structural details appear to be somewhat different. The tetrameric peptide bundle has helical tilts of 38°, while the data presented here and in Tian et al. (2002) on the intact protein suggest 25°. Potential reasons for such a difference are numerous, but the protocols for sample preparations are different, and of course, the truncation of the C and N terminal regions are likely to cause structural and functional perturbations as demonstrated for this protein (Tobler et al. 1999) and for the KcsA channel (Doyle et al. 1998; Zhou et al. 2001).
For hydrogen/deuterium exchange from the aqueous pore in samples prepared at pH 8.0, base-catalyzed exchange is not anticipated, because this is a proton channel (albeit probably in the closed state), and there is 20% negatively charged lipid in the surrounding bilayer lowering the effective pH at the surface of the bilayer (Krishtalik and Cramer 1995; Johnson et al. 2003). Therefore, the exchange is likely to be catalyzed by water (Connelly and McIntosh 1998) or by a relayed mechanism, such as the relayed imidic acid exchange mechanism (Eriksson et al. 1995), which has been implicated for exchange in transmembrane peptides (Cotten et al. 1999). Interestingly, even these remaining resonances from the transmembrane helix are exchanged in Figure 2C ▶. The amides nominally facing the lipid environment have been exposed to water. Even for a relayed exchange mechanism (Cotten et al. 1999) water exposure and the ability to stabilize a charge transiently are essential. Either water has escaped from the pore to make occasional excursions into the lipid environment or the helices make occasional rotational excursions about their axes to expose the amides to water that on average face the lipid environment. This latter possibility is supported by numerous cross-linking studies (e.g., Bauer et al. 1999), in which it has been shown that by extending the length of time for cross-linking that many more interhelical sites can be linked. Moreover, stabilizing a charge on the peptide/lipid interface is likely to be energetically very costly, and hence, very rare, and therefore, it is likely that the helices make occasional rotational excursions about their axis.
Secondary structure prediction and a helical wheel diagram (Fig. 3A ▶) suggests that residues 46–62 form a strongly amphipathic helix that could be associated with the membrane’s hydrophobic/hydrophilic interfacial region. Often such helices are illustrated as lying on the surface of the membrane. However, the hydrophobic/hydrophilic interface, as delineated by the fatty acid ester linkages, is buried quite deeply within the membrane. Such a position would protect amide sites from exchange, and the amphipathic nature of the helix would hinder helix rotation, greatly extending the exchange lifetimes of these sites, thereby providing an explanation for the slowly exchanging amides in the 40 to 100 ppm region of the spectra in Figure 2 ▶. This hypothesis is supported by trypsin cleavage that shows that this entire stretch of amino acids is protected from cleavage while solubilized in micelles (Kochendoerfer et al. 1999). It is important to recognize the reproducibility of the resonances in this set of spectra, especially between Figure 2, B and C ▶. This documents the reproducibility of these NMR spectra obtained on small quantities of protein in fluid lipid bilayers.
Figure 3.

Comparison of helical and PISA wheels for a tilt angle of 80° and for the amphipathic protein helical segment of M2 protein (Arg45–Gly62). (A) The hydrophilic residues are displayed in the upper half of the helical wheel (red) and the hydrophobic residues in the lower half (blue). The residues are marked by the positions of the Cα carbons and the rotational orientation is chosen to predict the most likely orientation in the hydrophobic/hydrophilic bilayer interface. (B) Simulated 80° PISA wheel with residue positions predicted from the orientation of the helical wheel, such that the hydrophilic portion of the PISA wheel is shown in red and the hydrophobic portion in blue.
The residual resonances in Figure 2, B and C ▶, are well fit by a PISA wheel characterizing an α-helix with a tilt of 80°. Without resonance assignments we hypothesize that the putative amphipathic α-helix described above is responsible for these resonances that exchange slowly. The helical wheel for this region of the protein is shown in Figure 3A ▶, and the corresponding PISA wheel with predicted resonance positions is given in Figure 3B ▶. The rotational orientation of the helical wheel is defined relative to the Cα carbon at the center of the hydrophilic portion of the wheel, where it is assigned to the rotational origin (0°). For a residue having Cα at the origin, the PISEMA 15N resonance is calculated and assigned as the rotational origin of the PISA wheel (0°). Again, it should be remembered that there is a variability of at least ±10 ppm and ±1 kHz for these resonances as described above, suggesting that resonance frequencies that appear to belong to a hydrophobic portion of the PISA wheel, may be in a hydrophilic portion of the helical surface. Moreover, because the helix does not lie parallel to the bilayer surface, the hydrophilic exposure of the helix will be greater at one end of the helix than at the other. Because the number of hydrophilic residues is greater at the C-terminal end of this helix it is likely that this end is positioned further into the head group region and the N-terminal region more deeply buried in the hydrophobic interstices of the bilayer.
A couple of resonances that are protected from H/D exchange appear to be in the hydrophilic domain as defined by the red region of the PISA wheel in Figure 3B ▶. Potentially these are the Leu46 and Ser50 resonances, both from the most buried end of the amphipathic helix. Moreover, almost all of the hydrophilic residues have amphipathic side chains such as arginine and lysine. These residues can “snorkel” to the polar regions of the bilayer from a position within the hydrophobic domain (de Planque et al. 1999; Strandberg et al. 2002). This further suggests the deep location of the helix within the membrane. The strong electrostatic attractions between positively charged snorkeling residues and negatively charged phosphates of the lipid head groups restricts the motion of an amphipathic helix (Hung et al. 1999), resulting in only small amplitude rotational motions of the helical backbone about the helix axis and resulting in resonances protected from extensive H/D exchange (Fig. 2C ▶).
Shown in Figure 4 ▶ is the PISEMA spectrum of 15N Phe-labeled M2 protein incorporated into uniformly aligned DMPC/DMPG bilayers as for the sample used to achieve the spectrum in Figure 2A ▶. There are five phenylalanine residues in M2, and four of them (47, 48, 54, 55) are located in this amphipathic helix subtending an arc on the helical wheel of just 120° (Fig. 3A ▶). The fifth phenylalanine is in the C terminus at position 91, and is likely to be in a loop. Most of the resonances are close to the 80° PISA wheel defined by the H/D exchange data in Figure 2 ▶, while a single resonance is far removed from this wheel, and we therefore assign it to Phe91. The position of this latter resonance in the PISEMA spectrum suggests that it is in a highly structured region of the protein, because both the chemical shift and dipolar coupling are far from isotropic values. The observation that most of the phenylalanine resonances are consistent with the 80° tilt PISA wheel confirms the assignment of this wheel to the amphipathic helix (residues 45–62). Second, these results qualitatively confirm the orientation of the helical wheel, and hence, the rotational orientation about the PISA wheel.
Figure 4.

PISEMA spectrum of 15N Phe-labeled M2 protein incorporated into uniformly aligned DMPC/DMPG lipid bilayers. The same 80° PISA wheel simulation from Figure 2 ▶ is superimposed on the spectrum.
A model for the backbone of residues Leu26–Gly62 of M2 protein can be developed based on these results (Fig. 5 ▶) and consistent with the previous proteolysis results (Kochendoerfer et al. 1999) and the functional assays of truncated proteins (Tobler et al. 1999). Although this protein functions as a tetramer, and although we have evidence that in our sample preparations the protein is tetrameric and forms an aqueous pore as mentioned earlier, we do not yet have the structural restraints to adequately constrain the tetrameric bundle as we have done for the M2–transmembrane peptide (Nishimura et al. 2002). Therefore, the model in Figure 5 ▶ shows only a monomer of this tetrameric structure. As with the peptide it is possible to state that the tetramer is fourfold symmetric or at least pseudosymmetric, because individual sites do not give rise to multiple resonances. The single resonance for Phe-91 is an example supporting this symmetry statement. Consequently, the model presented in Figure 5 ▶ of one monomer in the symmetric tetrameric bundle has the transmembrane helix tilted with respect to the bilayer normal by 25° and the amphipathic helix tilted by 80°. In addition, the rotational orientation is defined for the amphipathic helix, and this helix is positioned substantially within the hydrophobic region of the bilayer so that H/D exchange is hindered. As suggested by Lear, DeGrado, and coworkers (Kochendoerfer et al. 1999) this amphipathic helix may be involved in stabilizing the tetrameric bundle. However, unlike their model, which suggests that the helix is an extension of the transmembrane helix above the membrane surface and approximately parallel to the bilayer normal, we have shown that this helix is tilted at 80° to the normal and is approximately in the plane of the bilayer. From this position in the bilayer it could form stabilizing interactions with an adjacent helix in the tetrameric bundle through electrostatic interactions.
Figure 5.
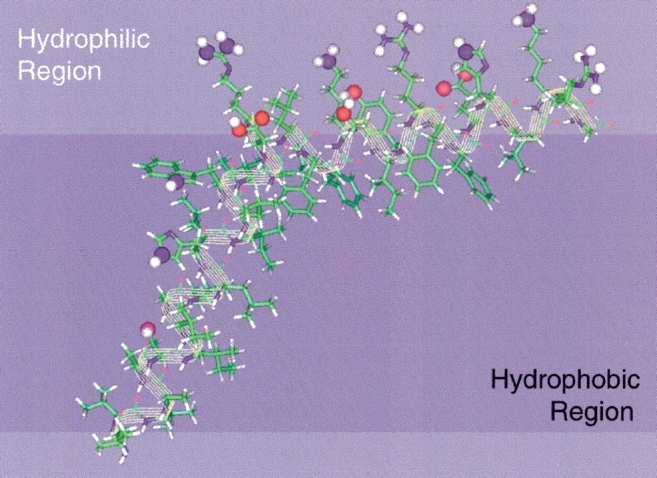
Structural model of the M2 transmembrane region (Leu26∼Asp44) and the amphipathic helix (Arg45–Gly62). The rotational orientation of the transmembrane helix is positioned such that the helix is viewed from the axis of the tetrameric pore; hence, the hydrophilic residues are shown on the exposed face. The orientation of the two helices with respect to each other is arbitrary other than the characterized tilts of the helices with respect to the bilayer normal. The hydrophilic residues of the amphipathic helix are capable of "snorkeling" well into the hydrophilic head group region of the lipid bilayer.
Materials and methods
M2 protein expression and isotope labeling
The expression plasmid pET37 was constructed based on plasmid pET22 and pET15, which were purchased from Novagen Inc. The cDNA encoding M2 protein from influenza A virus strain A/Udorn/72 (H3N2) has been modified by attaching —(CAC)6 for a His6-tag at the 3′ end. The Cys19 and Cys50 residues were mutated to serine, leaving only Cys17 available for forming a disulfide bond, which has been shown to play an important role in stabilizing the M2 tetramer (Sugrue and Hay 1991; Holsinger et al. 1995; Sakaguchi et al. 1997). For uniform 15N labeling, the expression plasmid was transformed into E. coli BL21(DE3) pLysS. Cells from a fresh single colony were used to inoculate 5 mL of LB media. All media contained 34 μg/mL chloramphenicol and 50 μg/mL carbenicillin. The cells were grown in a shaker overnight at 37°C, and each 2-mL culture was used to inoculate 50 mL of defined M9 media with 1 g/L 15NH4Cl (Cambridge Isotope Laboratories) as sole nitrogen source. For 15N Phe-specific labeling, the expression plasmid was transformed into a Phe auxotrophic strain of E. coli BL21(DE3). Similar to the uniform 15N labeling, cells from a fresh single colony were used to inoculate 5 mL of LB media, and a 2 mL overnight culture was used to inoculate 50 mL of LB media having 50 mg/L 15N-labeled Phe (Cambridge Isotope Laboratories). Cells in a 50-mL culture for uniform 15N labeling or 15N Phe labeling were grown at 37°C until an OD600 = 0.6–0.7 was achieved when, IPTG (isopropyl β-D-thiogalactopranoside) was added to a final concentration of 1 mM to induce expression of M2 protein. The cell growth was continued for another 4 h at 37°C. The cells from several 50 mL cultures were pooled and subsequently harvested by centrifugation and stored at −20°C overnight.
M2 protein purification and reconstitution into lipid bilayers
For cell lysis, the frozen cell pellet from 1 L of cell culture was suspended in 100 mL of binding buffer (50 mM Tris.HCl, 150 mM NaCl, pH 8.0, 50 μg/mL Lysozyme) at room temperature for 30 min, sonicated on ice until a very low viscosity of the suspension was achieved. The soluble fraction was removed by centrifugation (10,000 × g, 4°C, 20 min), and the pellet containing inclusion bodies was washed twice by suspending the pellet in 100 mL of the binding buffer without Lysozyme and sonication on ice. The supernatant was removed by centrifugation (10,000 × g, 4°C, 20 min) and the pellet was dissolved in 30 mL of the loading buffer (20 mM Tris.HCl, 50 mM NaCl, pH 8.0, 4 M Urea, 0.5% Octylglucoside [OG]). It was then put on ice for 2 h, transferred to a clean centrifuge tube for centrifugation (15,000 × g, 4°C, 20 min) to precipitate the insoluble cell debris. The supernatant was loaded onto a 10-mL precharged Ni-NTA affinity column with equilibrated resin. Fifty milliliters of loading buffer and 60 mL of wash buffer (20 mM Tris.HCl, 50 mM NaCl, pH 8.0, 30 mM imidazole, 30% glycerol [v/v]) were used sequentially to wash the column. Thirty milliliters of elution buffer (20 mM Tris.HCl, 50 mM NaCl, pH 8.0, 300 mM imidazole, 30% glycerol [v/v]) was used to elute the M2 protein from the column. The protein in the elution buffer was transferred to a dialysis bag with a 5-kD cutoff and dialyzed against 200 mL of buffer (50 mM Tris.HCl, 50 mM NaCl, pH 8.0, 30% glycerol [v/v]) at 4°C using three buffer changes. The contents of the dialysis bag were transferred to a clean tube and 200 mM OG was added from a stock solution to achieve a final concentration of 30 mM. DMPC/DMPG liposomes were prepared at a molar ratio of 4 : 1 and were added to the M2 protein solution in 30 mM OG achieving a lipid/protein molar ratio of 200 : 1. The solution was transferred into a dialysis bag with a 5-kD cutoff and dialyzed against 1000 mL of 50 mM Na2HPO4/NaH2PO4, pH 8.0 buffer with three buffer changes, then dialyzed against 10 mM Na2HPO4/NaH2PO4, pH 8.0 buffer with two more buffer changes. Proteoliposomes were pelleted by ultracentrifugation from the dialyzed solution (196,000 × g, 90 min) in a 70 Ti rotor (Beckman Instruments). The pellet was used for aligned sample preparation by suspending the 120-mg pellet in 2 mL HPLC grade H2O. The purity of the reconstituted M2 protein in DMPC/DMPG liposomes was checked using electrophoresis experiments as described previously (Tian et al. 2002).
PISEMA solid-state NMR experiments of M2 protein in lipid bilayers
The proteoliposomes of M2 protein in DMPC/DMPG were suspended in a final volume of 3 mL and were briefly sonicated in a bath sonicator, frozen in liquid nitrogen, and thawed at room temperature. After three sonication freeze–thaw cycles, aliquots of the proteoliposomes were spread onto the surface of 50 glass slides (dimensions 5.8 × 11.0 × 0.07 mm, Paul Marienfeld GmbH) with 60-μL aliquots on each slide. H2O was evaporated from the sample on glass slides by exposure of the glass slides to ambient room temperature for 12 h. The glass slides were then stacked into a glass tube (inner dimensions 6.0 × 6.0 × 15.0 mm, Wilmad) and equilibrated for 24 h at 43°C in a chamber containing a saturated solution of K2SO4, which provides an atmosphere of 93% relative humidity. The glass tube was then sealed using epoxy, incubated for 48 h at 43°C, and equilibrated for another 24 h at room temperature prior to insertion into the NMR probe. The solid-state NMR spectra were obtained at 30°C on a Chemagnetics CMX400 spectrometer, with a wide-bore 400/89 Oxford magnet (Oxford Instrument), using a home-built double resonance (1H/15N) probe with a solenoid coil having a square cross-section, 6 mm on a side. The cross-polarization (CP) Hahn spin-echo pulse sequence was used for 15N power match calibration for the 15N chemical shift /1H-15N dipolar coupling correlation PISEMA experiments. 15N chemical shifts were referenced to a saturated solution of 15NH4NO3 at 0 ppm. PISEMA spectra of the uniform 15N labeled and 15N Phe labeled M2 protein were obtained with a 1-msec contact time, 5.5 μsec 1H 90° pulse width, 60 kHz 1H decoupling power and the 2D data were acquired with 4000 accumulated FIDs using 256 complex data points in the t2 time domain and 18 real points in the t1 time domain. Data were processed with 200-Hz line broadening in the first dimension and a 90° sinebell window function in the second dimension using the spinsight software package and MATLAB scripts.
Hydrogen–deuterium exchange of uniform 15N-labeled M2 protein in aligned DMPC/DMPG lipid bilayers
Deuterium exchange of amide protons was accomplished by exposing a previously H2O hydrated aligned sample to a saturated D2O atmosphere at 43°C for 8 h (Cotten et al. 1999). The sample was resealed using epoxy and placed into the NMR probe for PISEMA data acquisition. After finishing the PISEMA experiment for the first round of H/D exchange, more extensive hydrogen–deuterium exchange was performed by exposing the sample to a saturated D2O atmosphere at 43°C for an additional 16 h and 20 μL of D2O was added to the sample. The sample was then resealed and placed back into the NMR probe for another PISEMA experiment.
PISA wheel simulation and tilt angle analysis
The magnetic field Bo direction was set parallel to the lipid bilayer normal. Averaged values of the chemical shift tensors (σ11 = 31.3 ppm, σ22 = 55.2 pm, σ33 = 201.8 ppm) from experimental data were used, as well as a motionally averaged value for the dipolar coupling constant of 10.375 kHz (Wang et al. 2000). A typical angle between the σ33 tensor element and ν// of the dipolar coupling of 17° was used, as experimentally confirmed (Wang et al. 2000). PISA wheels of a protein helix were calculated by rotating the helix about its axis while calculating the anisotropic dipolar and chemical shift observables. A series of simulated PISA wheels for different tilt angles of the helix were overlaid with the resonances in the PISEMA spectra of uniform 15N-labeled M2 protein before and after hydrogen–deuterium exchange as well as the 15N Phe-labeled M2 protein to determine the tilt angle of the transmembrane helix and interfacial amphipathic helix of the M2 protein with respect to the lipid bilayer normal. For a given tilt angle, the rotational orientation position on the PISA wheel was noted relative to a fixed position in the protein helical wheel. This fixed position is defined as the Cα atom at the top of the helix, more quantitatively located in the Bo/helix axis plane. Protein helical wheels were drawn in rotational angle steps of 100° per-residue from the primary sequence.
Acknowledgments
We gratefully acknowledge discussions with Dr. Riqiang Fu, Dr. Zhehong Gan on PISEMA experiments and MATLAB data processing scripts. Additionally, we thank Ashley Blue for maintenance of the CMX 400 MHz NMR spectrometer. This work was supported by the National Science Foundation (NSF) MCB 02-35774 (T.A.C.) and Public Health Services Research Grant R37-AI-70201(R.A.L.). R.A.L. is an Investigator of the Howard Hughes Medical Institute. The spectroscopy was performed at the National High Magnetic Field Laboratory supported by NSF Cooperative Agreement (DMR 0084173) and the State of Florida.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DMPC, dimyristoylphosphatidylcholine
DMPG, dimyristoylphosphatidylglycerol
PISA wheel, polar index slant angle wheel
NMR, nuclear magnetic resonance
M2-TMP, M2 transmembrane peptide
PISEMA, polarization inversion spin exchange at the magic angle
IPTG, isopropyl β-D-thiogalactopranoside
OG, β-octyl-glucoside
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03168503.
References
- Altenbach, C., Cai, K., Klein-Seetharaman, J., Khorana, H.G., and Hubbell, W.L. 2001. Structure and function in rhodopsin: Mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306–319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry 40 15483–15492. [DOI] [PubMed] [Google Scholar]
- Bauer, C.M., Pinto, L.H., Cross, T.A., and Lamb, R.A. 1999. The influenza virus M2 ion channel protein: Probing the structure of the transmembrane domain in intact cells by using engineered disulfide cross-linking. Virology 254 196–209. [DOI] [PubMed] [Google Scholar]
- Bechinger, B., Zasloff, M., and Opella, S.J. 1998. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 74 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhmakov, I.V., Geraghty, F.M., Ogden, D.C., Hayhurst, A., Antoniou, M., and Hay, A.J. 1996. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 494 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, J.J., Kaufman, J.D., Stahl, S.J., Wingfield, P.T., and Bax, A. 2002. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J. Am. Chem. Soc. 124 2450–2451. [DOI] [PubMed] [Google Scholar]
- Connelly, G.P. and McIntosh, L.P. 1998. Characterization of a buried neutral histidine in Bacillus circulans xylanase: Internal dynamics and interaction with a bound water molecule. Biochemistry 37 1810–1818. [DOI] [PubMed] [Google Scholar]
- Cortes, D.M., Cuello, L.G., and Perozo, E. 2001. Molecular architecture of full-length KcsA: Role of cytoplasmic domains in ion permeation and activation gating. J. Gen. Physiol. 117 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten, M., Fu, R., and Cross, T.A. 1999. Solid-state NMR and hydrogen-deuterium exchange in a bilayer-solubilized peptide: Structural and mechanistic implications. Biophys. J. 76 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, T.A. and Quine, J.R. 2000. Protein structure in anisotropic environments: Development of orientational constrains. Concepts Magn. Reson. 12 55–70. [Google Scholar]
- de Planque, M.R., Kruijtzer, J.A., Liskamp, R.M., Marsh, D., Greathouse, D.V., Koeppe 2nd, R.E., de Kruijff, B., and Killian, J.A. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 274 20839–20846. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., Morais Cabral, J., Pfuetzner, R.A., Kuo, A., Gulbis, J.M., Cohen, S.L., Chait, B.T., and MacKinnon, R. 1998. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 280 69–77. [DOI] [PubMed] [Google Scholar]
- Duff, K.C. and Ashley, R.H. 1992. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology 190 485–489. [DOI] [PubMed] [Google Scholar]
- Eriksson, M.A., Hard, T., and Nilsson, L. 1995. On the pH dependence of amide proton exchange rates in proteins. Biophys. J. 69 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, R. and Cross, T.A. 1999. Solid-state nuclear magnetic resonance investigation of protein and polypeptide structure. Annu. Rev. Biophys. Biomol. Struct. 28 235–268. [DOI] [PubMed] [Google Scholar]
- Grambas, S., Bennett, M.S., and Hay, A.J. 1992. Influence of amantadine resistance mutations on the pH regulatory function of the M2 protein of influenza A viruses. Virology 191 541–549. [DOI] [PubMed] [Google Scholar]
- Han, X., Bushweller, J.H., Cafiso, D.S., and Tamm, L.K. 2001. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8 715–720. [DOI] [PubMed] [Google Scholar]
- Holsinger, L.J. and Lamb, R.A. 1991. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology 183 32–43. [DOI] [PubMed] [Google Scholar]
- Holsinger, L.J., Nichani, D., Pinto, L.H., and Lamb, R.A. 1994. Influenza A virus M2 ion channel protein: A structure–function analysis. J. Virol. 68 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger, L.J., Shaughnessy, M.A., Micko, A., Pinto, L.H., and Lamb, R.A. 1995. Analysis of the posttranslational modifications of the influenza virus M2 protein. J. Virol. 69 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W., Lee, K.C., and Cross, T.A. 1993. Tryptophans in membrane proteins: Indole ring orientations and functional implications in the gramicidin channel. Biochemistry 32 7035–7047. [DOI] [PubMed] [Google Scholar]
- Huang, H.W. 2000. Action of antimicrobial peptides: Two-state model. Biochemistry 39 8347–8352. [DOI] [PubMed] [Google Scholar]
- Hung, S.C., Wang, W., Chan, S.I., and Chen, H.M. 1999. Membrane lysis by the antibacterial peptides cecropins B1 and B3: A spin-label electron spin resonance study on phospholipid bilayers. Biophys. J. 77 3120–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, P.M., Choy, W.Y., Lo, E.I., Chen, L., Forman-Kay, J.D., Raetz, C.R., Prive, G.G., Bishop, R.E., and Kay, L.E. 2002. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. 99 13560–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.E., Xie, M., Singh, L.M., Edge, R., and Cornell, R.B. 2003. Both acidic and basic amino acids in an amphitropic enzyme, CTP:Phosphocholine Cytidylyltransferase, dictate its selectivity for anionic membranes. J. Biol. Chem. 278 514–522. [DOI] [PubMed] [Google Scholar]
- Ketchem, R., Roux, B., and Cross, T.A. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure 5 1655–1669. [DOI] [PubMed] [Google Scholar]
- Ketchem, R.R., Hu, W., and Cross, T.A. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 261 1457–1460. [DOI] [PubMed] [Google Scholar]
- Kim, S. and Cross, T.A. 2002. Uniformity, ideality, and hydrogen bonds in transmembrane α-helices. Biophys. J. 83 2084–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendoerfer, G.G., Salom, D., Lear, J.D., Wilk-Orescan, R., Kent, S.B., and DeGrado, W.F. 1999. Total chemical synthesis of the integral membrane protein influenza A virus M2: role of its C-terminal domain in tetramer assembly. Biochemistry 38 11905–11913. [DOI] [PubMed] [Google Scholar]
- Krishna, A.G., Menon, S.T., Terry, T.J., and Sakmar, T.P. 2002. Evidence that helix 8 of rhodopsin acts as a membrane-dependent conformational switch. Biochemistry 41 8298–8309. [DOI] [PubMed] [Google Scholar]
- Krishtalik, L.I. and Cramer, W.A. 1995. On the physical basis for the cis-positive rule describing protein orientation in biological membranes. FEBS Lett. 369 140–143. [DOI] [PubMed] [Google Scholar]
- Kuo, A., Gulbis, J.M., Antcliff, J.F., Rahman, T., Lowe, E.D., Zimmer, J., Cuthbertson, J., Ashcroft, F.M., Ezaki, T., and Doyle, D.A. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300 1922–1926. [DOI] [PubMed] [Google Scholar]
- Lamb, R.A., Zebedee, S.L., and Richardson, C.D. 1985. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 40 627–633. [DOI] [PubMed] [Google Scholar]
- Mai, W., Hu, W., Wang, C., and Cross, T.A. 1993. Orientational constraints as three-dimensional structural constraints from chemical shift anisotropy: The polypeptide backbone of gramicidin A in a lipid bilayer. Protein Sci. 2 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi, F.M. and Opella, S.J. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi, F.M., Opella, S.J., Juvvadi, P., and Merrifield, R.B. 1999. Orientation of cecropin A helices in phospholipid bilayers determined by solid-state NMR spectroscopy. Biophys. J. 77 3152–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, K., Kim, S., Zhang, L., and Cross, T.A. 2002. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry 41 13170–13177. [DOI] [PubMed] [Google Scholar]
- Oh, K.J., Zhan, H., Cui, C., Hideg, K., Collier, R.J., and Hubbell, W.L. 1996. Organization of diphtheria toxin T domain in bilayers: A site-directed spin labeling study. Science 273 810–812. [DOI] [PubMed] [Google Scholar]
- Oh, K.J., Zhan, H., Cui, C., Altenbach, C., Hubbell, W.L., and Collier, R.J. 1999. Conformation of the diphtheria toxin T domain in membranes: A site-directed spin-labeling study of the TH8 helix and TL5 loop. Biochemistry 38 10336–10343. [DOI] [PubMed] [Google Scholar]
- Palczewski, K., Kumasaka, T., Hori, T., Behnke, C.A., Motoshima, H., Fox, B.A., Le Trong, I., Teller, D.C., Okada, T., Stenkamp, R.E., et al. 2000. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289 739–745. [DOI] [PubMed] [Google Scholar]
- Pinto, L.H., Dieckmann, G.R., Gandhi, C.S., Papworth, C.G., Braman, J., Shaughnessy, M.A., Lear, J.D., Lamb, R.A., and DeGrado, W.F. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. 94 11301–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quine, J.R. and Cross, T.A. 2000. Protein structure in anisotropic environments: unique structural fold from orientational constrains. Concepts Magn. Reson. 12 71–82. [Google Scholar]
- Sakaguchi, T., Tu, Q., Pinto, L.H., and Lamb, R.A. 1997. The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer. Proc. Natl. Acad. Sci. 94 5000–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder, C., Ford, C.M., Wharton, S.A., and Hay, A.J. 1994. Functional reconstitution in lipid vesicles of influenza virus M2 protein expressed by baculovirus: Evidence for proton transfer activity. J. Gen. Virol. 75 3477–3484. [DOI] [PubMed] [Google Scholar]
- Strandberg, E., Morein, S., Rijkers, D.T., Liskamp, R.M., van der Wel, P.C., and Killian, J.A. 2002. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry 41 7190–7198. [DOI] [PubMed] [Google Scholar]
- Sugrue, R.J. and Hay, A.J. 1991. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology 180 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, Q., Iqbal, M., and Cross, T.A. 1992. Determination of the 13C chemical shift and 14N electric field gradient tensor orientations with respect to the molecular frame in a polypeptide. J. Am. Chem. Soc. 114 5312–5321. [Google Scholar]
- Tian, C., Tobler, K., Lamb, R.A., Pinto, L.H., and Cross, T.A. 2002. Expression and initial structural insights from solid-state NMR of the M2 proton channel from influenza A virus. Biochemistry 41 11294–11300. [DOI] [PubMed] [Google Scholar]
- Tobler, K., Kelly, M.L., Pinto, L.H., and Lamb, R.A. 1999. Effect of cytoplasmic tail truncations on the activity of the M(2) ion channel of influenza A virus. J. Virol. 73 9695–9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosteson, M.T., Pinto, L.H., Holsinger, L.J., and Lamb, R.A. 1994. Reconstitution of the influenza virus M2 ion channel in lipid bilayers. J. Membr. Biol. 142 117–126. [DOI] [PubMed] [Google Scholar]
- Wang, C., Lamb, R.A., and Pinto, L.H. 1994. Direct measurement of the influenza A virus M2 protein ion channel activity in mammalian cells. Virology 205 133–140. [DOI] [PubMed] [Google Scholar]
- Wang, J., Denny, J., Tian, C., Kim, S., Mo, Y., Kovacs, F., Song, Z., Nishimura, K., Gan, Z., Fu, R., et al. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144 162–167. [DOI] [PubMed] [Google Scholar]
- Wang, J., Kim, S., Kovacs, F., and Cross, T.A. 2001. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci. 10 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener, M.C. and White, S.H. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L., Harroun, T.A., Weiss, T.M., Ding, L., and Huang, H.W. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov, S.D., Lindeberg, M., and Cramer, W.A. 1999. Kinetic description of structural changes linked to membrane import of the colicin E1 channel protein. Biochemistry 38 11325–11332. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Morais-Cabral, J.H., Kaufman, A., and MacKinnon, R. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414 43–48. [DOI] [PubMed] [Google Scholar]